
Abstract
Genetic engineering provides new opportunities for improving economically important traits in sugarcane cultivars. In this study, an efficient Agrobacterium-mediated transformation system that uses the bar gene (a herbicide resistance gene that is used in conjunction with the herbicide Basta) as a selection marker was developed. Using this transformation selection system, all of the resistant plants after selection were nearly 100% polymerase chain reaction (PCR) detection positive and showed herbicide resistance. Each gram of sugarcane calli used for transformation produced approximately 12 transgenic lines. It took approximately 4 months to generate transgenic plants that measured 10 cm in height for greenhouse transplantation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sugarcane is one of the most important commercial crops and is an important bio- energy crop. Sugarcane is cultivated in large areas of Indonesia and other tropical and subtropical countries, and it contributes to 60 to 70% of the annual sugar consumption worldwide (Manickavasagam et al. 2004; Suprasanna et al. 2011). In addition, it is used as raw material for ethanol production in Brazil, which is also the largest producer of cane sugar in the world (Snyman 2004). Similar to other food crops, the production of sugarcane is affected by biotic and abiotic stresses, such as disease, insects, and drought (Viswanathan and Rao 2011; Srikanth et al. 2011; Priji and Hemaprabha 2015). Most modern sugarcane cultivars are inter-specific hybrids and have complex genetic characteristics and low fertility, thus rendering genetic improvement through traditional breeding difficult. These cultivars are primary candidates for improvement through genetic engineering (Ingelbrecht et al. 1999). Transformation and transgene expression are key biotechnologies for sugarcane improvement (Dong et al. 2014). Electroporation (Arencibia et al. 1995), particle bombardment (Falco et al. 2000), and Agrobacterium-mediated transformation (Enríquez-Obregón et al. 1998) are the three main methods of successful sugarcane transformation. Particle bombardment has been used in more than 60% of the existing sugarcane transformation studies (Arvinth et al. 2010; Basnayake et al. 2011; Zhu et al. 2011; Rani et al. 2012; Xiong et al. 2012). Direct transformation procedures, such as particle bombardment and electroporation, have several disadvantages, including poor reproducibility, variable transgene copy number, and instability of the gene construct in the new host (Santosa et al. 2004). Agrobacterium-mediated transformation, by contrast, has advantages over other gene-delivery technologies. Successful sugarcane transformation via Agrobacterium has been reported but exhibited low efficiency (Zhangsun et al. 2007; Joyce et al. 2010; Mayavan et al. 2013). However, Dong et al. (2014) achieved high-efficiency Agrobacterium-mediated transformation for sugarcane. Desiccation treatment during co-cultivation played a critical role in improving sugarcane transformation efficiency for sugarcane transformation efficiency using Agrobacterium. However, the selection system that they used for the transformation was the pmi/mannose system. The pmi gene was thought to be a safety gene, but it was out of use after transformation. Several studies reported transgene silencing in sugarcane (Wei et al. 2003; Mudge et al. 2009). Some studies also reported the use of antibiotic-resistant genes, such as hptII and nptII (Zhangsun et al. 2007; Joyce et al. 2010; Kalunke et al. 2009), but these selection markers were also useless after transformation.
In this study, an efficient Agrobacterium-mediated transformation system that uses the bar gene as a selection agent for sugarcane was developed for use on an industrial scale. A bar/Basta selection system was successfully used in this transformation system. The bar gene is a herbicide resistance gene, and Basta is a herbicide. By using this transformation system, the detection rate of resistant plants following transformation was almost 100%. Each gram of sugarcane calli used during the transformation produced more than 12 transgenic lines. It took less than 4 months from callus infection to produce greenhouse-ready transgenic plants that measured 10 cm in height.
Material and Methods
Media Composition
-
M1: MS +30 g/L sucrose + 8 g/L agar +2.0 mg/L 2,4-D
-
M2: MS +30 g/L sucrose + 8 g/L agar +2.0 mg/L 6-BA
-
M3: MS +30 g/L sucrose + 8 g/L agar
-
M-Initiating: 1/5 strength MS medium + 30 g/L sucrose + 30 g/L glucose + 100 mM acetosyringone
-
M-Resting: M1+300 mg/L Timentin
-
M-Selection: M1+ 300 mg/L Timentin + 2.0 mg/L glufosinate-ammonium
-
M-Regeneration: M2+300 mg/L Timentin + 2.0 mg/L glufosinate-ammonium
-
M-Elongation: M3+300 mg/L Timentin + 2.0 mg/L glufosinate-ammonium
-
M-Rooting: M3+1 mg/L NAA+300 mg/L Timentin + 2.0 mg/L glufosinate-ammonium
Plant Material and Callus Induction
Plant material of the sugarcane variety ROC22 was obtained from a field in Hainan, China. Tops and tillers containing the immature leaf whorl were used as source material for embryogenic callus induction, which was initiated within 24 h of cutting and collection. Transverse sections of immature sugarcane leaf whorls were prepared essentially as described by Bower and Brich (1992). Transverse, 1-mm-thick sections were obtained from just above the meristem and placed on callus induction medium (M1) (Murashige and Skoog 1962). Callus cultures were maintained in the dark at 28 °C and subcultured on fresh medium every 2 weeks, for a total culture duration of approximately 45 days. Light yellow, compact calli were selected and fragmented before transformation.
Determination of Minimum Inhibitory Concentration of Basta
Transverse sections that were prepared as above for callus induction were inoculated in M1 medium modified with different concentrations (0, 0.5, 1.0, 1.5, 2.0, 2.5, and 3.0 mg/L) of Basta (200 g/L glufosinate-ammonium, Bayer, Germany) (Mayavan et al. 2013). The cultures were maintained in the dark at 28 °C for 14 days and then transferred to regeneration medium (M2) modified with different concentrations (0, 0.5, 1.0, 1.5, 2.0, 2.5 and 3.0 mg/L) of Basta. The Basta concentration with an inhibitory effect on secondary leaf development was chosen as the minimum inhibitory concentration and adopted for transformation selection. Every treatment contained 48 pieces of transverse sections in duplicates of five.
Binary Vectors and Agrobacterium Strain
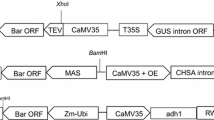
All of the binary vectors comprised a range of target genes and a selectable marker gene of the bar gene. The bar gene promoted by CaMV 35S promoter was introduced between the two XhoI sites of pCAMBIA3300. The binary vector pCAMBIA3300 contained the npt II gene for bacterial selection. The reporter gene green fluorescent protein gene (GFP) promoted by Ubi1 was introduced between the BamHI and SacI sites of pCAMBIA3300 (Fig. 1). The recombinant binary vectors were mobilized into Agrobacterium EHA105 via freeze-thawing with liquid nitrogen. The Agrobacterium strain was cultured on YEP (yeast extract 10 g/L, peptone 10 g/L, NaCl 5 g/L) medium containing the appropriate antibiotics (kanamycin 50 mg/L, streptomycin 50 mg/L, and rifampicin 10 mg/L). Five of the other vectors had the same construction as the bar-GFP vector, but the GFP gene was replaced by five other genes (synthetic Medicago truncatula defensins 4 (AD2), native Medicago truncatula defensins 4 with KDEL (AD3), synthetic Medicago truncatula defensins 4 with KDEL (AD4), inorganic pyrophosphatase gene (Ppase), sucrose:sucrose 1-fructosyl transferase gene (SST)).

Schematic representation of the binary vector used in sugarcane transformation. Bar gene promoted by CaMV 35S promoter and GFP gene or five other target genes (AD2, AD3, AD4, SST, Ppase) promoted by Ubi 1 promoter. CaMV 35S P: cauliflower mosaic virus 35S promoter; 35S poly A: cauliflower mosaic virus 35S poly A terminator; nos ter: nopaline synthase terminator; Ubi 1 P: maize Ubi 1 promoter
Initiation of Agrobacterium
Agrobacterium EHA105 cultures that harbored a vector comprising the visual gene GFP (Ubi1 promoter with an intron) and the selection marker gene bar (CaMV 35S promoter) were streaked on YEP medium containing antibiotics, as described above, and grown at 28 °C for 2 days. A single colony was selected and sub-cultured overnight on fresh YEP medium at 28 °C. Bacteria were collected after centrifugation, resuspended in a starter culture of liquid initiating medium (M-Initiating) and diluted to an optical density of approximately 0.6 at 600 nm. The starter culture was vortexed at 100 rpm for 2 h at 28 °C before infection.
Infection and co-Cultivation
The calli used for transformation were weighed before infection, placed in Petri dishes, air-dried for 1 h on a clean laboratory bench, and then transferred to an Erlenmeyer flask. All of the calli were heat-shocked in an incubator at 45 °C for 5 min in sufficient pre-warmed (45 °C) M-Initiating medium without Agrobacterium to cover the calli. The M-initiating medium was pipetted out, and sufficient infection medium with Agrobacterium starter culture was added and shaken gently (less than 100 revolutions) for 10 min at 28 °C. The calli/Agrobacterium mixture was vacuumed for 5 min at −27.5 mmHg of vacuum and then sonicated for 2 min in an ultrasonic cleaner. The mixture was shaken gently (fewer than 100 revolutions) for 10 min again in the dark. The Agrobacterium suspension was then pipetted out, and the calli were transferred to a Petri dish, blotted dry with filter paper to remove excess Agrobacterium suspension, and air-dried for approximately 30 min on the clean bench. The calli were then transferred to a new empty Petri dish, sealed with paraffin film, and then incubated in the dark at 22 °C for 3 days (Dong et al. 2014).
Selection of Infected Calli
After co-cultivation, the calli were transferred to resting medium (M-Resting) and stored in the dark at 28 °C for 7 days. Subsequently, all calli were transferred to selection medium (M-Selection) for 30 days in the dark at 28 °C. The selection medium was renewed once every 2 weeks during the 30 days of selection. Each Petri dish contained approximately 10 pieces of callus.
Regeneration, Elongation, and Rooting
At least two or three calli continued to grow well in each Petri dish after 30 days of selection. These resistant calli were transferred to regeneration medium (M-Regeneration) and cultivated in an illuminated incubator for 14 days at 30 °C ± 1 °C in 14 h of light each day. Transgenic healthy calli sprouted green buds during these 14 d. The green buds were then transferred to elongation medium (M-Elongation) and cultivated in an illuminated incubator for 20 days at 30 °C ± 1 °C in 14 h of light each day. After 20 days of elongation, buds grew to approximately 3 to 4 cm. A single shoot from each bud was transferred to rooting medium (M-Rooting) and cultivated in an illuminated incubator for 20 days at 30 °C ± 1 °C in 14 h of light each day. These shoots then grew to 6 to 7 cm in height and were sampled for molecular analysis and transferred to soil.
Visualization of GFP Expression
The resistant calli, shoots, and leaves were observed for GFP gene expression using fluorescence microscope equipment. A GFP 510-nm long-pass emission filter with a 480/40-nm excitation filter was used. Fluorescence images were captured with a Canon digital camera.
Total Genomic DNA Extraction and PCR Assay
A PCR assay was performed to detect the integration of the bar gene and to confirm the absence of Agrobacterium contamination in the transformed lines as the resistant plants grew to 8–10 cm in height. The PCR reactions were conducted using the primers specific to the bar, GFP, and VirG genes (Table 1). Vector plasmid was used as a positive control, and the total genomic DNA extracted from wild-type plants was used as a negative control.
Detection of PAT/bar Protein by QuickStix Strips
All of the resistant plants were sampled and tested by QuickStix strips (QuickStix Kit for Libertylink (bar) Cotton Leaf & Seed, Envirologix, USA). Approximately 10 mg of leaf tissue was taken from each resistant plant and pushed into the tapered bottom of a 1.5-ml Eppendorf tube. Wild-type plant tissue samples served as a negative control. A pestle was inserted into the tube, and the tissue was ground by rotating the pestle against the sides of the tube with twisting motions. This process was continued for 20–30 s or until the leaf tissue was well grounded, at which point 0.5 ml of extraction buffer was added. The grinding step was repeated to mix the tissue thoroughly with extraction buffer. The pestle was discarded, and QuickStix strips were inserted into the extraction tube. The strips were allowed to develop for 10 min before final assay interpretations were made.
Southern Blot Analysis
Southern blot analysis was performed to determine the integration and copy number of the bar gene. Approximately 10 μg of total genomic DNA that was extracted from transformed sugarcane plants was digested with EcoRI restriction enzyme, separated by electrophoresis agarose gel, and transferred onto a Hybond N+ membrane. A 558-bp digoxin-labeled DNA fragment corresponding to the bar gene was used as a probe for hybridization according to the instruction manual (DIG High Prime DNA Labeling and Detection Starter Kit I, Roche). The results were documented with a Canon color digital camera system.
Herbicide Sensitivity Assay
Herbicide (Basta, 200 g/L glufosinate-ammonium, YONON Biosciences Co., Ltd., Zhejiang, China) sensitivity assays were performed on transformed sugarcane plants containing single bar gene insertions under greenhouse conditions. According to the instruction manual for this herbicide, 0.8 g/L to 2.0 g/L was used in the sugarcane fields. Hence, 2.0 g/L was chosen to screen the resistance of the transformed lines in the greenhouse.
Estimated Transformation Efficiency
Cultivar ROC22 and six vectors with the bar selection marker gene with different target genes (GFP, Ppase, SST, AD2, AD3, AD4) were used to test the efficiency of the transformation system. The transformation process and medium used were as described above. Selection Efficiency (SE) and Transformation Efficiency (TE) formulas were used to estimate the transformation efficiency. SE = number of bar gene-PCR–positive shoots / number of resistant shoots obtained after rooting selection, or number of bar gene-PCR–positive shoots / weight (gram) of calli used for transformation. TE = number of both bar gene and target gene-PCR–positive shoots / number of resistant shoots or number of both bar gene and target gene-PCR–positive shoots / weight (gram) of the calli used for transformation.
Results
Plant Material and Callus Induction
ROC22 represents the largest acreage of sugarcane cultivars in China and is affected by serious diseases and insect pests. Therefore, considerable genetic improvement has been achieved in this cultivar. Young embryogenic calli (Fig. 2) of this cultivar were chosen as explants for genetic transformation because of their year-round availability, ease of handling, and efficiency of transformant selection. After 45 days of induction, light yellow, hard and compact calli (Fig. 2c) were obtained that were suitable for transformation.

Embryogenic callus induction of cultivar ROC22 for transformation. a. 15 days after induction: no embryogenic cells. b. 30 days after induction: tiny outgrowths of embryogenic callus. c. 45 days after induction: embryogenic callus suitable for transformation
Determination of Minimum Inhibitory Concentration of Basta
Basta has been identified as an efficient selective agent in monocot species (Fromm et al. 1990). At a concentration of 2.0 mg/L, Basta completely inhibited callus growth and secondary leaf formation (Fig. 3). Hence, 2.0 mg/L of Basta was chosen as the minimum inhibitory concentration to select transformed calli and plants.

Minimum inhibitory concentration of Basta. a. No-Basta medium; b. 0.5 mg/L Basta; c. 1.0 mg/L Basta; d. 1.5 mg/L Basta; e. 2.0 mg/L Basta; f. 2.5 mg/L Basta. All of the transverse sections grew well, and the secondary leaf emerged quickly after regeneration on 0 mg/L Basta medium. Callus growth and bud formation were inhibited in direct proportion to the increases in the concentration of Basta from 0.5 mg/L to 1.5 mg/L. At 2.0 mg/L or above, callus growth and secondary leaf formation were completely inhibited
Post-Transformation and Selection
To decrease the percentage of chimeras and escapes in plant transformation, stringent selection pressure must be employed (Darbani et al. 2007). Hence, we added 2.0 mg/L of Basta to all of the transformation media used, including the callus selection, regeneration, elongation, and rooting media.
After 30 days of callus selection in the dark, 2–3 or more pieces of new calli sprouted and showed significant resistance to 2.0 mg/L of Basta in each Petri dish (Fig. 4a). These resistant calli regenerated quickly and stayed healthy on the regeneration medium supplemented with 2.0 mg/L of Basta (Fig. 4b). All of the regenerated buds were transferred to elongation medium (Fig. 4c). After 20 days of elongation, a single shoot was selected from each of the buds and transferred to rooting medium. Transformed shoots grew fast and germinated healthy roots (Fig. 4d).

Selection of transformed sugarcane plants. a. After 30 days of dark selection, some resistant calli were healthy on the selection medium; b. after 14 days of regeneration, cultivation-resistant calli emerged quickly and were healthy; c. after 20 days of elongation, cultivation-resistant, dark-green shoots grew quickly; d. after 20 days of rooting cultivation, several branched roots emerged; the resistant shoots elongated to 10-cm in height and were sampled for DNA extraction and PCR analysis. All media used were supplemented with 2.0 mg/L of Basta
Visual Observation of GFP Gene Expression
The infected, resistant calli and resistant shoots were evaluated for GFP gene expression under a microscope during the transformation process (Fig. 5a–c). Roots, transected sections of shoot, and leaves of 1-y-old transformed plants from the greenhouse were evaluated for GFP gene expression (Fig. 5d–f). The GFP gene expression level was tested in all tissue types. Bright green fluorescence was observed in the transformed calli, leaves, and roots. Roots, transverse sections, and leaves of the wild type showed red auto fluorescence.

Observation of GFP gene expression. a. Infected callus after 3 days of co-cultivation showing transient GFP expression; b. resistant callus after 30 days of callus selection showing stable GFP expression; c. resistant buds after 14 days of regeneration showing stable GFP expression; d. root of transformed plants from greenhouse showing stable GFP expression; e. hand-cut section of transformed plants from greenhouse showing stable GFP expression; f. leaf of transformed plants from greenhouse showing stable GFP expression
PCR Assay of Potential Transformed Shoots
All resistant shoots were sampled and evaluated for bar gene and GFP gene integration by PCR following rooting cultivation. Bar gene–specific primers amplified a 558 bp fragment in the vector plasmid and resistant plants (Fig. 6a), and the GFP gene-specific primers amplified a 249-bp fragment in the vector plasmid and resistant shoots (Fig. 6b). Both the bar gene and the GFP gene were negative for wild-type sugarcane genomic DNA. Figure 6 shows 23 resistant plants, with only one (Fig. 6, lane 11) that was PCR negative for both the bar gene and the GFP gene.

PCR assay of transformed sugarcane plants. Lanes 1–23 contained genomic DNA of resistant plants; CK-: wild-type sugarcane genomic DNA; CK+: vector plasmid DNA. a. PCR amplification of bar gene from the genomic DNA of resistant plants; b. PCR amplification of the GFP gene from the genomic DNA of resistant plants; c. identification of Agrobacterium contamination from resistant plants via VirG gene amplification
To determine the absence of Agrobacterium in the resistant plants, VirG gene PCR detection was done using VirG gene-specific primers. No amplification was found in the resistant plants (Fig. 6c), which suggested that the resistant sugarcane plants from the rooting medium were free from Agrobacterium. This result also indicates that the PCR-amplified fragments emerged from the integrated bar gene and GFP gene in the plant genome. Therefore, only the PCR-positive resistant plants were transplanted to the greenhouse. The PCR-negative plants were discarded.
Detection of PAT/bar Protein by Quick Stix Strips
All resistant plants were sampled and tested with QuickStix strips (QuickStix Kit for Libertylink (bar) Cotton Leaf & Seed, Envirologix, USA). If the sample extraction contained the PAT/bar protein, a test line would develop on the membrane strip between the control line and the protective arrow tape. As Fig. 7 shows, most of the resistant plant sample extractions developed a test line; this QuickStix strip testing result was in complete concordance with the bar gene PCR analysis results. Thus, if the resistant plant was bar gene PCR-positive, the QuickStix strip testing was also positive.

Detection of PAT/bar protein by QuickStix strips. 1–23 resistant plant leaf tissue extraction samples; CK- wild-type leaf tissue extraction sample
Southern Blot Assay
Leaves of transformed shoots were sampled to determine the copy number of the integrated bar gene in transformed plant genomes by Southern blot. The DNA extracted from transformed sugarcane plants was digested with EcoRI, and a 558-bp digoxin-labeled DNA fragment corresponding to the bar gene was used as a probe. The transformed sugarcane shoots showed one to five integrated copies (Fig. 8).

Southern blot analysis of integration sites in transgenic sugarcane. Lane M: DNA marker ladder, which had been labeled (DSTM5000, DONGSHENG BIOTECH, Guangzhou, China); lane 1: wild-type; lanes 2–8: transformed sugarcane plant DNA samples; lane 9: EcoRI-digested plasmid DNA as positive control
Herbicide Sensitivity Assay
We sprayed 2.0 g/L of Basta (200 g/L glufosinate-ammonium, YONON Biosciences Co., Ltd., Zhejiang, China) on transformed and wild-type sugarcane plants under greenhouse conditions. Ten days later, wild-type sugarcane plants were growth-arrested, which resulted in death, but the transformed plants were not affected by Basta and grew healthily (Fig. 9).

Herbicide sensitivity assay. Transformed sugarcane shoots stayed healthy 2 weeks after Basta spraying, but the wild-type sugarcane plants died off
Transformation Efficiency
As shown in Table 2, a high level of transformation efficiency was achieved. Each gram of calli used for transformation produced approximately 12 lines of transformed sugarcane plants. Our results showed 97.4% of the resistant shoots were bar gene PCR-positive and 88.7% of the resistant shoots were PCR-positive for the bar and target genes. Target genes were lost more often than the bar gene was during the process of integration into the sugarcane genome, which led to a lower percentage of target gene PCR-positive plants compared with the bar gene-positive transformants.
Discussion
Increased sugarcane transformation efficiency was achieved using the bar/Basta transformation selection system in this study. Referring to relevant literature and discussing with some other experts, we knew that utilizing a suitable formation of calli was very important to increase the transformation efficiency. The embryonic calli were the best formation for transformation in our previous studies. Thus, after approximately 45 days of induction, the light yellow and compact embryonic calli were chosen for the initiation of transformation. Each gram of calli used for transformation produced approximately 12 lines of transformed sugarcane plants. The most suitable Basta concentration was chosen and supplemented in the media during the dark, regeneration, and rooting selection steps, thereby inducing high selection efficiency. Almost all of the resistant plants in the rooting medium were PCR-positive for the selection marker bar gene. Inadequate Basta concentration (< 1 mg/L) in the selection medium may induce a low PCR-positive percentage of the selection marker bar gene. An excessive Basta concentration (> 3 mg/L) may result in fewer resistant plants obtained and higher copy numbers of transformed shoots at the end. The production of transgenic plants takes fewer than 4 months from callus formation to greenhouse transplantation. This sugarcane transformation protocol resulted in a high proportion of low-copy-number (Manickavasagam et al. 2004; Suprasanna et al. 2011; Snyman 2004) transformed lines and approximately 15% high-copy-number (≥ 4) transformed lines. The selection marker bar genes were expressed stably and conferred excellent herbicide resistance in our later studies on transformed sugarcane ratoon generations. The high transformation efficiency and herbicide resistance demonstrate the broad utility of our transformation selection system in the application of biotechnology for sugarcane variety improvement.
Abbreviations
- MS:
-
Murashige and Skoog medium
- GFP:
-
Green fluorescent protein
- 2,4-D:
-
2,4-Dichlorophenoxyacetic acid
- 6-BA:
-
6-Benzylaminopurine
- NAA:
-
1-Naphthaleneacetic acid
- AD2:
-
Synthetic Medicago truncatula Defensins4
- AD3:
-
Native Medicago truncatula Defensins4 with KDEL
- AD4:
-
Nynthetic Medicago truncatula Defensins4 with KDEL
- Ppase:
-
Inorganic Pyrophosphatase Gene
- SST:
-
Sucrose:sucrose 1-fructosyl transferase gene
References
Arencibia A, Molina P, Dela Riva G, Selman-Housein G (1995) Production of transgenic sugarcane (Saccharum officinarum L.) plants by intact cell electroporation. Plant Cell Rep 14(5):305–309
Arvinth S, Arun S, Selvakesavan RK, Srikanth J, Mukunthan N, Ananda Kumar P, Premachandran MN, Subramonian N (2010) Genetic transformation and pyramiding of aprotinin-expressing sugarcane with cry1Ab for shoot borer(Chilo infuscatellus) resistance. Plant Cell Rep 29(4):383–395
Basnayake SWV, Moyle R, Brich RG (2011) Embryogenic callus proliferation and regeneration conditions for genetic transformation of diverse sugarcane cultivars. Plant Cell Rep 30(3):439–448
Bower R, Brich RG (1992) Transgenic sugarcane plants via microprojectile bombardment. Plant J 2(3):409–416
Darbani B, Eimanifar A, Stewart CN, Camargo WN (2007) Methods to produce marker free transgenic plants. Biotechnol J 2(1):83–90
Dong SJ, Delucca P, Geijskes RJ, Ke J, Mayo K, Mai P, Sainz M, Caffall K, Moser T, Yarnall M, Setliff K, Jain R, Rawls E, Jones MS, Dunder E (2014) Advances in agrobacterium-mediated sugarcane transformation and stable transgene expression. Sugar Tech 16(4):366–371
Enríquez-Obregón GA, Vázquez-Padrón RI, Prieto-Samsonov DL, De la Riva GA, Selman-Housein G (1998) Herbicide resistance sugarcane (Saccharum officinarum L.) plants by agrobacterium-mediated transformation. Planta 206(1):20–27
Falco MC, Neto AT, Ulian EC (2000) Transformation and expression of a gene for herbicide resistance in a Brazilian sugarcane. Plant Cell Rep 19(12):1188–1194
Fromm ME, Morrish F, Armstrong C, Williams R, Thomas J, Klein TM (1990) Inheritance and expression of chimeric genes in the progeny of transgenic maize plants. Bio/Technology 8:833–838
Ingelbrecht IL, Irvine JE, Mirkov TE (1999) Posttranscriptional gene silencing in transgenic sugarcane: dissection of homology-dependent virus resistance in a monocot that has complex poly-ploid genome. Plant Physiol 119(4):1187–1197
Joyce P, Kuwahata M, Turner N, Lakshmanan P (2010) Selection system and co-cultivation medium are important determinants of Agrobacterium-mediated transformation of sugarcane. Plant Cell Rep 29(2):173–183
Kalunke RM, Kolge AM, Babu KH, Prasad DT (2009) Agrobacterium mediated transformation of sugarcane for borer resistance using Cry1Aa3 gene and one-step regeneration of transgenic plants. Sugar Tech 11(4):355–359
Manickavasagam M, Ganapathi A, Anbazhagan VR, Sudhakar B, Selvaraj N, Vasudevan A, Kasthurirengan S (2004) Agrobacterium-mediated genetic transformation and development of herbicide-resistant sugarcane (Saccharum species hybrids) using axillary buds. Plant Cell Rep 23(3):134–143
Mayavan S, Subramanyam K, Arun M, Rajesh M, Kapil Dev G, Sivanandhan G, Jaganath B, Manickavasagam M, Selvaraj N, Ganapathi A (2013) Agrobacterium tumefaciens-mediated in planta seed transformation strategy in sugarcane. Plant Cell Rep 32(10):1557–1574
Mudge RS, Osabe K, Casu RE, Bonnett GD, Manners J, Brich RG (2009) Efficient silencing of reporter transgenes coupled to known functional promoters in sugarcane, a highly polyploid crop species. Planta 229(3):549–558
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Plant Physiol 15(3):473–497
Priji PJ, Hemaprabha G (2015) Sugarcane specific drought responsive candidate genes belonging to ABA dependent pathway indentified from basic species clones of Saccharum sp. and Erianthus sp. Sugar Tech 17(2):130–137
Rani K, Sandhu SK, Gosal SS (2012) Genetic augmentation of sugarcane through direct gene transformation with OsglyII gene construct. Sugar Tech 14(3):229–236
Santosa DA, Hendroko R, Farouk A, Greiner R (2004) A rapid and highly efficient method for transformation of sugarcane callus. Mol Biotechnol 28(2):113–119
Snyman SJ (2004) Sugarcane transformation. Transgenic Crops World 2004:103–114
Srikanth K, Subramonian N, Premachandran MN (2011) Advances in transgenic research for insect resistance in sugarcane. Trop Plant Biol 4(1):52–61
Suprasanna P, Patade VY, Desai NS, Devarumath RM, Kawar PG, Pagariya MC, Canapathi A, Manickavasagam M, Badu KH (2011) Biotechnological developments in sugarcane improvement: an overview. Sugar Tech 13(4):322–335
Viswanathan R, Rao GP (2011) Disease scenario and Management of Major Sugarcane Diseases in India. Sugar Tech 13(4):336–353
Wei HR, Wang ML, Moore PH, Albert HH (2003) Comparative expression analysis of two sugarcane polyubiquitin promotersand flanking sequences in transgenic plants. J Plant Physiol 160:1241–1251
Xiong Y, Jung JH, Zeng QC, Gallo M, Altpeter F (2012) Comparison of procedures for DNA coating of micro-carriers in the transient and stable biolistic transformation of sugarcane. Plant Cell 112(1):95–99
Zhangsun DT, Luo SL, Chen RK, Tang KX (2007) Improved agrobacterium-mediated genetic transformation of GNA transgenic sugarcane. Biologia 62(4):386–393
Zhu YJ, McCafferty H, Osterman G, Lim S, Agbayani R, Lehrer A, Schenck S, Komor E (2011) Genetic transformation with untranslatable coat protein gene of sugarcane yellow leaf virus reduces virus titers in sugarcane. Transgenic Res 20(3):503–512
Acknowledgements
Mr. Wenzhi Wang received financial support from Hainan Natural Science Foundation of Hainan Province of China (20163122). Ms. Shuzhen Zhang received financial support from National Natural Science Foundation of China (31371687).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Hugo A. Campos
Rights and permissions
About this article

Cite this article
Wang, W.Z., Yang, B.P., Feng, C.L. et al. Efficient Sugarcane Transformation via bar Gene Selection. Tropical Plant Biol. 10, 77–85 (2017). https://doi.org/10.1007/s12042-017-9186-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12042-017-9186-7