
Abstract
Peanut seed oil is an important commodity worldwide and breeding efforts have been to improve both the quality and quantity of oil produced. Identifying sources of variation and elucidating the genetics of oil concentration and quality in peanut is essential to advancing the development of improved genotypes. The objective of this study was to discover QTLs for oil traits in an advanced backcross population derived from a cross between a wild-species derived amphidiploid, TxAG-6, and a cultivated genotype, Florunner. A BC1F1 population was developed for genetic mapping and an advanced backcross BC3F6 population was phenotyped in three environments and genotyped using SSR markers. Composite interval mapping results identified three genomic regions associated with oil concentration in a combined analysis. Marker PM36, associated with oil concentration and multiple fatty acids in this study, mapped directly to a HD-ZIP transcription factor in diploid Arachis genome sequences. For fatty acid concentrations, results suggested 17 QTLs identified in two or more environments, 15 of which were present across environments. Fourteen genomic regions on 13 linkage groups contained significant QTLs for more than one trait, suggesting that same genes or gene families are responsible for multiple phenotypes. QTLs and the genes identified in this study could be effective tools in marker-assisted breeding targeted at pyramiding seed oil alleles from wild-species while minimizing introgression of non-target chromatin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cultivated peanut (Arachis hypogaea L.) oil derived from seed cotyledons has been utilized for centuries. In China, India, and much of Africa, most of the peanut seed produced is crushed directly for oil consumption. In the United States, peanut oil is often a by-product produced from seed deemed unacceptable for the edible market, which is the primary consumer of peanut seed. Recently, non-food usage of vegetable oils has increased concurrently with worldwide demand for biodiesel. Limiting factors on the use of peanut oil for biodiesel include high cost due to its functionality as a cooking oil and quality. Peanut breeders have attempted to increase peanut oil concentration to satisfy the edible cooking oil market and reduce costs for biodiesel production (Wilson et al. 2013b; Wilson et al. 2013c). Breeding efforts have also been directed toward lowering oil concentration to reduce fat content (Isleib et al. 2004).
Oil concentration in peanut is a quantitatively inherited trait (Wilson et al. 2013b; Wilson et al. 2013c), controlled by multiple genes (Wilson et al. 2013c) and environmental factors (Baring et al. 2013). The Kennedy pathway, an important route to triacylglycerol (TAG) biosynthesis in plants, is catalyzed by several enzymes, including acyl-CoA: diacylglycerol acyltransferase (DGAT). DGAT catalyzes the final step in the pathway and this step is considered rate limiting in plants (Lung and Weselake 2006; Zheng et al. 2008). Multiple DGAT gene sequences have been identified in peanut (Saha et al. 2006; Burow et al. 2014a) and soybean (Glycine max), a close leguminous relative (Eskandari et al. 2013b). A positive correlation exists between DGAT activity and soy oil accumulation (Lardizabal et al. 2008) and a DGAT2 gene-based single nucleotide polymorphism (SNP) marker is associated with oil biosynthesis in soy (Eskandari et al. 2013a). The relationship between oil concentration and DGAT gene-based markers has not been established in peanut.
Although genetic diversity is limited among cultivated tetraploid peanut genotypes, oil concentration and other important agronomic traits such as root-knot nematode [Meloidogyne arenaria (Neal) Chitwood] resistance can be improved using genes derived from wild diploid relatives via an amphidiploid introgression pathway (Simpson 1991; Simpson and Starr 2001; Wilson et al. 2013c; Burow et al. 2014b). Recent studies examining the genetic basis of inheritance have identified specific QTLs or chromosome regions associated with oil concentration in peanut (Gomez et al. 2009; Sarvamangala et al. 2011; Pandey et al. 2014). However, QTLs for oil-related traits derived from wild peanut species have not been identified. Identifying these QTLs could greatly increase the efficiency of wild species derived gene introgression through selection of targeted regions from the amphidiploid parent in advanced backcross progeny. Advanced backcross analysis, as proposed by Tanksley and Nelson (1996), has been utilized to locate and transfer specific QTLs for a variety of traits from wild or unadapted donor genotypes into elite germplasm (Bernacchi et al. 1998a; Bernacchi et al. 1998b; Fulton et al. 2000; Tian et al. 2006; Jing et al. 2010).
The physical properties of vegetable oil are primarily determined by the composition of its building blocks, fatty acids. These molecules are components of membrane phospholipids and are stored as TAGs in higher plants. In the past 25 years, breeders have developed peanut cultivars with improved oil fatty acid composition. Specifically, increasing the ratio of oleic acid (18:1), a monounsaturated fatty acid, to linoleic acid (18:2), a polyunsaturated fatty acid (high O/L), has important human health benefits (O’Byrne et al. 1997; Vassiliou et al. 2009). High oleic content also decreases the rate of oxidation over time, thus improving the shelf life of edible peanut products (López et al. 2001) and vegetable-oil derived biodiesel (Graef et al. 2009). Two homoeologous genes, ahFAD2A and ahFAD2B, code for the enzyme fatty acid desaturate (FAD2) which catalyzes the conversion of oleic acid to linoleic acid (Moore and Knauft 1989; Jung et al. 2000). Single nucleotide changes in each of the ahFAD2A and the ahFAD2B genes block this conversion and lead to the high oleic phenotype (Jung et al. 2000, López et al. 2000). However, genetic studies indicate that other modifier genetic regions also affect the ratio of these fatty acids (López et al. 2001; Isleib et al. 2006).
Other fatty acids comprise approximately 20 % of peanut oil. The relative proportions of these fatty acids are also important from a nutritional standpoint as they vary in degree of saturation (Ros and Mataix 2006) and are a critical consideration when peanut oils are esterified for biodiesel (Davis et al. 2009; Ramos et al. 2009). There is currently only one published study on inheritance of these fatty acids in peanut (Wang et al. 2015), and specific pathway data are lacking.
In the present study, an advanced backcross population (BC3F6) derived from crosses between recurrent tetraploid parent ‘Florunner’ component line UF439-16-10-3-2, henceforth referred to as ‘Florunner’(Norden et al. 1969), and donor parent TxAG-6 (Simpson et al. 1993), a wild species derived amphidiploid, was used to address the following objectives: (1) identify additive QTLs for oil concentration and quality traits across three environments and (2) determine the effect of DGAT and ahFAD2 gene-based SNPs on oil concentration and quality.
Results
Phenotypic Data
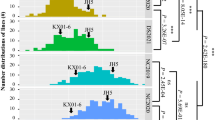
Phenotypic means for TxAG-6 and Florunner at College Station in 2012 are presented in Table 1. Notable differences are present for oil, palmitic, oleic, linoleic, eicosenoic, behenic, and lignoceric concentrations between the parents. Histograms indicated normal or nearly normal distributions and quantitative modes of inheritance for fatty acid phenotypes in combined BC3F6 data (Fig. 1). Broad variation was present for most phenotypes in the BC3F6 population across environments (Table 2). Broad-sense heritability estimates for fatty acids in combined data ranged from 0.14 for lignoceric acid to 0.94 for linoleic acid, suggesting large variation in the degree of genetic contribution to phenotypes. Estimates of broad-sense heritability were high for oil concentration in all three environments and combined data (Table 2).

Distribution of eight fatty acids and oleic to linoleic ratio (O/L ratio) in an advanced backcross population derived from a cross between Florunner and TxAG-6 across environments. P-values for distribution curves were generated using a Shapiro-Wilk test, where a significant P-value indicates a normal distribution. Values on the x-axis are the percentages of that particular fatty acid, or the oleic:linoleic fatty acid ratio, as appropriate
Phenotypic data for oil quality traits revealed strong negative associations across environments between palmitic and oleic acid, palmitic acid and O/L ratio, stearic and linoleic acid, oleic and linoleic acid, oleic and lignoceric acid, and linoleic acid with O/L ratio (Table 3). Strong positive correlations were observed across environments for palmitic and stearic acid, stearic and arachidic acid, stearic and behenic acid, oleic acid and O/L ratio, arachidic and behenic acid, eicosenoic and behenic acid, eicosenoic and lignoceric acid, and behenic and lignoceric acid. Oil concentration was positively correlated with oleic acid and O/L ratio across environments, and negatively correlated with linoleic acid. Our previous research identified a negative correlation between oil concentration and O/L ratio in a different population (Wilson et al. 2013a).
Marker Polymorphisms and Genetic Map
A genetic linkage map consisting of 91 SSR markers on 22 linkage groups covering a map distance of 1321.9 cM was constructed using the BC1F1 mapping population (Fig. 2). Average distance between markers was 14.5 cM and the number of markers on linkage groups ranged from two to ten. A total of 149 primers scored were polymorphic between TxAG-6 and Florunner and segregated in the BC3F6 population. Of these, 105 were scored as dominant markers and 44 were scored as co-dominant.

Map locations of QTLs for oil concentration and fatty acids in three environments and a combined data set using composite interval mapping (CIM) analysis. Env 1 denotes the College Station, TX environment, Env 2 denotes Lubbock, TX, and Env 3 denotes Brownfield, TX
QTL Analysis
Single–factor ANOVA identified three, eight, five, and thirteen QTLs associated with oil concentration at College Station, Lubbock, Brownfield, and combined across environments, respectively (Table 4). Across environments, ten QTLs from TxAG-6 increased oil concentration, while four QTLs decreased it. In the combined analysis, total negative additive effects for TxAG-6 alleles were 53.4 g/kg while total positive additive effects equaled 119.3 g/kg. The QTLs on LG5 near PM36 and LG6 near TC7A02 increased oil concentration and accounted for 29 to 78 % of the phenotypic variation observed. Both favorable alleles were inherited from TxAG-6. Total phenotypic variance of QTLs for oil concentration in Brownfield and across environments suggested multiple markers were linked (Table 4); therefore, CIM was performed to ensure QTLs were independent.
Results from CIM revealed a major favorable QTL for oil concentration on LG6 near TC7A02 in all environments and in the combined data set (Table 5). Additional genomic regions on LG5 were positively associated with oil concentration in Brownfield and in the combined analysis, including a QTL near PM36. Total phenotypic variance explained (PVE) by these markers ranged from 18 to 25 % across environments (Table 5).
For fatty acid concentration, 48 unique QTLs were identified using single-marker analysis (SMA) (Table 4). Of these, 11 QTLs (6 unique) were associated with stearic acid and one QTL was associated with lignoceric acid. For stearic acid, a QTL tagged by TC6E01 contributed 30, 53, and 12 % of total phenotypic variation in College Station, Lubbock, and Brownfield, respectively. In College Station and Lubbock, 20 QTLs were detected at each environment for fatty acid composition and O/L ratio, while in Brownfield, 11 QTLs were detected. A total of 42 QTLs were present in the combined data for fatty acid composition. Using CIM, results suggested 17 QTLs in two or more environments; 15 of which were present across environments (Table 5).
Including significant single-environment QTLs, a total of 14 genomic regions on 13 linkage groups contained QTLs for more than one trait using CIM (Fig. 2). Two distinct regions on LG5 controlled multiple traits, including a region flanked by TC11A02 and PM36. A combination of seven traits over three environments mapped to this region. In several instances, highly correlated oil phenotypes mapped to the same genomic region. Stearic and arachidic acids, which were significantly correlated in all three environments, mapped to the same genomic regions on LG3, LG20, LG21, and LG22 (Fig. 2 and Supplemental Table 1). Closely related traits including O/L ratio, linoleic acid, and oleic acid mapped to the same genomic region on LG9.
Of the six markers linked to phenotypic trait (s) and mapped to the public Arachis genome sequences, three primer sequences mapped to a single gene or genomic-coordinates (Table 6). TC1A02 and TC7A02 mapped to a transporter and vacuolar sorting protein, while the PM36 sequence mapped directly to a GLABRA 2 (GL2) HD-ZIP transcription factor designated Aradu.SEJ3V in A. duranensis (Fig. 3).

Location of simple sequence repeat (SSR) marker PM36 and gene model Aradu.SEJ3V on Arachis duranensis linkage group A05 using the peanutbase.org genome browser
Two-way epistatic interactions were detected for oil concentration in all three environments and in the combined analysis (Table 7). Across environments, 15 interactions were highly significant for oil concentration and five interactions were observed in the combined analysis. For fatty acid composition, a total of 53 epistatic interactions were significant across environments. The number of epistatic interactions observed across environments ranged from 0 for behenic acid to 15 for stearic acid. As expected in a highly inbred population, an additive model explained most variation present in LOD scores for all observed interactions.
Discussion
Genetic diversity in cultivated peanut is limited due a genetic bottleneck during tetraploidization and modern breeding techniques focused on deriving new varieties from a very limited germplasm base. This narrow genetic base has hampered efforts to construct dense genetic maps and improve important traits, such as oil quantity and disease resistance (Burow et al. 2013). Breeders have turned to wild relatives in other polyploid crops such as upland cotton (Gossypium hirsutum) for trait introgression (Percival et al. 1999; Mergeai 2006). In peanut, amphidiploid introgression pathways, including the one used for development of TxAG-6, are an important mechanism for introducing novel traits. Previous attempts to identify QTLs for oil quantity and quality traits have focused on populations derived from cultivated germplasm. This study is the first designed to discover QTLs using SSR and gene-based markers in an advanced backcross segregating population derived from a high-oil synthetic amphidiploid.
The majority of the makers scored in the BC3F6 population were scored dominant due to the absence of heterozygotes; therefore, QTLs and epistatic interactions detected in this study were overwhelmingly additive. Utilizing a BCnF1 population for genotyping as proposed by Tanksley and Nelson (1996) would have greatly increased the frequency of heterozygotes, thus allowing for the detection of dominant QTLs and dominant epistatic QTL interactions. However, all cultivated peanut varieties grown in the U.S. are highly inbred genotypes; hence, only additive genetic effects are of practical importance to most peanut geneticists.
The preponderance of evidence from this population and from other populations grown in different environments indicates oil concentration in peanut seed cotyledons is a highly heritable quantitative trait (Sarvamangala et al. 2011; Wilson et al. 2013b, c). The number of markers identified for oil concentration through CIM increased with broad-sense heritability across environments, underscoring a direct relationship between phenotypic and marker data.
A novel QTL for oil concentration, tagged by SSR marker TC7A02, was the only major QTL identified herein significant across all environments using single-marker ANOVA and CIM. This positive allele derived from TxAG-6 is perhaps the most important wild-species derived marker governing the high-oil phenotype in peanut. In addition, a high number of mapped loci were associated with multiple traits using CIM (Fig. 2). Many of these closely mapped fatty acids were also significantly correlated (Table 3) and are tightly linked in pathways controlling fatty acid synthesis in oilseed crops (Barker et al. 2007). This indicates that the same genes, gene families, and/or tightly linked genes are responsible for multiple phenotypes.
This study is the second to reveal an association between a DGAT gene-based marker and oil concentration in peanut. The DGAT2 allele from TxAG-6 was positively associated with oil concentration in Lubbock and in the combined analysis, explaining 7 and 2 % of phenotypic variation, respectively. Opportunities exist for additional studies aimed at identifying additional DGAT genes in peanut and confirming their effect on oil concentration and quality in peanut and transformed constructs.
The significance of marker PM36 on LG5 in determining oil concentration in peanut is clear based on our data and results from other studies (Selvaraj et al. 2009; Sarvamangala et al. 2011). Our manuscript is the first study in peanut to reveal that the genomic region flanking PM36 is a part of the 5′ untranslated region (UTR) of the GL2 HD-ZIP transcription factor in Arachis. Previous studies from oilseeds Brassica napus (Chai et al. 2010), and Glycine max (Liu et al. 2014) including Arabidopsis thaliana (Shen et al. 2006; Shi et al. 2012), suggest the GL2 family of HD-ZIP transcription factors negatively regulate seed oil production. Bands of PM36 detected in TxAG-6 (212 bp) and Florunner (218 bp) differed by 6 bp. This difference in the 5′ end of the HD-ZIP sequence may cause discrepancies in transcriptional regulation between these genotypes. Specifically, the deletion of 6 bp in TxAG-6 may negatively regulate this HD-ZIP transcription factor, leading to higher oil accumulation in TxAG-6. The accumulation of fatty acid chains in oilseeds resulting in high oil phenotypes is likely a function of genes coding for enzymes involved in TAG biosynthetic pathways and the regulation of these genes at the transcriptional level (Liu et al. 2014). Additional research is needed to determine a direct correlation between the expression of HD-ZIP transcription factors and TAG accumulation in Arachis.
TC7A02 and TC1A02 also mapped to unique genomic coordinates on the 3′ end of the genes annotated in the reference sequence. Annotation of the genes to which these markers mapped suggest their possible involvement in transport of metabolites and proteins. Vacuolar sorting proteins such VSP9 are thought to play a role in endosomal trafficking (Xiang et al. 2013), and genes coding proteins containing VSP9 domains are required for multiple plant functions in Arabidopsis (Goh et al. 2007). Further investigation is needed to establish the precise role of protein transporting genes in the movement of enzymes catalyzing fatty acid synthesis from endomembrane systems in Arachis and their role in seed development. This study lays the groundwork for fine mapping, sequencing, and discovery of genes and regulatory networks at LG5 and other linkage groups associated with seed oil traits.
Fatty acid heritability estimates were highly variable in this study. In the combined data set, lowly heritable fatty acids including palmitic, behenic, and lignoceric acids had relatively few significantly associated QTLs using ANOVA and CIM compared with more heritable fatty acids. Molecular and phenotypic observations in this study confirm our hypothesis from a previous manuscript regarding the importance of environmental effects in determining observed phenotypes of certain minor fatty acids, such as behenic and lignoceric (Wilson et al. 2013a). Modifying the concentration of these lowly heritable fatty acids in peanut oil though traditional breeding techniques will likely be difficult.
We observed multiple QTLs for oleic and linoleic acids and O/L ratio across all environments using ANOVA and CIM. As expected, a common genomic region was associated with oleic acid, linoleic acid, and O/L ratio. QTLs discovered from two-way ANOVA in this study and previous studies (Sarvamangala et al. 2011; Wang et al. 2015) support the hypothesis proposed by López et al. (2001) and Isleib et al. (2006) stating modifier genes other than ahFAD2A and ahFAD2B affect O/L ratio.
The ahFAD2A and ahFAD2B SNPs in this study are unique and specifically designed to differentiate two low-oleic parents, TxAG-6 and Florunner (Burow et al. 2014a). The effect the FAD2B allele on O/L ratio was only significant across environments using a P value of 0.001 for SMA. However, the ahFAD2A allele was associated with O/L ratio at a significance level of P ≤ 0.07 at College Station, Lubbock, and across environments (data not shown). Analysis of a small population consisting of fewer than 100 individuals limits the statistical power available to identify minor effect QTLs and can lead to the overestimation of genetic effects of major QTLs (Beavis 1998). Therefore, a larger population size combined with a less stringent P-value could result the discovery of additional significant QTLs, such as ahFAD2 SNPs and O/L ratio.
Epistatic interactions are key factors underpinning the intricate genetic systems governing quantitative traits in crop species. These interactions are difficult for breeders to manipulate because they are difficult to measure, often sensitive to environment, and typically explain only a small amount of phenotypic variation (Bernardo 2010). Herein, multiple epistatic interactions were identified at a stringent statistical threshold, but there were no interactions identified consistently for any single trait across all environments. However, it is notable that almost 38 % of all significant epistatic interactions identified for fatty acid composition across environments involved one or more markers from LG5. Potentially, this additive epistatic variation could be fixed in inbred lines using markers for selection. In practice, this would be a difficult task for breeders due to the sensitivity of these interactions to environment.
Conclusions
Phenotypic observations including heritability and correlation analysis emphasize the limits associated with improving oil concentration and fatty acid composition. Our data indicate strong phenotypic associations and genetic linkages among many fatty acids and between oil concentration and fatty acid composition across diverse environments. These relationships likely cannot be altered through conventional breeding techniques or marker assisted selection. However, paired comparisons indicate that most measured phenotypic associations for oil quality and quantity are not fixed and are sensitive to environment and/or genetic background. Identifying desirable-trait markers present in multiple environments that are not tightly linked with undesirable markers could greatly improve selection efficiency through pyramiding multiple positive effect QTL allele linked markers. It is expected that transcriptome- and whole-genome sequencing of peanut may lead to the discovery of a multitude of gene sequences and gene-based markers, which will aid in the unraveling of the complex gene networks governing quantitatively inherited traits.
Methods
Plant Materials
A single backcross population was developed for genetic mapping using a Florunner (Norden et al. 1969) component line (UF439-16-10-3-2) and synthetic amphidiploid TxAG-6 (Simpson 1991). This BC1F1 population consisting of 78 individuals was previously used to create a restriction fragment length polymorphism (RFLP) linkage map (Burow et al. 2001) and an SSR linkage map (Gomez et al. 2008). Two additional backcrosses to Florunner were completed to produce a BC3 population representing a subset of the original BC1F1 mapping population as described by Burow et al. (2014b). BC3F6 populations were derived from BC1F1 plants through single-seed descent.
A total of 90 BC3F6 breeding lines were planted in a randomized complete block design consisting of twin 10 ft. rows and two replications at a field site near Brownfield, TX in 2010. These 90 BC3F6 breeding lines were planted in 12 in. diameter clay pots containing two plants with two replications in a greenhouse at College Station, TX and Lubbock, TX in 2012. Phenotyping was performed for oil concentration, palmitic acid (16:0), stearic acid (18:0), oleic acid (18:1), linoleic acid (18:2), arachidic acid (20:0), eicosenoic acid (20:1), behenic acid (22:0) lignoceric acid (24:0), and O/L ratio on seed produced at each environment. For oil concentration, a 10 g sample of sound mature kernels was randomly selected from each plot at Brownfield, TX and each individual plant at the greenhouse environments. These samples were measured using nuclear magnetic resonance (NMR) as described previously (Wilson et al. 2013b; Wilson et al. 2013c). Fatty acid methyl esters (FAME) of extracted peanut oil were prepared similar to Jungman (2000). Fatty acid composition of FAME solutions was measured using gas chromatography using protocols developed by Wilson et al. (2013a). Oil was extracted from a composite of five seeds from each replication because preliminary data (not shown) indicated there was not a significant replication effect for any fatty acid measured using a limited, random sample of BC3F6 lines within each environment.
DNA Isolation
Ninety BC3F6 were planted in the greenhouse at Texas A&M AgriLife Research and Extension, Lubbock. Young leaf samples were harvested from each of the plants and were stored at -80 °C upon freezing with liquid nitrogen. DNA was extracted using the Qiagen DNAeasy (Valencia, CA) mini plant kit as described by manufacturer’s protocol. The quality and quantity of DNA was assessed using a Nano Drop-1000 spectrophotomoter and 1 % agarose gel.
SSR Genotyping
A total of 152 SSR primers pairs utilized by Gomez et al. (2008) and Belamkar et al. (2011) were tested for polymorphisms between parents TxAG-6 and Florunner. Polymorphic SSR primers were genotyped on 90 BC3F6 lines, along with the two parents of the cross. Polymerase chain reaction (PCR) amplifications were performed using a three-primer system incorporating specific fluorescent dye-labeled primers as described by Belamkar et al. (2011). Each 10 μl reaction consisted of 10–20 ng of template DNA in TE buffer pH 8 .0, 1X PCR buffer, 2 mM MgCl2, 0.5 μl of 10 mM forward and M13-tagged reverse primers, an M13 reverse primer 5′-GACGTTGTAAAACGACGGCC-3′ with a 5′ dye fluorescent dye (D2, D3, or D4), 0.5 U Taq polymerase, and 1.25 μl of 2 mM dNTPs. Primer amplifications were completed in an MJ Research PTC 100 Thermal Cycler (BioRad, Hercules CA) touchdown strategy similar to Belamkar et al. (2011). The PCR protocol used was as follows: 3 m at 94 °C; 20 touchdown cycles of 30 s at 94 °C, 60s at 65–55 °C (dropping 0.5 °C per cycle), 60 s extension at 72 °C; and 15 cycles of 30 s at 94 °C, 60s at 55 °C, 60 s at 72 °C; followed by 5 cycles of 30 s at 94 °C, 60s at 49 °C, 60 s at 72 °C; then 10 m at 72 °C, followed by a soak at 4 °C. The five cycles with the 49 °C annealing temperature were added to enhance incorporation of the dye-labeled primer (Schuelke 2000). Amplified products were detected on a Beckman Coulter CEQ 8000 Genetic Analysis System.
SNP Genotyping
DGAT gene based sequences developed by Burow et al. (2014a) and ahFAD2A and ahFAD2B sequences derived from transcriptome data (Chopra et al. 2015) were used to construct Kompetitive Allele Specific PCR (KASP) single nucleotide polymorphism (SNP) primers. For each putative varietal SNP, two allele-specific forward primers and one common reverse primer were designed (LGC Genomics, Hoddesdon, UK). Genotyping reactions were performed on a LightCycler 480 (Roche, Branford, CT) in a final volume of 10 μl containing 1X KASP Reaction Mix (LGC Genomics, Hoddesdon, UK), 0.14 μl Assay mix, and 10–20 ng genomic DNA. The following cycling conditions were used: 15 min at 94 °C; 10 touchdown cycles of 20 s at 94 °C, 60s at 65–57 °C (dropping 1.0 °C per cycle); 26 cycles of 20 s and 94 °C, 60 s at 57 °C, and read at 37 °C for 5 s. Fluorescence detection of the reactions was performed using a built- in scanner and the data were analyzed using the LightCycler 480 software (Roche, Branford, CT).
QTL Analysis
Composite interval mapping was performed using Rqtl package v.3.1.2 from R (Broman et al. 2003) using genetic distances from a BC1F1 SSR map (Gomez et al. 2008) and markers scores from the BC3F6 population. Single maker analysis was performed on the BC3F6 population using the SSR makers mapped to the BC1F1 population and additional polymorphic SSR and SNP markers.
SSR sequences from six loci linked to one or more phenotypic traits were mapped to the publicly available genome sequences for the A (Arachis duranensis) and B genome (Arachis ipaënsis) progenitor species of cultivated peanut (Bertioli et al. 2016). Two selected markers, PM36 and IPAHM 103 have previously been linked to oil accumulation in Arachis (Selvaraj et al. 2009; Sarvamangala et al. 2011), while the loci tagged by TC7A02 is unique to this study. Other loci selected for mapping were linked to multiple fatty acids in different environments.
Statistical Analysis
Single marker analysis was performed with Proc GLM of SAS ver. 9.2 (SAS Institute, Cary, NC) with LSMEANS estimates for oil and fatty acid concentrations as the dependent variables and genotypic scores as independent variables in each environment. A separate analysis of combined data from all environments that included an environment x marker effect as a dependent variable was also performed. An R2 value was generated for each marker and phenotype combination to estimate the proportion of phenotypic variance accounted for by the marker. For CIM, a LOD threshold of 3.3 was used to correspond to a genome-wide P-value of 0.0001 and a LOD score of 2.3 was used for a chromosomal P-value of 0.0001 (Larson and Mayland, 2007). PVE scores were estimated using the formula h2 = 1–10-2LOD/n from R-qtl, where n = the number of individuals scored in the BC3F6 population. Epistatic interactions were calculated using a 2-way scan using the scantwo function in Rqtl. Three odds ratios were calculated for the full model as detailed by Brothers et al. (2013). In addition, these interactions were subject to a two-way ANOVA in SAS Proc GLM (SAS Institute, Cary NC) with phenotypic data as the response variable to determine significance at a P-value of 0.001.
Estimated genetic variance components were computed using the GLM procedure of SAS. Broad-sense heritability estimates for oil concentration and fatty acid concentrations across environments were calculated using the following formula: H2 = Vg / [Vg + (Vge / e) + (Ve / re)], where Vg, Vge, and Ve refer to genotypic variance, genotype x environment variance and residual variance, respectively. Coefficients ‘e’ and ‘re’ refer to environments and replications within environments.
Broad-sense heritability in each environment was calculated for oil concentration using the following formula: H2 = Vg / (Vg + Ve). There was one combined replication for fatty acids in each environment; therefore, broad-sense heritability was only calculated across environments. Pearson’s correlation coefficients for fatty acids and oil concentration were derived using Proc CORR of SAS.
References
Baring MR et al. (2013) Variability of total oil content in peanut across the state of Texas. J. Crop Improv 27:125–126
Barker GC et al. (2007) Novel insights into seed fatty acid synthesis and modification pathways from genetic diversity and quantitative trait loci analysis of the Brassica C genome. Plant Physiol 144:1827–1842
Beavis, WD (1998) QTL analyses: power, precision, and accuracy. In: Paterson AH, editor. Molecular dissection of complex traits CRC Press Inc, Boca Raton pp 145–162
Belamkar V et al. (2011) A first insight into population structure and linkage disequilibrium in the US peanut minicore collection. Genetica 139:411–429
Bernacchi D et al. (1998a) Advanced backcross QTL analysis in tomato I. Identification of QTLs for traits of agronomic importance from Lycopersicon hirsutum. Theor Appl Genet 97:381–397
Bernacchi D et al. (1998b) Advanced backcross QTL analysis of tomato II. Evaluation of near-isogenic lines carrying single-donor introgressions for desirable wild QTL-alleles derived from Lycopersicon hirsutum and L. pimpinellifolium. Theor Appl Genet 97:170–180
Bernardo, R (2010) Breeding for quantitative traits in plants. Stemma Press, Woodbury, MN
Bertioli DJ et al. (2016) The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. doi:10.1038/ng.3517
Broman KW, Wu H, Sen Ś, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890
Brothers AN et al. (2013) Genetic architecture of floral traits in Iris hexagona and Iris fulva. J Hered 104:853–861
Burow MD, Simpson CE, Starr JL, Paterson AH (2001) Transmission of genetics of chromatin from a synthetic amphidiploid to cultivated peanut (Arachis hypogaea L.): broadening the gene pool of a monophyletic polyploid species. Genetics 159:823–837
Burow, M.D et al (2013) Marker-Assisted selection for biotic stress resistance in peanut. In: Varshney RK, and Tuberosa R, editors. Translational genomics for crop breeding: biotic stress, Vol I. John Wiley & Sons Ltd, Chichester. doi:10.1002/9781118728475.ch8
Burow, MD et al (2014a). Identification of additional FAD2 genes plus DGAT genes in peanut and mapping QTLs for fatty acid composition in peanut. 46th Annual Meeting of the American Peanut Research and Education Society, San Antonio, Texas. 10 July 2014. Paper 72
Burow MD et al. (2014b) Introgression of homeologous quantitative trait loci (QTLs) for resistance to the root-knot nematode [Meloidogyne arenaria (Neal) Chitwood] in an advanced backcross-QTL population of peanut (Arachis hypogaea L.). Mol. Breeding 34:393–406
Chai G et al. (2010) Brassica GLABRA2 genes: analysis of function related to seed oil content and development of functional markers. Theor. Appl. Genet 120:1597–1610
Chopra R et al. (2015) Next-generation transcriptome sequencing, SNP discovery and validation in four market classes of peanut, Arachis hypogaea L. Mol. Genetics and. Genomics 290:1169–1180
Davis JP, Geller D, Faircloth WH, Sanders TH (2009) Comparisons of biodiesel produced from unrefined oils of different peanut cultivars. J Am Oil Chem Soc 86:252–236
Eskandari M, Cober ER, Rajcan I (2013a) Genetic control of soybean oil I: QTL and genes associated with seed oil accumulation in RIL populations derived from crossing moderately high-oil parents. Theor Appl Genet 126:483–495
Eskandari M, Cober ER, Rajcan I (2013b) Using the candidate gene approach for detecting genes underlying seed oil concentration and yield in soybean. Theor Appl Genet 126(7):1839–1850
Fulton TM et al. (2000) Advanced backcross QTL analysis of a Lycopersicon esculentum x Lycopersicon parviflorum cross. Theor Appl Genet 100:1025–1042
Goh T et al. (2007) VPS9a, the common activator for two distinct types of Rab5 GTPases, is essential for the development of Arabidopsis thaliana. Plant Cell 19:3504–3515
Gomez, SM et al (2008) Towards and integrated SSR/RFLP map of tetraploid peanut. Third International Conference of the Peanut Research Community, ICRASAT, Hyderabad, Andhra Pradesh, India. 4–8 Nov. 2008. Paper 21
Gomez SM, Narayana M, Schubert AM, Ayers JL, Baring MR, Burow MD (2009) Identification of QTLs for pod and kernel traits in cultivated peanut by bulked segregant analysis. Electr J Biotech 12(2):1–10
Graef G et al. (2009) A high-oleic and low-palmitic-acid soybean: agronomic performance and evaluation as a feedstock for biodiesel. Plant Biotechnology J. 7:411–421
Isleib TG, Pattee HE, Giesbrecht FG (2004) Oil, sugar, and starch characteristics in peanut breeding lines selected for low and high oil content and their combining ability. J Agric Food Chem 52:3165–3168
Isleib TG, Wilson RF, Novitzky WP (2006) Partial dominance, pleiotropism, and epistasis in the inheritance of the high-oleate trait in peanut. Crop Sci 46:1331–1335
Jing Z et al. (2010) QTL analysis of yield-related traits using an advanced backcross population derived from common wild rice (Oryza rufipogon L). Mol. Plant Breed 1:2–10
Jung S, Powell G, Moore K, Abbott A (2000) The high oleate trait in the cultivated peanut [Arachis hypogaea L.] II. Molecular basis and genetics of the trait. Mol Gen Genet 263:806–811
Jungman, B (2000) The effect of fatty acid profiles on peanut seed germination at low soil temperatures. M.S. thesis., Texas Tech Univ., Lubbock
Lardizabal K et al. (2008) Expression of Umbelopsis ramanniana DGAT2A in seed increases oil in soybean. Plant Physiol 148:89–96
Larson SR, Mayland HF (2007) Comparative mapping of fiber, protein, and mineral content QTLs in two interspecific Leymus wild rye full-sib families. Mol Breeding 20:331–347
Liu YF et al. (2014) Soybean GmMYB73 promotes lipid accumulation in transgenic plants. BMC Plant Biol 14:–73
López Y et al. (2000) Isolation and characterization of the Δ12-fatty acid desaturase in peanut (Arachis hypogaea L.) and search for polymorphisms for the high oleate trait in Spanish market-type lines. Theor Appl Genet 101:1131–1138
López Y, Smith OD, Senseman SA, Rooney WL (2001) Genetic factors influencing high O/L acid content in Spanish market-type peanut cultivars. Crop Sci 41:51–56
Lung SC, Weselake RJ (2006) Diacylglycerol acyltransferase: a key mediator of plant triacylglycerol synthesis. Lipids 41:1073–1088
Mergeai G (2006) Cotton improvement through interspecific hybridization. Cahiers. Agri 15:135–143
Moore KM, Knauft DA (1989) The inheritance of high oleic acid in peanut. Jour Hered. 80:252–253
Norden AJ, Lipscomb RW, Carver WA (1969) Registration of ‘Florunner’ peanuts. Crop Sci 9:850
O’Byrne DJ, Knauft DA, Shireman RB (1997) Low fat monounsaturated rich diets containing high-oleic peanuts improve serum lipoprotein profiles. Lipids 32:687–695
Pandey MS et al. (2014) Identification of QTLs associated with oil content and mapping FAD2 genes and their relative contribution to oil quality in peanut (Arachis hypogaea L. BMC Genet 15:–133
Percival AE, Wendel JF, Stewert JM (1999) Taxonomy and germplasm resources. In: Smith CW, Cothren JT (eds) Cotton: Origin, history, technology, and production. John Wiley & Sons, New York, pp. 33–63
Ramos MJ et al. (2009) Influence of fatty acid composition of raw materials on biodiesel properties. Bioresource Tech 100:261–268
Ros E, Mataix J (2006) Fatty acid composition of nuts – implications for cardiovascular health. British. J Nutr 96:S29–S35
Saha S, Enugutti B, Rajakumari S, Rajasekharan R (2006) Cytosolic triacylglycerol biosynthetic pathway in oilseeds. Molecular cloning and expression of peanut cytosolic diacylglycerol acyltransferace. Plant Physiol 141:1533–1543
Sarvamangala C, Gowda MVC, Varshney RK (2011) Identification of quantitative trait loci for protein content, oil content and oil quality in groundnut (Arachis hypogaea L. Field Crops Res 122:49–59
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nature Biotech 18:233–234
Selvaraj MG et al. (2009) Identification of QTLs for pod and kernel traits in cultivated peanut by bulked segregant analysis. Electronic J. Of. Biotech 12. doi:10.2225/vol12
Shen B, Sinkevicius KW, Selinger DA, Tarczynski MC (2006) The homeobox gene GLABRA2 affects seed oil content in Arabidopsis. Plant Mol Biol 60:377–387
Shi L et al. (2012) Arabidopsis glabra2 mutant seeds deficient in mucilage biosynthesis produce more oil. Plant J 69:37–46
Simpson CE (1991) Pathways for introgression of pest resistance into Arachis hypogaea L. Peanut Sci. 18:22–26
Simpson CE, Starr JL (2001) Registration of ‘COAN’ peanut. Crop Sci 41:918
Simpson CE et al. (1993) Registration of ‘TxAG-6’ and ‘TxAG-7’ peanut germplasm. Crop Sci 33:–1418
Tanksley SD, Nelson JC (1996) Advanced backcross QTL analysis: a method for the transfer of valuable QTLs from unadapted germplasm into elite breeding lines. Theor Appl Genet 92:191–203
Tian F et al. (2006) Construction of introgression lines carrying wild rice (Oryza rufipogon Griff.) segments in cultivated rice (O. sativa L.) background and characterization of introgressed segments associated with yield-related traits. Theor. Appl. Genet 112:570–580
Vassiliou EK et al. (2009) Oleic acid and peanut oil high in oleic acid reverse the inhibitory effect of insulin production of the inflammatory cytokine TNF-alpha both in vitro and in vivo systems. Lipids Health Dis 8:–25
Wang ML et al. (2015) Genetic mapping of QTLs controlling fatty acids provided insights into genetic control of fatty acid synthesis pathway in peanut (Arachis hypogaea L.). Plos One. doi:10.1371/journal.pone.0119454
Wilson JN et al. (2013a) Generation means analysis of fatty acid composition in peanut. J. Crop Improv 27:430–443
Wilson JN et al. (2013b) Diallel analysis of oil production components in peanut (Arachis hypogaea L.). Int J Agro. doi:10.1155/2013/975701
Wilson JN et al. (2013c) Generation means analysis of oil concentration in peanut. J. Crop Improv 27:85–95
Xiang L, Etxeberria E, Van den Ende W (2013) Vacuolar protein sorting mechanisms in plants. FEBS J 280:979–993. doi:10.1111/febs.12092
Zheng P et al. (2008) A phenylalanine in DGAT is key determinant of oil content and composition in maize. Nat Genet 40:367–372
Acknowledgments
This work was funded by USDA/NIFA award TEX08835 to MDB, National Peanut Board award #329/TX-99 to MRB, MDB, and CES, and through support provided by the Office of Agriculture, Research and Policy, Bureau of Food Security, U.S. Agency for International Development, under the terms of Award No. AID-ECG-A-00-07-0001 to The University of Georgia as management entity for the U.S. Feed the Future Innovation Lab on Peanut Productivity and Mycotoxin Control. The opinions expressed herein are those of the author(s) and do not necessarily reflect the views of the U.S. Agency for International Development.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Ray Ming
Key message: QTLs, some with large phenotypic effects, were observed for oil and fatty acid concentrations. Marker PM36 mapped directly to a HD-ZIP transcription factor in diploid Arachis genome sequences.
Electronic supplementary material
Table S1
(DOCX 18 kb)
Rights and permissions
About this article

Cite this article
Wilson, J.N., Chopra, R., Baring, M.R. et al. Advanced Backcross Quantitative Trait Loci (QTL) Analysis of Oil Concentration and Oil Quality Traits in Peanut (Arachis hypogaea L.). Tropical Plant Biol. 10, 1–17 (2017). https://doi.org/10.1007/s12042-016-9180-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12042-016-9180-5