
Abstract
Wheat hybrid necrosis is caused by the interaction of two dominant complementary genes, Ne1 and Ne2, located on chromosome arms 5BL and 2BS, respectively. The sequences of Ne1 or Ne2 have not yet been identified. It is also not known whether Ne1 and Ne2 are structural or regulatory genes. Understanding the proteomic pathways may provide a knowledge base for protecting or maximizing the photosynthesis capacity of wheat. Using DIGE and MALDI-TOF-TOF MS, the flag leaf protein patterns of the two unique \(\hbox {F}_{14}\) near-isogenic line siblings (NILs), the necrotic ShunMai 12Ah (Ne1 Ne1 Ne2 Ne2) and the normal ShunMai 12Af (Ne1 Ne1 ne2 ne2) were compared. Due to the presence or absence of Ne2, (i) three protein spots were expressed or disappeared, (ii) seven RuBisCO-related proteins were altered significantly, and (iii) 21 photosynthesis/glucose related proteins were changed significantly. Three hypotheses were deduced, (i) Ne1 may also encode protein(s), (ii) genetic maladjustment of RuBisCO could lead to early leaf death, and (iii) interactions between nuclear genes and chloroplast genes could determine photosynthetic traits. Our hypothetical model presents the RuBisCO pathway of hybrid necrosis in wheat and explains how Ne1 and Ne2 interact at molecular level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The world is facing the challenge of producing more wheat on less land and using less water with fewer chemicals (http://www.rothamsted.ac.uk/). Genetic enhancement of the net photosynthetic productivity of wheat represents the most cost-effective and environment-friendly method for increasing sustainable global food security. The 20:20 wheat\(^{{\textregistered }}\) focusses on photosynthetic productivity trying to increase wheat yield potential to 20 t/ha within the next 20 years (http://www.rothamsted.ac.uk/).
Wheat hybrid necrosis is literally a genetic early leaf death (GELD) that dramatically reduces photosynthetic longevity and productivity. Progressive lethal or semi-lethal necrosis of \(\hbox {F}_{1}\) plants in some wheat hybrids is brought about by the two dominant complementary genes (Kostyuchenko 1936; Caldwell and Compton 1943), Ne1 and Ne2 (Hermsen 1963a, b), located on chromosome arms 5BL and 2BS, respectively (Chu et al. 2006). While Ne1 and Ne2 have been reported and characterized, their sequences have not been cloned (Chu et al. 2006) and their primary products are yet to be identified. It is also not clear whether Ne1 and Ne2 are structural or regulatory genes (Pan et al. 2009). When either Ne1 or Ne2 is present alone in a wheat genotype (e.g. Ne1Ne1ne2ne2 or ne1ne1Ne2Ne2), the plant is normal. But when found together, hybrid necrosis occurs. How does the two nonharmful genes trigger GELD, has remained unknown. The molecular causation of wheat hybrid necrosis has long been a major puzzle since 1929 (Naskidashvili et al. 2010). Understanding the proteomics pathway of the GELD is expected to enrich our knowledge base at a molecular level for protecting or even maximizing the photosynthetic longevity and productivity of wheat. The purpose of this study was to ascertain whether Ne2 encodes protein(s) by using DIGE and MALDI-TOF-TOF MS.
Materials and methods
The two near-isogenic line siblings (NILs) of Ne2: ShunMai 12Ah and ShunMai 12Af
The logical way to understand the proteomic behaviour of a gene is to observe the effects of silencing that gene, i.e. to shut off or knock out the gene, and then to see what happens. As the sequences of Ne1 and Ne2 are not known yet, the molecular gene-silencing techniques could not be used to shut off Ne1 or Ne2. The development of NILs with different compositions of Ne genes is crucial for further identification of their encoded products and molecular functions. Therefore, we spent about 20 years to create the two \(\hbox {F}_{14}\) NILs: the necrotic ShunMai 12Ah of the homozygous dominant genotype Ne1Ne1Ne2Ne2, vs the normal ShunMai 12Af of the Ne1Ne1ne2ne2 genotype (Pan et al. 2015a). Theoretically, in the \(\hbox {F}_{14}\) population, the necrotic ShunMai 12Ah and the normal ShunMai 12Af have 99.99% genetic similarity. Their most distinct phenotypic difference in the growing season is that each leaf (from the very first leaf to the last flag leaf) of the necrotic ShunMai 12Ah died in 18–22 days, while each leaf of the normal ShunMai 12Af remained green for 52–56 days (Pan et al. 2015a). These contrasting materials are crucial for ascertaining whether Ne2 encodes protein(s) or not.

Comparisons of the sampled plants and flag leaves. (a) One representative plant of the necrotic ShunMai 12Ah on the sampling day; the flag leaves of some stems were still totally green, the other stem–leaves of each stem were all totally necrotic due to the Ne1Ne1Ne2Ne2 genotype. (b) One representative plant of the normal ShunMai 12Af on the sampling day; each stem has 3–5 green leaves including the flag leaf due to Ne1Ne1ne2ne2 genotype that is lacking Ne2. Each leaf (from the very first leaf to the last flag leaf) of the necrotic ShunMai 12Ah died in 18–22 days. In contrast, each leaf of the normal ShunMai 12Af remained green for 52–56 days. (c) What the sampled flag leaves looked like on the sampling day. The top half of each fully green flag leaf was sampled for protein analysis.
Leaf sampling
The representative plants at the sampling day are shown in figure 1. When the flag leaves of the necrotic ShunMai 12Ah (figure 1a) had fully extended and were totally green while the other stem–leaves were completely dead, the top halves (figure 1c) of the six totally green flag leaf blades were rinsed with distilled water, then cut/wrapped/labelled as one replicate and placed immediately in liquid nitrogen. Three replicates from the necrotic ShunMai 12Ah and three replicates from the normal ShunMai 12Af (figure 1b), were sampled and labelled as 12Ah1X, 12Ah2X, 12Ah3X, 12Af1X, 12Af2X and 12Af3X, respectively.
Protein preparation
Each sample was ground into fine powder in liquid nitrogen from which \(\sim \)1 g was transferred into a tube containing 25 mL of trichloroacetic acid / acetone (1:9, v/v) as well as 65 mM DDT, and the protein was then precipitated at −20\({^{\circ }}\hbox {C}\) for 1 h. The sample tubes were centrifuged at 10,000 rpm for 45 min and the supernatants were discarded. Twenty-five mL of acetone was added to each precipitate and the tube was stored at −20\({^{\circ }}\hbox {C}\) for 1 h and centrifuged again at 10,000 rpm at \(4{^{\circ }}\hbox {C}\) for 45 min. The precipitate was then vacuum dried, weighed and stored at −80\({^{\circ }}\hbox {C}\). A total of 200 mg of protein sample was added to 0.5 mL of DIGE lysis buffer containing 7 M urea, 2 M thiourea, 4% (w/v) CHAPS and 0.2% (v/v) immobilized pH gradient (IPG) buffer. The protein concentration in the supernatant was quantified by the Bradford method using Bio-Rad protein assay reagent. Aliquots of the protein samples were stored at −80\({^{\circ }}\hbox {C}\) until use in proteomic analysis.
Protein labelling using DIGE dyes
The protein samples were labelled using fluorescent CyDyes\(^{\mathrm{TM}}\) (Cy2, Cy3, and Cy5) developed for DIGE (GE Healthcare, Little Chalfont, UK). The experimental design using the three-dye approach is illustrated in table 1. Fifty \(\mu \)g of an internal standard (IS) containing an equal amount of the six sample proteins was labelled with 400 pmol Cy2. Fifty \(\mu \)g of each sample was labelled with 400 pmol Cy3 or Cy5. A dye swap between 12Ah and 12Af protein samples was carried out to avoid artifacts due to preferential labelling. Protein samples were maintained on ice and fluorescently labelled in the dark for 30 min. The reaction was then quenched by incubating with 1 \(\mu \)L of 10 mM L-lysine (GE Amersham Biosciences) on ice in the dark for 15 min.

(a) The representative 2D-DIGE image (Gel1). (b) The 31 significantly upregulated protein spots (arrow towards the number) and the 24 significantly downregulated protein spots (arrow towards the spot) in the normal ShunMai 12Af compared to the necrotic ShunMai 12Ah on a representative 2D gel.
2D-DIGE
For each gel, Cy2-labelled, Cy3-labelled, and Cy5-labelled proteins (\(50\,\mu \hbox {g}\) each) were pooled and an equal volume of rehydration buffer (8 M urea, 4% CHAPS, 2% DTT and 2% IPG buffer pH 3–10) was added so that the final concentration of DTT and IPG buffer was 1%. The pooled protein samples were subjected to isoelectric focussing (IEF) carried out by the Ettan IPGphor IEF system (GE Amersham) and the nonlinear IPG strips (pH 3–10, 13 cm, GE Healthcare). The IPG strips were rehydrated for 12 h in \(250\,\mu \hbox {L}\) of rehydration buffer containing the protein samples. IEF was performed in four steps: 30 V for 12 h, 500 V for 1 h, 1000 V for 1 h, and 8000 V for 8 h. After the IEF run was completed, the gel strips were equilibrated under gentle shaking for 15 min in equilibration buffer (50 mM Tris-HCl (pH 8.8), 6 M urea, 2% SDS, 30% glycerol, and 1% DTT). This step was repeated using the same buffer with 4% iodoacetamide in place of 1% DTT. The strips were then subjected to the second-dimensional electrophoresis after transfer onto 12.5% SDS-polyacrylamide gels. The second-dimension gels were cast between low fluorescent Pyrex glass plates (\(16\,\hbox {cm}\times 14\,\hbox {cm}\times 1\,\hbox {mm}\), GE Healthcare) to minimize background fluorescence during scanning. Electrophoresis was performed using the Hofer SE 600 system (GE Amersham) at 15 mA per gel for 30 min, followed by 30 mA per gel until the bromophenol blue reached the end of the gel.
Image acquisitions, analysis and processing
The gels were scanned using a Typhoon FLA 9000 Biomolecular Imager (GE Healthcare). Excitation and emission wavelengths for Cy2, Cy3, and Cy5 were 488/520, 532/580 and 633/670 nm, respectively. Gels were scanned at \(100\,\mu \hbox {m}\) resolution and the photo multiplier tube voltage was set to values ranging between 500 and 700 V to ensure maximum pixel intensity of 50,000 and 80,000 pixels for the three dyes.
Spot identification, background elimination, point matching and differential analysis of the protein spots were all completed using 2D differential in-gel analysis and biological variance analysis (DeCyder) software module; GE. The biological variation analysis module was used for integral matching of the internal standard and all the gel samples were subjected to comparative cross-gel statistical analysis based on spot volumes with a t test. P value \({<}0.05\) was considered statistically significant.
2D gel excision, tryptic digestion
Protein extracts were separated on preparative gels and proteins of interest were recovered from the gels for identification. Proteins (\(400\,\mu \hbox {g}\)) from 12Af1X and 12Ah2X were resolved on separate preparative polyacrylamide gels and were visualized by staining with a modified silver staining methods compatible with subsequent mass spectrometric analysis. All the differentially expressed spots (\({>}1.5\)-fold) were selected and excised manually from the two preparative gels. Each protein spot of interest cut from the preparative gels was destained for 20 min in 30 mM potassium ferricyanide/100 mM sodium thiosulphate (1:1 v/v) and washed with Milli-Q water until the gel pieces were destained, then was incubated in 0.2 M \(\hbox {NH}_{4}\hbox {HCO}_{3}\) for 20 min and then lyophilized. Each spot was digested overnight in \(5\,\hbox {ng}\,\mu \hbox {L}^{-1}\) trypsin in 25 mM \(\hbox {NH}_{4}\hbox {HCO}_{3}\). The peptides were extracted thrice with 60% ACN/0.1% TFA. The extracts were pooled and dried completely by a vacuum centrifuge.

The 3D visualization of the three missing proteins 986, 682 and 671 in the normal ShunMai 12Af compared to the necrotic ShunMai 12Ah.
MALDI-TOF-TOF MS analysis
MALDI-TOF-TOF instrument (4800 proteomics analyser) parameters were set using the 4000 Series Explorer software (Applied Biosystems, Foster City, USA). The MS spectra were recorded in reflector mode in a mass range from 800 to 4000 with a focus mass of 2000. MS used a CalMix5 standard to calibrate the instrument (ABI 4700 calibration mixture). For one main MS spectrum 25 subspectra with 125 shots per subspectrum were accumulated using a random search pattern. For MS calibration, autolysis peaks of trypsin (\([\mathrm{M}+\mathrm{H}]+842.5100\) and 2211.1046) were used as internal calibrates, and up to 10 of the most intense ion signals were selected as precursors for MS/MS acquisition, excluding the trypsin autolysis peaks and the matrix ion signals. In MS/MS positive ion mode, for one main MS spectrum 50 subspectra with 50 shots per subspectrum were accumulated using a random search pattern. The collision energy was 2 kV, the collision gas was air, and the default calibration was set using the Glu1-Fibrinopeptide B (\([\mathrm{M}+\mathrm{H}]+1570.6696\)) spotted onto Cal 7 positions of the MALDI target. Combined peptide mass fingerprinting PMF and MS/MS queries were performed by using the MASCOT search engine 2.2 (Matrix Science, Boston, USA) embedded into GPS-explorer software 3.6 (Applied Biosystems) on the NCBI database Viridiplantae with the following parameter settings: 100 ppm mass accuracy, trypsin cleavage one missed cleavage allowed, carbamidomethylation set as fixed modification, oxidation of methionine was allowed as variable modification, MS/MS fragment tolerance was set to 0.4 Da. A GPS explorer protein confidence index \(\ge \)95% was used for further manual validation.
Results
Fifty-five differentially expressed protein spots
Each 2D gel detected 1267 reproducible protein spots (\(n=9\)). DIGE acquisition (figure 2a) and 2D DeCyder software analysis revealed 24 significantly downregulated and 31 upregulated protein spots in the normal ShunMai 12Af compared to the necrotic ShunMai 12Ah (figure 2b).
Three protein spots disappeared due to the absence of Ne2
The three protein spots 986, 682, and 671, which were readily detectable in the necrotic ShunMai 12Ah, were absent in the normal ShunMai 12Af that did not contain Ne2 (figure 3). This qualitative difference suggests that Ne2 could encode one or two or three of those proteins (986, 682 and 671) because it was reported (Caldwell and Compton 1943) that the loss of Ne genes could be resulted from chromosomal aberration (Pan et al. 2015a). It is rational to suppose that protein spots 986, 682 and 671 might be located on the same lost fragment of chromosome 2BS.
The top 10 proteins matched in the database by spot 986 were all large subunits (LSUs) of dicotyledon. Because Ne2 is a nuclear gene, it could not encode the LSUs. In addition, the gels gave protein 986 the molecular weight of \({\sim }15\) kD which is of the correct order for the small subunits (SSUs) (12–18 kD) and is much less than that of the LSU (52–55 kD) (Mehta et al. 1992). Therefore protein 986 was first deduced to be a heterogeneous SSU of RuBisCO or at least to be involved closely with RuBisCO.
Identifications of the 32 differentially expressed proteins
The top 32 significantly changed proteins were analysed by MALDI-TOF-TOF MS and the database-matched identifications are summarized in table 2. The other 23 changed proteins that were not analysed by MALDI-TOF-TOF MS are listed in table 3.
The 21 photosynthesis\(\varvec{/}\)glucose-related proteins
Of the 32 identified proteins, 21 were photosynthesis/glucose-related proteins (of which seven proteins were upregulated and 14 proteins were downregulated significantly in the normal ShunMai 12Af), six were Met/Cys/Adenine-metabolism related proteins, three were disease-resistance related proteins, and two were antioxidant-related proteins (figure 4).
The seven RuBisCO-related proteins
Seven of the 21 photosynthesis/glucose related proteins are involved in RuBisCO. Proteins 480 (RuBisCO activase), 544 (RuBisCO activase) and 700 (SSU precursor), were upregulated, while proteins 612 (RuBisCO activase), 632 (SSU) and 896 (SSU) were downregulated significantly, and in contrast, protein 986 (LSU) was absent in the normal ShunMai 12Af. It is now clear that the subunits of the RuBisCO were altered by proteins 700, 632, 896 and 986, and the activity of RuBisCO activase system were changed by proteins 480, 544, and 612 in the normal ShunMai 12Af that lacked Ne2.
There are three unique findings when we sum up all these results: (i) the two NILs, the necrotic ShunMai 12Ah of the homozygous dominant genotype Ne1Ne1Ne2Ne2 vs the normal ShunMai 12Af of the Ne1Ne1ne2ne2 genotype; (ii) proteins 986, 682 and 671 were absent in the normal ShunMai 12Af due to the absence of Ne2; (iii) the composition and activity of the RuBisCO were evidently altered due to the absence of Ne2.
Discussion
Hypothetic RuBisCO maladjustment model
The three unique points mentioned above encouraged us to postulate the following hypothetical RuBisCO maladjustment model. The nuclear gene Ne2 could encode protein 986 that represents at least one copy of the SSUs. That copy encoded by Ne2 might be maladjusted to the LSUs encoded by Ne1’s rbcL. Such maladjustment might increase the oxygenase activity and the production of superoxide species such as \(\hbox {H}_{2}\hbox {O}_{2}\) in a cumulative and dynamic way, then affected the other germane proteins changing, finally leading to the early leaf death (figure 5).
Evolutionarily speaking, common wheat is an allohexaploid species (AABBDD) derived from natural hybridizations among at least three diploid ancestor grasses. In the nature of things, common wheat must have at least three types of chloroplast-genomes (A, D and B). Therefore the interactions between different chloroplast rbcL genes and different nuclear rbcS genes certainly lead to different RuBisCO effects. The bifunctional holoenzyme RuBisCO is composed of eight LSUs encoded by the chloroplast rbcL genes and eight SSUs encoded by the nuclear rbcS genes. That is to say, the production of RuBisCO needs the cooperation or complementation of nuclear genes and chloroplast genes. RuBisCO activity is not always balanced for optimal crop productivity (Mehta et al. 1992). The rbcS gene family of Triticum has more than 10 copies located at different sites on the genome, the wheat RuBisCOs are a collection of heterogeneous holoenzyme composed of different SSUs (Dean and Dunsmuir 1989). The B type cytoplasm demonstrates higher RuBPCase activity than those of D type cytoplasm (Evans and Austin 1986). In fact, a large number of interspecies hybrids featured necrotic leaves in their \(\hbox {F}_{1}\) plants. For example, one of the interspecies hybrids, the \(\hbox {F}_{1}\) plants of T. turgidum durum (BBAA)/Aegilops tauschii (DD) featured necrotic leaves (Kenan-Eichler et al. 2011), that might have implied a maladjustment between the AABB’s LSUs and the DD’s SSUs.

Functional categorization of the identified proteins. Twenty-one are photosynthesis/glucose-related proteins. Six are Met/Cys/Adenine metabolism-related proteins. Three are disease resistance-related proteins. Two are antioxidant-related proteins.
Chloroplast degeneration is the consequence of wheat hybrid necrosis
The microscopic observations confirmed that chloroplast degeneration as a consequence of ‘hybrid necrosis’ in wheat (Toxlpeus and Hermsen 1964). We also observed that the mesophyll cells of the necrotic ShunMai 12Ah died neatly following the chloroplast \(\rightarrow \) chromoplast \(\rightarrow \) gerontoplast steps (figures 6 and 7). The cell death process confirmed that morphologically chloroplasts died first. These observations also strongly support the hypothetical RuBisCO maladjustment model that leads to the chloroplast-pathway cell death.

Hypothetical RuBisCO maladjustment model. If any SSU were maladjusted to the LSUs, the balance of carboxylase/oxygenase activity of RuBisCO would be changed. If the photorespiration increased to a certain level, accumulated superoxide species such as \(\hbox {H}_{2}\hbox {O}_{2}\) would destroy the chloroplasts directly, which would cause the disastrous conglomeration of mesophyll cell content and lead to the so-called necrosis. \(\hbox {H}_{2}\hbox {O}_{2}\) is also used as a potent antimicrobial agent when cells are infected with a pathogen, and that would activate the antioxidant and/or immune systems causing hypersensitive response (HR).

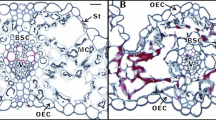
Light microscopic images of the dying mesophyll cells in pinnate-shape. Initially, plasmolysis startes; subsequently, cytoplasm shrinkage/condensation; finally, cytoplasm exhausted empty cells, the cell wall was maintained and unchanged. Vacuoles and condensation of nucleus could not be seen (for method details see Pan et al. 2015b).

SEM images of the dying mesophyll cells. Fme, faveolate-mesophyll of wheat leaf blade; Sr, stoma room; PTs, plasmolysis and cytoplasm shrinkage started; cCm, condensed cytoplasmic mass; eC, empty cells with the cell wall (W) remained and unchanged; Ec, ergastic crystals (for method details see Pan et al. 2015b).
Rethinking the autoimmunity model
Plant hybrid necrosis could result from autoimmunity, perhaps as a pleiotropic effect of the evolution of genes that are involved in pathogen response (Bomblies and Weigel 2007). Tsn1–ToxA interactions are associated with wheat necrosis and photosynthesis pathways (Farisa et al. 2010). To compare with those two hypothetic models on plant hybrid necrosis, in our study, proteins 276, 693 and 755 were all related to disease resistance; protein 682 was also associated with plant defense responses and closely related to the immune systems. Because protein 682 was also lost in the normal ShunMai 12Af, we may well assume that Ne2 encoded protein 682, and postulate an autoimmune model. However, we have to consider the following six points carefully. (i) It is known that Ne2 is tightly linked to Lr13, which in turn tightly linked to Lr23. Both Lr13 and Lr23 confer effective rust fungus resistance (Pukhalskiy et al. 2000). Because proteins 682 and 693 are pathogen-related (PR) proteins, they are more likely to be the product of Lr13 and Lr23 tightly linked to Ne2, rather than the products of Ne2 itself. (ii) Protein 682 was matched to glucan endo-1,3-beta-D-glucosidase (a polypeptide of 335 AA in wheat), which is responsible for the hydrolysis of beta-glucans, a process that does not produce any superoxide species that should increase dramatically in hybrid necrosis leaves (Khanna-Chopra et al. 1998). (iii) An autoimmune process should not occur only in leaves. Glucan endo-1,3-beta-D-glucosidase (protein 682) in wheat is not only in the chlorenchyma but also in root cells. However, the root cells are all normal in the necrotic plants of wheat (Caldwell and Compton 1943). Thus, even if the autoimmune process occurs only in the chlorenchyma, the question arises as to why the necrosis always starts at the leaf tip of the oldest leaf and gradually proceeds to the leaf base, rather than starting at all parts of the leaf blade simultaneously. (iv) \(\hbox {H}_{2}\hbox {O}_{2}\) is also used as a potent antimicrobial agent when cells are infected by pathogens, which could also lead to the hypersensitive cell death. (v) Glucan endo-1,3-beta-D-glucosidase in wheat cannot change the structure of LSUs or SSUs. The maladjusted RuBisCO can produce and accumulate superoxide species that can also activate resistance responses including glucosidase. (vi) In an autoimmune process, antibodies must occur. However, no new protein spots, i.e. no antibodies were detected in the necrotic \(\hbox {F}_{1}\)s of the parents Pan555 and Zheng891 (Jiang et al. 2008; Pan et al. 2009). These six arguments make us favour the hypothetical RuBisCO maladjustment model.
To further confirm this model, we urgently need to try: (i) test the other eight NILs hoping to obtain the Ne1 NILs to confirm if Ne1 also encodes protein(s) related to RuBisCO, (ii) separate and compare the RuBisCOs from the parents, ShunMai 12Ah, and ShunMai 12Af, to see if they are truly different in their SSUs and LSUs, (iii) sequence 986, 682 and 671 to reveal their AA sequences, (iv) sequence Ne2 and Ne1.
Conclusion
Ne2 is a structural gene that encodes protein(s)
Based on the confirmed conclusion that Ne2 is a dominant gene, we can also deduce that Ne2 is a regulatory gene that encodes miRNA which targets certain protein(s). If that is the case, the presence of Ne2 should have caused the absence of its target protein spots 986, 682 and 671, and the absence of Ne2 should have caused the presence of its target protein spots 986, 682 and 671. The two \(\hbox {F}_{14}\) NIL siblings with a different composition of the Ne2 gene, and the 2D and 2D-DIGE techs did reveal the conclusive relationship that the presence or absence of Ne2 caused the presence or absence of protein spots 986, 682 and 671. Thus it is rational to conclude that Ne2 is a structural gene that encodes protein(s).
The RuBisCO maladjustment model needs to be confirmed further
If further investigations on Ne1 and/or sequencings confirm the hypothetical RuBisCO maladjustment model, it would help scientists to radically improve the photosynthetic efficiency of wheat by modifying the RuBisCO genes and/or the assembly patterns (Liu et al. 2010) to make more efficient use of light, \(\hbox {CO}_{2}\) and water to increase sustainable global food security.
References
Bomblies K. and Weigel D. 2007 Hybrid necrosis: autoimmunity as a potential gene-flow barrier in plant species. Nat. Rev. Genet. 8, 382–393.
Caldwell R. M. and Compton L. E. 1943 Complementary lethal genes in wheat, causing a progressive lethal necrosis of seedlings. J. Hered. 34, 67–70.
Chu C. G., Faris J. D., Friesen T. L. and Xu S. S. 2006 Molecular mapping of hybrid necrosis genes Ne1 and Ne2 in hexaploid wheat using microsatellite markers. Theor. Appl. Genet. 112, 1374–1381.
Dean C. P. and Dunsmuir P. 1989 Structure, evolution and regulation of rbcS genes in higher plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 40, 415–439.
Evans J. R. and Austin R. B. 1986 The specific activity of RuBisCO in relation to genotype in wheat. Planta 167, 344–350.
Farisa J. D., Zhang Z. C., Lu H. J., Lua S., Reddy L., Cloutier S. et al. 2010 A unique wheat disease resistance-like gene governs effector-triggered susceptibility to necrotrophic pathogens. Proc. Natl. Acad. Sci. USA 1, 6.
Hermsen J. G. T. 1963a Sources and distribution of the complementary genes for hybrid necrosis in wheat. Euphytica 12, 147–160.
Hermsen J. G. T. 1963b The genetic basis of hybrid necrosis in wheat. Genetica 33, 245–287.
Jiang Q. Y., Chen H., Pan X. L., Pan Q. Y., Shi Y. H., Li X. R. et al. 2008 Proteomic analysis of wheat (Triticum aestivum L.) hybrid necrosis. Plant Sci 175, 394–401.
Kenan-Eichler M., Leshkowitz D., Tal L., Noor E., Melamed-Bessudo C., Feldman M. et al. 2011 Wheat hybridization and polyploidization results in deregulation of small RNAs. Genetics 188, 263–272.
Khanna-Chopra R., Dalal M. G., Kumar P., Laloraya M. 1998 A genetic system involving superoxide causes \(\text{ F }_{1}\) necrosis in wheat (T. aestivum L.). Biochemi. Biophysiol. Res. Commun. 248, 712–715.
Kostyuchenko I. A. 1936 The premature perishing of the hybrids in wheat crosses. Bull. Appl. Bot. Genet. Plant Breed. Ser. V-A Wheat 19, 127–137 (in Russian).
Liu C., Young A. L., Windhof A. S., Bracher A., Saschenbrecker S., Rao B. V. et al. 2010 Coupled chaperon action in folding and assembly of hexadecameric Rubisco. Nature 463, 197–202.
Mehta R. A., Fawcett T. W., Porath D. and Mattoo A. K. 1992 Oxidative stress causes rapid membrane translocation and in vivo degradation of ribulose-1,5-bisphosphate carboxylase/oxygenase. J. Biochem. 267, 2810–2816.
Naskidashvili P., Naskidashvili M., Naskidashvili I., Gakharia N. 2010 Revealing genes of hybrid necrosis and red hybrid chlorosis in crosses of varieties of georgian wheat and the importance of these genetic phenomena for selection and theoretical research. Bull. Georgian Natl. Acad. Sci. 4, 145–150.
Pan X. L., Jiang Q. Y., Pan Q. Y., Wen X. F., Shi Y. H., Wang Y. J. et al. 2009 Proteomic analysis of hybrid necrosis in wheat (Triticum aestivum) leaves. Funct. Plant Biol. 36, 251–259.
Pan X. L., Pan S. R., Shi Y. H., Pan Q. Y., Zhang L., Pan T. Y. et al. 2015a Registration of the wheat line ShunMai yyAh for hybrid necrosis. J. Plant Register. 9, 407–410.
Pan X. L., Pan S. R., Zhang G. Y., Shi Y. H., Zhang L., Yao M. P. et al. 2015b Anatomy of wheat flag leaf blade. Wheat Res. 36, 1–33 (in Chinese).
Pukhalskiy V. A., Martynov S. P. and Dobrotvorskaya T. V. 2000 Analysis of geographical and breeding-related distribution of hybrid necrosis genes in bread wheat (Triticum aestivum L.). Euphytica 114, 233–240.
Toxlpeus H. and Hermsen J. G. T. H. 1964 Chloroplast degeneration as a consequence of “hybrid necrosis” in wheat. Euphytica 13, 29–32.
Acknowledgements
We acknowledge the National Natural Science Foundation of China (grant no. 30971786) for financial support. All the protein tests were done in the labs of Shanghai Applied Protein Technology. We thank Drs Fred Magdoff (USA), Derek Smith (UK), Ming-Bo Wang (Australia) for help in searching for the age-old references on wheat hybrid necrosis and/or polishing the manuscript. We also thank Ms Lisa Brennan, English Teaching Centre, University of Liverpool, for the careful editing.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Pan S. R., Pan X. L., Pan Q. Y., Shi Y. H., Zhang L., Fan Y. and Xue Y. R. 2017 Ne2 encodes protein(s) and the altered RuBisCO could be the proteomics leader of hybrid necrosis in wheat (Triticum aestivum L.). J. Genet. 96, xx–xx]
Corresponding editor: Arun Joshi
Rights and permissions
About this article

Cite this article
Pan, S.R., Pan, X.L., Pan, Q.Y. et al. Ne2 encodes protein(s) and the altered RuBisCO could be the proteomics leader of hybrid necrosis in wheat (Triticum aestivum L.). J Genet 96, 261–271 (2017). https://doi.org/10.1007/s12041-017-0771-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12041-017-0771-1