
Abstract
The Eosin Y photocatalyzed Biginelli protocol has been established by a cascade one-pot three-component reaction of primary alcohols, α-ketoester, and urea to provide pharmacologically promising 3,4-dihydropyrimidin-2(1H)-ones in high yields. The key benefits of the present scheme are the capability to allow operational simplicity, readily available substrates, straightforward workup and high yields. This Eosin Y based photocatalytic approach can permit conquering traditional metal-catalyzed reactions in a sustainable manner, thus delivering economic and environmental rewards.
Graphical abstract
An environmentally benign protocol for the synthesis of 3,4-DHPMs has been developed using inexpensive molecular oxygen, visible light as green energy source and eosin Y as photoreceptor-cum-sensitizer. The utilized photoreceptor revealed its unique properties in rapid intersystem crossing, high photo and chemical stability, ease of separation and high catalytic efficiency.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The gradually increasing demand for greener methodology for concurrent chemical synthesis has enforced chemists to develop atomic economically and environmentally benign synthetic routes for producing well usable chemicals.1 Visible-light-assisted transformations have especially attracted growing interest due to their green and beneficial properties, sustainability, readily availability and ease of handling.2 In addition, compared to the conventional catalytic protocols, photo-catalysis under visible-light irradiation has been revealed as a powerful synthetic tool that produces mild and eco-friendly organic conversions.3,4,5,6 Exhilarate by this, various dyes and metal-complexes; bearing ruthenium and iridium, are reported as photocatalysts in the last couple of years especially.7,8,9,10,11,12,13,14,15,16
The controlled oxidation of alcohols is one of the important transformations in organic synthetic chemistry as their products play an important intermediate role in the formation of fine chemicals, important agrochemicals, pharmaceutical entities and other high-value products.17,18,19 Oxidation of primary aromatic alcohols are mostly achieved using rather strong oxidizing agents, that are toxic and hazardous to the environment i.e. hyperchlorite, permanganate, etc. and expensive noble metal catalysts including Au, Pt, Pd.20,21,22,23,24,25 As the alternative route, oxygen plays an important role as an excellent oxidant because of prevention of toxic, hazardous and stoichiometric by-products.26 Based on the perspective, various homogenous and heterogeneous metal catalysts have been reported. In equality, transition-metal free photocatalysts are greener and striking, because of inexpensive, easy departure from the reaction mixture and non-poisonous.27,28 So far, several photocatalytic methods have also been reported for the oxidation of primary aromatic alcohols.29,30,31,32,33,34
Notably, 3,4-dihydropyrimidin-2(1H)-one (DHPMs) are the core structural motifs for many potentially active biological molecules such as calcium channel blockers, ant-inflammatory and antitumor.35 DHPMs are identified as encouraging anticancer agents (Figure 1) especially monastrol, responsible to block the bipolar-mitotic-spindle in mammalian cells that results in triggering the arrest of G2/M mitotic phase further leading to cell apoptosis.36,37

Some DHPM derivatives with anticancer activity.
Various methods have been published in the literature for the composite of 3,4-dihydropyrimidinones by using ultrasonic irradiation, microwaves, ionic liquids, Thermal methods and metal catalysts (i.e. copper (II) sulfamate, Dendrimer-PWA).38,39,40,41,42,43,44,45,46,47,48 These methods and catalysts mentioned above have the common drawbacks of difficult work-up, lower product yield, noxious and steep catalysts, acidic circumstance and long-time reactions.49
The reported literature prompted us to explore a tandem cascade methodology for the fabrication of DHPMs utilizing primary aromatic alcohols. For a tandem cascade approach, a photooxidative system is required to be established that is selective and high yielding.
Here, we developed a greener and environmentally benign protocol for the synthesis of 3,4-DHPMs using molecular oxygen,28,50,51,52 visible light irradiation as a green energy source,53 eosin Y as photoreceptor and sensitizer, silver nitrate as an add-on photoreaction enhancer and inorganic salt K2S2O8 as a strong oxidizing agent.54 Eosin Y revealed unique properties like as rapid intersystem crossing to the lowest triplet state, high photo and chemical stability, ease of separation from the reaction mixture and high catalytic efficiency.55 This strategy embraces two distinct features involving activation of the system using visible light and initial activation of the dye through light absorption followed by system activation. Our investigated style has a prominent quality like easy workup, inexpensive catalyst, simple filtration, high yield and easy scalability. Our approach combines a dye i.e. Eosin Y, a light energy acceptor, with an electron acceptor photocatalyst, silver nitrate.
2 Experimental
2.1 General information and materials
General standard methods were used to purify and dry the solvents. Reagents and solvents (procured from Spectrochem, Aldrich, Acros and Merck) were used as such without added purification unless otherwise required. TLC (Analytical thin layer chromatography) was performed on Merck Kieselgel-60 F-254. Silica-gel 100-200 mesh was used to perform column chromatography. M.P. (Melting points) were recorded on Mel-Temp apparatus in capillary tubes and are uncorrected. Proton NMR spectra were attained at Bruker spectrometer (400 MHz) using CDCl3 as solvent (7.26 ppm- referenced to residual chloroform) or d6-DMSO (2.50 ppm – referenced to residual and 3.34 ppm – referenced to residual water in DMSO-d6). Chemical shift values are articulated in ppm (parts per million) downfield with respect to TMS. Coupling constant values (J values) are presented in Hz. 13C NMR spectra were obtained at 75 MHz in using Bruker spectrometers using CDCl3 as solvent (77.0 ppm – referenced to residual chloroform) or d6-DMSO (39.5 ppm – referenced to residual DMSO). Perkin Elmer (Spectrum-II) used for IR spectra. Mass spectrophotometer (Brucker-micrOTOF-QII) used for mass spectra.
2.2 Experimental procedures
2.2.1 General procedure of the synthesis of 3,4-dihydropyrimidin-2(1H)-ones
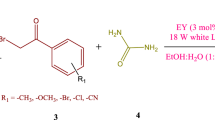
Alcohol 1b (1.0 mmol), α-ketoester 2b (1.0 mmol) and urea 3b (1.2 mmol) was dissolved in a mixture of acetonitrile and water (1:1) at room temperature in the presence of air bubble. Eosin Y (1.0 mmol), Silver nitrate (2.0 mol%) and potassium persulphate (1.0 mmol) was added and the reaction mixture was stirred for 48 h under visible light at room temperature. The reaction was monitored using TLC. After the completion of reaction, the reaction mixture was partitioned between water and ethyl acetate. The separated organic layer was washed with saturated brine solution, dried over anhydrous sodium sulfate, concentrated in vacuo to afford compounds DHPM with excellent yields (upto 88%). The compounds DHPM were further purified by using column chromatography over silica gel with the mixture of ethylacetate/hexane to get the pure DHPMs.
Ethyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate 4a: Yield 88%; white solid, M.p. 203–204 °C; IR (ATR) ν cm−1 3243 (N-H), 1701 (C=O), 1638 (C=C). 1H NMR (400 MHz, DMSO-d6) δ 9.22 (1H, s, NH), 7.75 (1H, s, NH), 7.27 (5H, m, ArH), 5.15 (1H, d, J = 4.0 Hz, CH), 3.98 (2H, q, J = 15.2, 8.0 Hz, CH2), 2.26 (3H, s, CH3), 1.10 (3H, t, J = 8.0 Hz, CH3); 13C NMR (75 MHz, DMSO-d6) δ 165.9, 153.1, 148.3, 145.3, 129.1, 128.2, 127.8, 98.2, 60.2, 55.5, 19.0, 14.7. MS m/z 261 (M+1); Anal. Calc. for C14H16N2O3: C, 64.60; H, 6.20; N, 10.76; found: C, 64.59; H, 6.23; N, 10.73.
Methyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate, 4b: Yield 80%; White solid; M.p. 208–210 °C; IR (ATR) ν cm−1 3228 (N-H), 1697 (C=O), 1653 (C=C).1HNMR (400 MHz, DMSO-d6) δ 9.20 (1H, s, NH), 7.70 (1H, s, NH), 7.29 (5H, m, ArH), 5.13 (1H, d, J = 4.0 Hz, CH), 3.70 (s, OCH3), 2.28 (3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ 164.3, 152.7, 148.9, 145.1, 128.9, 128.2, 127.5, 100.2, 55.6, 54.1, 15.7. MS m/z247 (M+1); Anal. Calc. for C13H14N2O3: C, 63.40; H, 5.73; N, 11.38; found: C, 63.42; H, 5.76; N, 11.33.
Ethyl 4-(4-chlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate, 4c: Yield 75%; white solid; M.p. 213-215 °C; IR (ATR) ν cm−1 3239 (N-H), 1701 (C=O), 1638 (C=C).1H NMR (400 MHz CDCl3) δ 7.98 (s,1H, NH), 5.81 (s, 1H, NH), 7.27-7.33 (m, 4H, ArH), 5.41 (s, 1H, CH), 4.10 (2H, q, CH2), 2.38 (3H, s, CH3), 1.21 (3H, t, CH3); 13C NMR (75MHz, CDCl3) δ 165.4, 153.0, 146.3, 142.1, 133.7, 128.9, 128.0,101.1, 60.2, 55.17, 18.7; MS m/z 296 (M+2); Anal. Calc. for C14H15ClN2O3: C, 57.05; H, 5.13; N, 9.50; found: C, 57.04; H, 5.18; N, 9.42.
Methyl 4-(4-chlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate, 4d: Yield 73%; white solid; M.p. 180-181 °C; IR (ATR) ν cm−1 3225 (N-H), 1706 (C=O), 1635 (C=C).1H NMR (400 MHz DMSO-d6) δ 9.30 (s,1H, NH), 7.72 (s, 1H, NH), 7.39 (m, 4H, ArH), 5.12 (s, 1H, CH), 3.59 (s, 3H, OCH3), 2.25 (s, 3H, CH3); 13C NMR (75MHz, DMSO-d6) δ 165.3, 152.8, 149.5, 132.8, 132.3, 129.5, 128.4, 128.0, 127.7, 98.9, 51.5, 51.4, 18.7; MS m/z 282 (M+2); Anal. Calc. for C13H13ClN2O3: C, 55.62; H, 4.67; N, 9.98; found: C, 55.64; H, 4.71; N, 9.94.
Ethyl 4-(4-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate, 4e: Yield 85%; light brown solid; M.p. 205-206 °C; IR (ATR) ν cm−1 3227 (N-H), 1705 (C=O), 1643 (C=C).1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, 1H, NH), 8.30 (s, 1H, NH), 7.30 (m, 2H, ArH), 6.79 (m, 2H, ArH), 5.25 (s, 1H, CH), 3.95 (2H, q, J = 16.0, 8.0 Hz, CH2), 3.84 (s, 3H, Ar-OCH3), 2.30 (3H, s, CH3), 1.09 (3H, t, J = 8.0 Hz, CH3). 13C NMR (75MHz, DMSO-d6) δ 165.6, 160.5, 153.8, 134.5, 127.9, 113.8, 106.6, 55.8, 52.5, 52.9, 19.3; MS m/z291 (M+1); Anal. Calc. for C15H18N2O4: C, 62.06; H, 6.25; N, 9.65; found: C, C, 62.08; H, 6.28; N, 9.60.
Methyl 4-(4-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate, 4f: Yield 82%; light brown solid; M.p. 187-188 °C; IR (ATR) ν cm−1 3226 (N-H), 1708 (C=O), 1653 (C=C). 1H NMR (400 MHz, DMSO-d6) δ 10.24 (s, 1H, NH), 8.57 (s, 1H, NH), 7.36 (m, 2H, ArH), 6.83 (m, 2H, ArH), 5.28 (s, 1H, CH), 3.83 (s, 3H, Ar-OCH3), 3.45 (s, 3H, OCH3), 2.30 (s, 3H, CH3). 13C NMR (75MHz, DMSO-d6) δ 166.0, 160.8, 154.2, 134.8, 127.4, 113.2, 106.5, 56.5, 51.8, 51.2, 19.9; MS m/z 277 (M+1); Anal. Calc. for C14H16N2O4: C, 60.86; H, 5.84; N, 10.14; found: C, 60.90; H, 5.81; N, 10.10.
Ethyl 6-methyl-2-oxo-4-(p-tolyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate 4g: Yield 78%; light brown solid; M.p. 209-210 °C; IR (ATR) ν cm−1 3241 (N-H), 1700 (C=O), 1641 (C=C).1H NMR (400 MHz, DMSO-d6,) δ 10.36 (s, 1H, NH), 8.47 (s, 1H, NH), 7.20 (m, 2H, ArH), 6.72 (m, 2H, ArH), 5.31 (s, 1H, CH), 3.91 (2H, q, J = 16.0, 8.0 Hz, CH2), 2.32 (3H, s, CH3), 2.21 (s, 3H, Ar-CH3), 1.11 (3H, t, J = 8.0 Hz, CH3); 13C NMR (75MHz, DMSO-d6) δ 165.2, 152.7, 151.1, 139.9, 134.7, 129.9, 128.5, 107.7, 53.9, 51.7, 21.0, 19.1; MS m/z275 (M+1); Anal. Calc. for C15H18N2O3: C, 65.68; H, 6.61; N, 10.21; found: C, 65.70; H, 6.66; N, 10.19.
Methyl 6-methyl-2-oxo-4-(p-tolyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate 4h: Yield 80%; light brown solid; M.p. 214-215 °C; IR (ATR) ν cm−1 3245 (N-H), 1703 (C=O), 1632 (C=C).1H NMR (400 MHz, DMSO-d6,) δ 10.50 (s, 1H, NH), 8.35 (s, 1H, NH), 7.01 (m, 4H, ArH), 5.20 (s, 1H, CH), 3.54 (s, 3H, OCH3), 2.28 (s, 3H, CH3), 2.24 (s, 3H, Ar-CH3); 13C NMR (75MHz, DMSO-d6) δ 165.3, 152.2, 151.0, 139.6, 134.0, 129.2, 128.4, 107.0, 53.3, 51.5, 21.7, 19.5; MS m/z 261 (M+1); Anal. Calc. for C14H16N2O3: C, 64.60; H, 6.20; N, 10.76; found: C, 64.53; H, 6.23; N, 10.68.
Ethyl 4-(4-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate, 4i: Yield 82%; white solid; M.p. 230-232 °C; IR (ATR) ν cm−1 3229 (N-H), 1706 (C=O), 1639 (C=C).1H NMR (400 MHz DMSO-d6) δ 9.53 (s,1H, NH), 7.84 (s, 1H, NH), 7.13 (m, 2H, ArH), 6.79 (m, 2H, ArH), 5.10 (s, 1H, CH), 3.87 (2H, q, J = 16.0, 8.0 Hz, CH2), 2.28 (3H, s, CH3), 1.07 (3H, t, J = 8.0 Hz, CH3); 13C NMR (75MHz, DMSO-d6) δ 165.1, 152.7, 149.8, 132.4, 132.8, 129.7, 128.1, 128.0, 127.7, 98.7, 51.7, 51.4, 18.9; MS m/z277 (M+1); Anal. Calc. for C14H16N2O4: C, 60.86; H, 5.84; N, 10.14; found: C, 60.88; H, 5.94; N, 10.08.
Methyl 4-(4-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate, 4j: Yield 80%; white solid; M.p. 240-242 °C; IR (ATR) ν cm−1 3231 (N-H), 1704 (C=O), 1636 (C=C).1H NMR (400 MHz DMSO-d6) δ 9.43 (s,1H, NH), 7.77 (s, 1H, NH), 7.00 (m, 4H, ArH), 5.08 (s, 1H, CH), 3.60 (s, 3H, OCH3), 2.27 (s, 3H, CH3); 13C NMR (75MHz, DMSO-d6) δ 166.5, 152.9, 149.8, 132.5, 132.4, 129.7, 128.9, 128.4, 127.8, 98.7, 51.7, 51.6, 18.6; MS m/z263 (M+1); Anal. Calc. for C13H14N2O4: C, 59.54; H, 5.38; N, 10.68; found: C, 59.55; H, 5.47; N, 10.60.
Ethyl 6-methyl-2-oxo-4-propyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate, 4k: Yield 5%; White solid; M.p. 154-156 °C IR (ATR) ν cm−1 3246 (N-H), 1708 (C=O), 1632 (C=C).1HNMR (400 MHz, CDCl3) δ7.65 (1H, s, NH), 5.60 (1H, s, NH), 4.25 (1H, t, CH), 4.11 (2H, q, CH2), 2.22 (3H, s, CH3), 1.64 (4H, m, CH2-CH2), 1.21 (t, 3H, -CH3), 0.85 (t, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ165.9, 154.2, 146.5, 101.7, 59.9, 51.4, 39.1, 18.6, 17.6, 14.3.MS m/z227 (M+1); Anal. Calc. for C11H18N2O3: C, 58.39; H, 8.02; N, 12.38; found: C, 58.43; H, 8.14; N, 12.31.
Ethyl 4-ethynyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate, 4l: Yield 22%; White solid; IR (ATR) ν cm−1 3247 (N-H), 1705 (C=O), 1635 (C=C).1HNMR (400 MHz, DMSO-d6) δ 9.30 (1H, s, NH), 7.69 (1H, s, NH), 5.03 (1H, s, CH), 3.90 (2H, q, J = 16.0, 8.0 Hz, CH2), 3.16 (1H, s, CH), 2.27 (3H, s, CH3), 1.25 (t, 3H, -CH3); 13C NMR (75 MHz, DMSO-d6) δ 167.1, 150.4, 147.9, 106.5, 81.1, 72.9, 65.7, 45.2, 17.4, 15.1. MS m/z209 (M+1); Anal. Calc. for C10H12N2O3: C, 57.68; H, 5.81; N, 13.45; found: C, 57.66; H, 5.85; N, 13.39.
Ethyl 4-(3-nitrophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate, 4m: Yield 72%; yellow solid; M.p. 228-230 °C; IR (ATR) ν cm−1 3333 (N-H), 1707 (C=O), 1621(C=C).1H NMR (400 MHz DMSO-d6) δ9.38 (s,1H, NH), 8.16 (s, 1H, NH), 7.6-8.10 (m, 4H, ArH), 5.31 (s, 1H, CH), 4.0 (2H, q, CH2), 2.28 (3H, s, CH3), 1.11 (3H, t, CH3); 13C NMR (75MHz, DMSO -d6) δ165.5, 152.2, 149.9, 148.2, 147.4133.4, 130.7, 122.8, 121.4,98.8, 59.8, 54.0, 18.3, 14.4.
3 Results and Discussion
The exploration was started by performing the reaction of benzyl alcohol (1a), ethyl acetoacetate (2a), urea (3a) and K2S2O8 (1 eq.) in acetonitrile/water (1:1) mixture under an open atmosphere and in a dark place at room temperature. The entire substrate was unreacted (Table 1, entry 1) and did not proceed at all even after 48 h. The above testing reaction was also performed at an elevated temperature of 50 °C but could not enhance the result of the reaction. The above test reaction was further studied in the presence of silver nitrate which does not afford any product (Table 1, entry 2). Silver nitrate was replaced with silver acetate and TiO2 but the formation of the product may not be realized (Table 1, entry 3). Following, we examined a similar investigation in visible light (source: white LED bulb), which enabled the formation of traces of the final product on spending 48 h with 1a (Table 1, entry 4). Besides, a similar model reaction was conducted using Eosin Y as photocatalyst (1 mol%), which provided the synthesis of desired 3,4-DHPM 4a was obtained in 48 h with 45% yield under photoreaction (Table 1, entry 5). The characterization of 4a was furnished by 1H NMR, 13C NMR, Mass-Spectrum and IR spectral studies, and found to be matched identically with the previously reported compounds.
The above outcome was extremely encouraging, for further optimization of the reaction to get an elevated yield of required product 4a. Subsequently, the template reaction was executed by varying amounts of photocatalyst Eosin Y, which does not improve the yield of the wanted product 4a (Table 1, entry 6). We used an organic dye Eosin Y as a photo-catalyst to initiate the reaction, which leads to the dehydrogenation of alcohol into desired carbonyl compound.53 Eosin Y worked as photocatalyst in the reaction. Then, we performed the reaction with varying amounts of K2S2O8 which revealed a decline in the yield of the desired product 4a (Table 1, entries 7 & 8). Potassium persulphate (K2S2O8) used in this protocol is not a photocatalyst, but photolysis of S2O82− to generate sulphate radical anion (SO4. −), which acts as a strong oxidizing agent in an aqueous system.54 The activity of K2S2O8 also depends on the amount of K2S2O8 used in the reaction. The reaction with a high amount of K2S2O8 reduced the desired yield by over-oxidation of alcohol into a carboxylic acid.
To improve the effectiveness of this reaction, we examined the altering amount of AgNO3 commencing 1.0 to 3.0 mol % (Table 1, entries 9-11). It was detected that 2.0 mol % was found as the best possible protocol, which facilitated the yield of the avidity product 4a to 88% in 40 h (Table 1, entry 10). Further increase in the quantity of silver nitrate could not get better yield (Table 1, entry 11). Silver nitrate helps in increasing the oxidation in reaction.56 The role of silver is to activate the molecular oxygen by adsorbing on their surface. It also enhanced the efficiency of eosin Y under the aqueous phase.57,58,59
Afterwards, we carefully evaluated the model reaction with different solvent systems such as DMSO, ethanol, H2O, chloroform, CH3CN and found that the CH3CN/H2O mixture was the most suitable solvent for this reaction as it increases the yield to 88% (Table 1, entries 12-16). Acetonitrile is a good solvent for photo-oxidation.60 It does not only possess strong polarity but also have a good dissolvent capacity of oxygen. To find out the impact of other photocatalysts, we examined the model reaction with different organic photocatalysts (Table 1, entries 17 & 18), which did not enhance the yield of the product.
Hence, the evaluated eosin Y (1.0 mol %), K2S2O8 (1.0 equiv.), AgNO3 (2.0 mol %) were the best choices with visible light irradiation at room temperature under an oxygen atmosphere.
With the optimized reaction conditions in hand, the substrate coverage of this photocatalytic oxidation system was explored (Scheme 1). Based on our initial efforts to obtain the high efficiency of photocatalytic conversion into the desired product, different primary aromatic and aliphatic alcohols were evaluated (Table 2). All the substituted benzyl alcohols with electron-donating and electron-withdrawing groups were easily utilized in this cascade approach in getting substituted 3,4-DHPMs in high yields (Table 2, compound 4a-4j, 4m). Electron-releasing substituents at para, position on the phenyl group were found to be efficient in accelerating the reaction, while electron-withdrawing groups substituents at meta and para position on phenyl group needed longer reaction times for their optimized conversions. Compared with the primary aromatic alcohols, primary aliphatic alcohols are found to be very less reactive.

Synthesis of various derivatives of 3,4-dihydropyrimidin-2(1H)-ones.
We evaluated various derivatives by using different types of primary alcohol (Benzyl alcohol, 4-chlorobenzyl alcohol, propargyl alcohol, methanol and butanol etc.) and α-ketoester (ethyl acetoacetate and methyl acetoacetate) in the reaction (Scheme 1). We used benzyl alcohol with ethyl acetoacetate and urea under similar reaction conditions, which gave 88% yield (4a) and reaction accomplished in 48 h (entry 1, Table 2).
Further benzyl alcohol treated with methyl acetoacetate and reaction conditions remained same which obtained 80% yield of the product (4b) in 48 h (entry 2, Table 2). We also found that both methyl acetoacetate and ethyl acetoacetate under similar optimized conditions gave good to excellent yields between 73-88% with aromatic alcohols (Table 2, entries 4c-4j, 4m). Further, we also treated aliphatic alcohols under similar reaction condition with ethyl acetoacetate that yielded in poor (Table 2, entries 4k-4l) even after an extended duration of time up to 72 h.
A plausible mechanism has been proposed for the in-situ oxidation of alcohol and the formation of 3,4-DHPMs which is summarized in Scheme 2. The sulphate radical anion (SO4. −) acts as an oxidizing agent under photo-irradiative conditions.54,67 It accepts one electron from 3EY* forming sulphate anion (SO42−) and converts it into radical cation (EY+.) Subsequently, EY+• accepts an electron from benzyl alcohol (1b) to regenerate EY and produce benzyl alcohol radical cation (5, Scheme 2). Further, benzyl alcohol radical (6) is formed due to the removal of a proton from 5.53 The Ag(I) activates the molecular oxygen (O2) and transforms it into radical anion (O2−.)58 that further accepts proton form superoxide radical (.OOH). The .OOH transforms 6 into carbonyl compound (7) (Scheme 2).17

Proposed mechanism step-I in-situ oxidation of primary alcohol.
The eosin Y gets involve in both the reductive and oxidative quenching cycles.68,69 The eosin Y activates both 7 and β-keto ester (2b) by donating and accepting one electron respectively. The activated aldehyde (8) further interacts with urea to form imine (11) and releases a molecule of H2O. The activated β-keto ester (9) attacks on imine (11) to form 3,4-DHPM by releasing water molecule (Scheme 3).47,70

Proposed mechanism step-II formation of 3,4-DHPMs.
4 Conclusions
We have disclosed a robust, efficient, and domino multicomponent cascade novel protocol to design 3,4-dihydropyrimidin-2(1H)-one derivative utilizing Biginelli reaction of primary alcohols using visible-light as green energy source. The key features of the present protocol include the capability to allow an operational simplicity, readily available substrates, straightforward workup, and high yields of the products. The synthetic efficacy and practicality of this Eosin Y based photocatalytic approach can allow in capacitating conventional metal-catalyzed reactions and could be rousing towards functionalization of a broad variety of C-C, and C-N bonds in a sustainable manner.
References
Zhou Q Q, Zou Y Q, Lu L Q and Xiao W J 2019 Visible-light-induced organic photochemical reactions through energy-transfer pathways Angew. Chem. Int. Edit. 58 1586
Schultz D M and Yoon T P 2014 Solar synthesis: prospects in visible light photocatalysis Science 343 6174
Prier C K, Rankic D A and MacMillan D W 2013 Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis Chem. Rev. 113 5322
Xi Y, Yi H and Lei A 2013 Synthetic applications of photoredox catalysis with visible light Org. Bio. Chem. 11 2387
Welin E R, Le C, Arias-Rotondo D M, McCusker J K and MacMillan D W 2017 Photosensitized, energy transfer-mediated organometallic catalysis through electronically excited nickel (II) Science 355 380
Kainz Q M, Matier C D, Bartoszewicz A, Zultanski S L, Peters J C and Fu G C 2016 Asymmetric copper-catalyzed CN cross-couplings induced by visible light Science 351 681
Lang X, Ma W, Chen C, Ji H and Zhao J 2014 Selective aerobic oxidation mediated by TiO2 photocatalysis Acc. Chem. Res. 47 355
Tsukamoto D, Shiraishi Y, Sugano Y, Ichikawa S, Tanaka S and Hirai T 2012 Gold nanoparticles located at the interface of anatase/rutile TiO2 particles as active plasmonic photocatalysts for aerobic oxidation J. Am. Chem. Soc. 134 6309
Hering T, Slanina T, Hancock A, Wille U and König B 2015 Visible light photooxidation of nitrate: the dawn of a nocturnal radical Chem. Comm. 51 6568
Yoon T, Ischay M and Du J 2010 Visible light photocatalysis as a greener approach to photochemical synthesis Nat. Chem. 2 527
Zuo Z, Ahneman D T, Chu L, Terrett J A, Doyle A G and MacMillan D W 2014 Merging photoredox with nickel catalysis: coupling of α-carboxyl sp3-carbons with aryl halides Science 345 437
Narayanam J M, Tucker J W and Stephenson C R 2009 Electron-transfer photoredox catalysis: development of a tin-free reductive dehalogenation reaction J. Am. Chem. Soc. 131 8756
Cuthbertson J D and MacMillan D W 2015 The direct arylation of allylic sp 3 C–H bonds via organic and photoredox catalysis Nature 519 74
Hari D P, Schroll P and König B 2012 Metal-free, visible-light-mediated direct C–H arylation of heteroarenes with aryl diazonium salts J. Am. Chem. Soc. 134 2958
Ghosh I, Ghosh T, Bardagi J I and König B 2014 Reduction of aryl halides by consecutive visible light-induced electron transfer processes Science 346 725
Meng Q Y, Zhong J J, Liu Q, Gao X X, Zhang H H, Lei T, et al. 2013 A cascade cross-coupling hydrogen evolution reaction by visible light catalysis J. Am. Chem. Soc. 135 19052
Ji X, Chen Y, Paul B and Vadivel S 2019 Photocatalytic oxidation of aromatic alcohols over silver supported on cobalt oxide nanostructured catalyst J. All. Comp. 783 583
Enache D I, Edwards J K, Landon P, Solsona-Espriu B, Carley A F, Herzing A A, et al. 2006 Solvent-free oxidation of primary alcohols to aldehydes using Au-Pd/TiO2 catalysts Science 311 362
Pillai U R and Sahle-Demessie E 2003 Oxidation of alcohols over Fe3+/montmorillonite-K10 using hydrogen peroxide App. Cat. A Gen. 245 103
Ding J, Xu W, Wan H, Yuan D, Chen C, Wang L, et al. 2018 Nitrogen vacancy engineered graphitic C3N4-based polymers for photocatalytic oxidation of aromatic alcohols to aldehydes App. Cat. B Env. 221 626
Lee J and Lee J C 2018 An efficient oxidation of alcohols by aqueous H2O2 with 1, 3-dibromo-5, 5-dimethylhydantoin Lett. Org. Chem. 15 895
ten Brink G J, Arends I W and Sheldon R A 2000 Green, catalytic oxidation of alcohols in water Science 287 1636
Mori K, Hara T, Mizugaki T, Ebitani K and Kaneda K 2004 Hydroxyapatite-supported palladium nanoclusters: a highly active heterogeneous catalyst for selective oxidation of alcohols by use of molecular oxygen J. Am. Chem. Soc. 126 10657
Marko I E, Giles P R, Tsukazaki M, Brown S M and Urch C J 1996 Copper-catalyzed oxidation of alcohols to aldehydes and ketones: an efficient, aerobic alternative Science 274 2044
Fu R, Yang Y, Ma X, Sun Y, Li J, Gao H, et al. 2017 An efficient, eco-friendly and sustainable one-pot synthesis of 3, 4-dihydropyrimidin-2 (1H)-ones directly from alcohols catalyzed by heteropolyanion-based ionic liquds Molecules 22 1531
Dai Y, Ren P, Li Y, Lv D, Shen Y, Li Y, et al. 2019 Solid base Bi24O31Br10(OH)δ with active lattice oxygen for the efficient photo-oxidation of primary alcohols to aldehydes Angew. Chem. Int. Edit. 58 6265
Schilling W, Riemer D, Zhang Y, Hatami N and Das S 2018 Metal-free catalyst for visible-light-induced oxidation of unactivated alcohols using air/oxygen as an oxidant ACS Cat. 8 5425
Zhang Y, Schilling W, Riemer D and Das S 2020 Metal-free photocatalysts for the oxidation of non-activated alcohols and the oxygenation of tertiary amines performed in air or oxygen Nature Prot. 15 822
Zhang X, Rakesh K, Ravindar L and Qin H L 2018 Visible-light initiated aerobic oxidations: a critical review Green Chem. 20 4790
Yu X, Wang L and Cohen S M 2017 Photocatalytic metal–organic frameworks for organic transformations CrystEngComm 19 4126
Chen B, Wang L and Gao S 2015 Recent advances in aerobic oxidation of alcohols and amines to imines ACS Cat. 5 5851
Fan W, Yang Q, Xu F and Li P 2014 A visible-light-promoted aerobic metal-free C-3 thiocyanation of indoles J. Org. Chem. 79 10588
Mitra S, Ghosh M, Mishra S and Hajra A 2015 Metal-free thiocyanation of imidazoheterocycles through visible light photoredox catalysis J. Org. Chem. 80 8275
Yadav A K and Yadav L D S 2015 Visible-light-mediated difunctionalization of styrenes: an unprecedented approach to 5-aryl-2-imino-1, 3-oxathiolanes Green Chem. 17 3515
Cui Y, Li C and Bao M 2019 Deep eutectic solvents (DESs) as powerful and recyclable catalysts and solvents for the synthesis of 3, 4-dihydropyrimidin-2 (1H)-ones/thiones Green Pro. Syn. 8 568
Mayer T U, Kapoor T M, Haggarty S J, King R W, Schreiber S L and Mitchison T J 1999 Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen Science 286 971
Russowsky D, Canto R M F, Sanches S A, D’Oca M G, De F Â, Pilli R A, et al. 2006 Synthesis and differential antiproliferative activity of Biginelli compounds against cancer cell lines: monastrol, oxo-monastrol and oxygenated analogues Bio. Chem. 34 173
Chen X H, Xu X Y, Liu H, Cun L F and Gong L Z 2006 Highly enantioselective organocatalytic Biginelli reaction J. Am. Chem. Soc. 128 14802
Rahman M, Majee A and Hajra A 2010 Microwave-assisted Brønsted acidic ionic liquid-promoted one-pot synthesis of heterobicyclic dihydropyrimidinones by a three-component coupling of cyclopentanone, aldehydes, and urea J. Het. Chem. 47 1230
Shen Z L, Xu X P and Ji S J 2010 Brønsted base-catalyzed one-pot three-component Biginelli-type reaction: an efficient synthesis of 4, 5, 6-triaryl-3, 4-dihydropyrimidin-2 (1 H)-one and mechanistic study J. Org. Chem. 75 1162
Moosavifar M 2012 An appropriate one-pot synthesis of dihydropyrimidinones catalyzed by heteropoly acid supported on zeolite: an efficient and reusable catalyst for the Biginelli reaction Comp. Ren. Chim. 15 444
Ramos L M, Ponce L T A Y, dos Santos M R, de Oliveira H C, Gomes A F, Gozzo F C, et al. 2012 Mechanistic studies on lewis acid catalyzed biginelli reactions in ionic liquids: evidence for the reactive intermediates and the role of the reagents J. Org. Chem. 77 10184
Fu R, Yang Y, Lai W, Ma Y, Chen Z, Zhou J, et al. 2015 Efficient and green microwave-assisted multicomponent Biginelli reaction for the synthesis of dihydropyrimidinones catalyzed by heteropolyanion-based ionic liquids under solvent-free conditions Synth. Comm. 45 467
Mansoor S S, Shafi S S and Ahmed S Z 2016 An efficient one-pot multicomponent synthesis of 3, 4-dihydropyrimidine-2-(1H)-ones/thiones/imines via a Lewis base catalyzed Biginelli-type reaction under solvent-free conditions Arab. J. Chem. 9 S846
Mohammadi B and Behbahani F K 2018 Recent developments in the synthesis and applications of dihydropyrimidin-2 (1H)-ones and thiones Mol. Div. 22 405
Safaei G J, Tavazo M and Mahdavinia G H 2018 Ultrasound promoted one-pot synthesis of 3, 4-dihydropyrimidin-2 (1H)-ones/thiones using dendrimer-attached phosphotungstic acid nanoparticles immobilized on nanosilica Ultra Sono. 40 230
Harsh S, Kumar S, Sharma R, Kumar Y and Kumar R 2020 Chlorophyll triggered one-pot synthesis of 3, 4-dihydropyrimidin-2 (1H)-ones via photo induced electron transfer reaction Arab. J. Chem. 13 4720
Wang J, Li Y, Peng Y and Song G 2014 Silver nitrate-catalyzed selective air oxidation of benzylic and allylic alcohols to corresponding aldehydes or ketones J. Chin. Chem. Soc. 61 517
Ghosh S, Saikh F, Das J and Pramanik A K 2013 Hantzsch 1, 4-dihydropyridine synthesis in aqueous ethanol by visible light Tetrahedron Lett. 54 58
Choudhary V R, Dhar A, Jana P, Jha R and Uphade B S 2005 A Green process for chlorine-free benzaldehyde from the solvent-free oxidation of benzyl alcohol with molecular oxygen over a supported nano-size gold catalyst Green Chem. 7 768
Nikitas N F, Tzaras D I, Triandafillidi I and Kokotos C G 2020 Photochemical oxidation of benzylic primary and secondary alcohols utilizing air as the oxidant Green Chem. 22 471
Prebil R, Stavber G and Stavber S 2014 Aerobic oxidation of alcohols by using a completely metal-free catalytic system Eur. J. Org. Chem. 2014 395
Yang X J, Zheng Y W, Zheng L Q, Wu L Z, Tung C H and Chen B 2019 Visible light-catalytic dehydrogenation of benzylic alcohols to carbonyl compounds by using an eosin Y and nickel–thiolate complex dual catalyst system Green Chem. 21 1401
Ghosh P P, Mukherjee P and Das A R 2013 Triton-X-100 catalyzed synthesis of 1, 4-dihydropyridines and their aromatization to pyridines and a new one pot synthesis of pyridines using visible light in aqueous media RSC Adv. 3 8220
Srivastava V, Singh P K and Singh P P 2019 Eosin Y catalysed visible-light mediated aerobic oxidation of tertiary amines Tetrahedron Lett. 60 151041
Jeena V and Robinson R S 2012 Convenient photooxidation of alcohols using dye sensitised zinc oxide in combination with silver nitrate and TEMPO Chem. Comm. 48 299
Alwan D B 2016 Effect of solvent type and annealing temperature on efficiency for Eosin-y dye sensitized solar cells Ir. J. Sci. 57(4A) 2429
Nagaraju P, Balaraju M, Reddy K M, Prasad P S and Lingaiah N 2008 Selective oxidation of allylic alcohols catalyzed by silver exchanged molybdovanado phosphoric acid catalyst in the presence of molecular oxygen Cat. Comm. 9 1389
Beier M J, Hansen T W and Grunwaldt J D 2009 Selective liquid-phase oxidation of alcohols catalyzed by a silver-based catalyst promoted by the presence of ceria J. Cat. 266 320
Reimers J R and Hall L E 1999 The solvation of acetonitrile J. Am. Chem. Soc. 121 3730
Fu N Y, Yuan Y F, Cao Z, Wang S W, Wang J T and Peppe C 2002 Indium (III) bromide-catalyzed preparation of dihydropyrimidinones: improved protocol conditions for the Biginelli reaction Tetrahedron 58 4801
Heravi M M, Derikvand F and Bamoharram F F 2005 A catalytic method for synthesis of Biginelli-type 3, 4-dihydropyrimidin-2 (1H)-one using 12-tungstophosphoric acid J. Mol. Cat. A Chem. 242 173
Mohamadpour F and Lashkari M 2018 Three-component reaction of β-keto esters, aromatic aldehydes and urea/thiourea promoted by caffeine, a green and natural, biodegradable catalyst for eco-safe Biginelli synthesis of 3, 4-dihydropyrimidin-2 (1H)-ones/thiones derivatives under solvent-free conditions J. Serb. Chem. Soc. 83 673
Yao B J, Wu W X, Ding L G and Dong Y B 2021 Sulfonic acid and ionic liquid functionalized covalent organic framework for efficient catalysis of the Biginelli reaction J. Org. Chem. 86 3024
Karimi J Z and Moaddeli M S 2012 Synthesis of 3, 4-dihydropyrimidin-2 (1H)-ones and their corresponding 2 (1H) thiones using trichloroacetic acid as a catalyst under solvent-free conditions ISRN Org. Chem. 2012 474626
Tu S, Fang F, Zhu S, Li T, Zhang X and Zhuang Q 2004 A new Biginelli reaction procedure using potassium hydrogen sulfate as the promoter for an efficient synthesis of 3, 4-dihydropyrimidin-2 (1H)-one J. Het. Chem. 41 253
Hori H, Yamamoto A, Hayakawa E, Taniyasu S, Yamashita N, Kutsuna S, et al. 2005 Efficient decomposition of environmentally persistent perfluorocarboxylic acids by use of persulfate as a photochemical oxidant Environ. Sci. Technol. 39 2383
Figg C A, Hickman J D, Scheutz G M, Shanmugam S, Carmean R N, Tucker B S, et al. 2018 Color-coding visible light polymerizations to elucidate the activation of trithiocarbonates using Eosin Y Macromolecules 51 1370
Meyer A U, Straková K, Slanina T and König B 2016 Eosin Y (EY) photoredox-catalyzed sulfonylation of alkenes: scope and mechanism Chem. A Eur. J. 22 8694
Devthade V, Kamble G, Ghugal S G, Chikhalia K H and Umare S S 2018 Visible light-driven Biginelli reaction over mesoporous g-C3N4 Lewis-base catalyst Chem. Select 3 4009
Author information
Authors and Affiliations
Corresponding authors
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
KUMAR, G., BHARGAVA, G., KUMAR, Y. et al. Eosin Y photocatalyzed access to Biginelli reaction using primary alcohols via domino multicomponent cascade: an approach towards sustainable synthesis of 3,4-dihydropyrimidin-2(1H)-ones. J Chem Sci 134, 44 (2022). https://doi.org/10.1007/s12039-022-02039-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12039-022-02039-z