
Abstract
The acid-base properties of the Patented Blue V dye were studied by spectrophotometry and tristimulus colourimetry. The mechanism of protonation of Patented Blue V has been investigated with semi-empirical and DFT methods. The quantum chemical calculations of total energy defined the most stable isomer for each protonated form in water solution. In addition to thermodynamic parameters, the condensed Fukui function and molecular electrostatic potential were successfully used as reactivity descriptors for the determination of the most favorable site for protonation. Moreover, for the explanation of the structure of the most deprotonated form of the dye in highly basic medium, the weak intramolecular interactions were investigated with the reduced density gradient function. The semi-empirical calculations of absorbance spectra explained the changing of the colour of the dye for the different protonated states. It has been shown that the dominant form of the Patented Blue V was the electro-neutral form, and the molar absorptivity (\(\upvarepsilon _{639}=1.06\times 10^{5}\,\hbox {dm}^{3}\cdot \hbox {mol}^{-1}\cdot \hbox {cm}^{-1})\) increases with the increase of the dielectric permittivity of the solvent. The replacement of polar solvents by less polar ones is causing a bathochromic shift of the absorption band of the dye, the value of which is correlated with the value of the Hansen parameter.
Graphical Abstract
SYNOPSIS The acid-base properties of the Patented blue V dye were studied by spectrophotometry and tristimulus colourimetry. The mechanism of protonation of Patented Blue V has been investigated with semi-empirical and DFT methods. The ionization constants of the dye functional groups were determined and the most probable protonation/deprotonation scheme was proposed.

Similar content being viewed by others
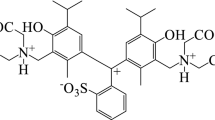

Avoid common mistakes on your manuscript.
1 Introduction
Synthetic food dyes are used more often than natural ones to enhance the appearance of food by giving it artificial colour. The safety of these dyes is highly controversial and depends on nature and concentration; they may be allergens and carcinogens.[1] Food dyes, such as Patented Blue V (PBV), are widely used for dyeing alcoholic and non-alcoholic beverages, confectionery and pharmaceuticals. However, an important task is the establishment of the physical, chemical and acid-base characteristics of PBV in solution. The characteristic of the acid-base properties is a proton-transfer reaction equilibrium constant (\(\hbox {pK}_{a})\).[2] In addition, \(\hbox {pK}_{a}\) is included in the adsorption, distribution, and metabolism (ADME) phenomena profile along with other physicochemical properties that determine the major pharmacokinetic and pharmacodynamic parameters of molecules.[2, 3] The direction and intensity of protolytic process can be estimated by \(\hbox {pK}_{a}\) value. In turn, it makes possible to determine the state of acid-base equilibrium, charge, ratio of the ionic-molecular forms of the substance. To the best of our knowledge, despite the use of this dye, data on its acid-base properties are not described in detail. It might be due to the peculiarities of the PBV. The dye is a polyfunctional organic compound with functional groups that are similar in acidic properties and with overlapping absorption spectra of its equilibrium forms. In such cases, standard methods such as potentiometry and spectrophotometry are uninformative. In some ways, the use of various chemometric algorithms allows to achieve better results.[4] Of equal interest is the use of tristimulus colourimetry approaches, which have proven themselves in the study of complexation, the development of methods for single-time determination of analytes and the creation of test methods.[5] Tristimulus colourimetry is a reliable method for the determination of the dissociation constants of all functional groups of flavonoids such as morin and quercetin.[6, 7] It has been shown that aggregation processes in the solution of 4-hydroxystyryl dyes can be observed by means of tristimulus colourimetry and ionization and hydroxylation constants of the dyes have been determined using chromaticity functions.[8]
The aim of this work is to study acid-base, spectrophotometric and solvatochromic properties of Patented Blue V colourant by spectrophotometry with chemometric tools, tristimulus colourimetry methods and DFT calculations.
2 Experimental and computational
The PBV food dye was obtained from Merck and purified by further recrystallization from methanol. The stock solution of the dye with a concentration of \(1\times 10^{-3}\,\hbox {mol}\cdot \hbox {dm}^{-3}\) was prepared by dissolving the reagent in distilled water, and dissolving in methanol for studying solvatochromism. The chemicals used in this work were of the analytically pure grade.
Electronic absorption spectra were recorded using SF-56 spectrophotometer in the wavelength range 380–780 nm in 1 cm quartz cells. The pH of the solutions was controlled with an ESL-63-07 glass electrode coupled with an EVL-1M3 silver-silver chloride reference electrode on an I-160 potentiometer.
In the study of solvatochromism, \(0.04\,\hbox {cm}^{3}\) of \(1\times 10^{-3}\hbox {mol}/\hbox {dm}^{3}\) dye solution in methanol and 5 \(\hbox {cm}^{3}\) of the organic solvent were added to the graduated test tubes, mixed and the absorption spectra were recorded in quartz cells (\(\hbox {l} = 1\,\hbox {cm}\)) on SF- 56 spectrophotometer. The \(\hbox {pK}_{\mathrm{a}}\) determination technique by the tristimulus colourimetry method was described in our previous investigation.[6,7,8] The electronic absorption spectra of the solutions were registered and the CIELAB coordinates of colours were calculated from the array of spectrophotometry data. The parameter SCD (specific colour discrimination) was calculated by the equation:
where, SCD – specific colour discrimination; \(S'\), \(S''\) – colour saturation of the studied solutions at \(\hbox {pH}^\prime \) and \(\hbox {pH}''\) accordingly, \(\Delta \hbox {pH} = \hbox {pH}'- \hbox {pH}''\); \(\Delta S = {\vert }S'- S''{\vert }\).
The colour saturation function was calculated by the formula:
where, A and B are colour coordinates in the CIELAB system.
For the spectrophotometric \(\hbox {pK}_{\mathrm{a}}\) determination, obtained electronic absorption spectra were processed using the SpectroCalc-H5A program. The \(\hbox {pK}_{\mathrm{a}}\) calculation algorithm is based on iteration methods and multiple linear regression analysis by the least squares method.[4]
All geometries described in this study were pre-optimized with the PM7 semi-empirical method.[9] This decision is based on the ability of PM7 to consider weak interactions that are present between phenyl rings in Patented Blue V (PBV). Also, in our previous studies, it has been found that PM7 can model similar organic compounds containing C, H, N, O, S very well.[10,11,12] Next step was the geometry optimization in water continuum COSMO solvation model[13] with the DFT B97-D3/6-31+G(d,p) method[14] that includes the Grimme’s DFT-D3 dispersion correction.[15, 16] Single-point energies were computed with evaluation of larger 6-311++G(d,p) basis set that contains diffuse functions for all atoms. The choice of B97-D3 functional was substantiated by literature data that was found to be the optimal choice for faithful representation of weak interactions and hydrogen bonds.[17]
The wavefunction for analysis of partial charges, electrostatic potential, and Fukui function was generated at TPSS/6-31G(d,p) level. We have chosen TPSS[18] functional as it was recently demonstrated that it gives best electron densities among other GGA of meta-GGA functionals.[19] Double-zeta basis set was chosen as it gives reliable electrostatic potential[20] and predictive power of reactivity descriptors obtained with this basis set is satisfactory.[21] We did not add diffuse functions since the Mulliken population analysis is incompatible with diffusion functions.
MOPAC2016 was used for PM7 calculations with L-BFGS optimizer.[22] The ORCA 3.0.3 package was used for DFT B97-D3, TPSS and ZINDO/S calculations.[23] To reduce the computational time, the resolution-of-identity (RI) technique was turned on.[24] The default settings of programs were used throughout the calculations; the only exception being the ORCA program, where the grid accuracy was increased with “Grid3 FinalGrid5” keywords. Calculations of electrostatic potential and reduced density gradient function (RDG)[25] were performed with Multiwfn 3.3.8.[26] Avogadro[27], Gabedit[28] and VMD[29] software were used for molecular systems building, input files preparation and visualization.
3 Results and Discussion
3.1 Acid-base equilibria of Patented Blue V in aqueous solutions
The absorption spectra of the PBV are illustrated in Figure 1. Apparently, in a strongly acidic medium (10 M \(\hbox {H}_{2}\hbox {SO}_{4})\) in the absorption spectrum of PBV, one low-intensity absorption band with a maximum at 415 nm is observed. Under such condition, the dye might exist in a protonated form. At pH3, an absorption band appears in the absorption spectrum with a maximum at 640 nm, the intensity of which increases with increasing pH. In alkaline medium at \(\hbox {pH}>10\), a hypsochromic shift of the absorption band at 10 nm is observed, which is probably caused by the dissociation of the hydroxyl group of PBV. The dye irreversibly decolourizes in a strongly alkaline medium at 2 M NaOH, which is associated with the central atom hydroxylation and vanishing of conjugation between all three phenyl rings.

Absorption spectra of PBV aqueous solution at different acidities: 1 – 10 M \(\hbox {H}_{2}\hbox {SO}_{4}\); 2 – pH 3; 3 – pH 9; 4 – pH 10; 5 – 2 M NaOH; \(\hbox {C}=2{\cdot }10^{-5}\,\hbox {M}\); \(\hbox {l}=1\,\hbox {cm}\).
For the acid-base equilibria study, the obtained dataset of absorption spectra of PBV was processed in SpectroCalc-H5A program (Table 1) or by tristimulus colourimetry method. The SCD values were plotted as a function of the medium pH (Figure 2).

Change in the specific colour discrimination of PBV aqueous solution depending on the medium acidity.

The protonation stages calculated for Patented Blue V using the B97-D3/6-311++G(d,p)//B97-D3/6-31+G(d,p) methods.
As shown, four peaks for PBV are observed on the \(\hbox {SCD} = \hbox {f(pH)}\) curve, indicating the presence of five acid-base forms of the dye in solution. The maximum pH values corresponding to the \(\hbox {pK}_{\mathrm{a}}\) values of the respective acid-base equilibria of the functional groups of PBV (Table 1).
As shown in Table 1, the \(\hbox {pK}_{\mathrm{a}}\) determined by the tristimulus colourimetry method and spectrophotometry satisfactorily correlate with each other. For the determination of the most probable protonation/deprotonation forms for the PBV dye, a series of quantum chemical calculations were carried out. Semi-empirical and DFT studies on similar dyes containing C, H, N, O, S have previously been reported in the literature.[10, 30, 31] PBV contains two sulfo groups (strongly acidic), one OH phenolic group (weakly acidic) and two diethylamino groups, which also could act as an acid when protonated. Due to the presence of non-identical reactivity centers in the dye, it is important to consider the protonation/deprotonation of each acidic group and nitrogen atoms in order to determine the preferred protonation site. Also, the presence of the neighboring sulfo group in the ortho position near central carbon atom that connected with three phenyl rings increases the number of possible conformers through the possible benzoxathiole cycle formation (Figure 3, structures I, II). Moreover, at each dissociation stage, deprotonation of each sulfo group has to be considered separately. In this study, 27 structures at different protonated states (see electronic Supplementary Information) were considered. The water-phase geometries of all proposed structures of the dye were optimized with DFT B97-D3/6-31+G(d,p) method. Single-point energies obtained at DFT B97-D3/6-311++G(d,p) level of theory were used to determine the most stable state that characterized by the minimal absolute energy.
Figure 3 illustrates the step-by-step protonation mechanism of PBV and the most stable structures for different protonated states. We started this investigation from totally deprotonated form V with next protonation of each nucleophilic center. Wherever possible, both benzoxathiole and non-cyclic forms were checked. As shown, the V state exists in non-cyclic form and there is a conjugated system of two N, N-diethylaniline moieties. Next reaction with hydroxide in the strongly basic medium is irreversible and leads to the triphenylmethanol VI. The protonation of V gives the phenol IV, which in the next protonation step is transformed to anilinium derivative III. Despite the conjugation between two aniline fragments, at the next protonation step, the dianilinium system II is formed with simultaneous cyclization of central carbon atom with neighboring \({\hbox {SO}_{3}}^{-}\) group. At the last stage, the protonation of sulfo group in ortho position to phenolic OH is predicted.

Molecular electrostatic potential of deprotonated state V. Values of minima (red dots) on the ESP surface are presented in kcal/mol.

Reduced density gradient (RDG) iso-surface of two forms of state VI.
Also, for investigation of the protonation/deprotonation steps, an alternative approach that is based on reactivity descriptors were used. Thus, the molecular electrostatic potential (ESP)[20] (Figure 4) and condensed Fukui function (CFF)[32] (Table 2) were used as reactivity descriptors in prediction of the most active sites in the form V. Analysis of minima of ESP iso-surface indicates that point M (Figure 4) has the lowest value of \(-257\,\hbox {kcal/mol}\). The distance between M and O28 is 1.94 Å, whereas the M-O64 and M-O66 distances correspond to 2.22 Å and 2.16 Å, respectively. Obviously, the closer position of ESP minimum to O28 indicates that this oxygen atom is the most active centre in protonation of state V.
According to CFF (Table 2), in case of a nucleophilic attack of form V, the most reactive atom is C1, and the reaction with nucleophile like hydroxyl anion will lead to the formation of triphenylmethanol VI. While for electrophilic attack (like protonation) O28 atom is the most reactive site, and in the case of protonation, the phenolic group will form.
State VI can also exist in two forms with different reciprocal position of tertiary alcohol hydroxyl and neighbouring sulfo group. DFT calculations show that position of OH and \({\hbox {SO}_{3}}^{-}\) groups on the same side give more stable structure. To understand this behavior, we have calculated the reduced density gradient (RDG) function that shows the presence of O-\(\hbox {H}{\cdot }{\cdot }{\cdot }\hbox {O}\)-S hydrogen bond (Figure 5). Blue region between the oxygen of sulfo group and hydrogen of hydroxyl of PBV indicates the presence of hydrogen bonding. Red areas indicate the repulsion effect in cyclic systems. Green and brown areas indicate dispersion interactions.
In the case of state IV, the CFF indicates that one of the nitrogen atoms (\(\hbox {CFF}^{-}\) value is 0.0429) is the most reactive centre in protonation reaction (Table 3). The formation of anilinium cation III (Figure 3) is preferable before protonation of sulfo groups. It is in total agreement with a comparison of corresponding total energies.

Minor forms of states I, II and III. The difference in Gibbs free energy (in kcal/mol) upon the most stable form and the amount (in %) of each form in the mixture are shown in brackets.
Analysis of the \(\hbox {CFF}^{-}\) for state III shows the limitations of this approach. Thus, the CFF shows that sulfo group (S29-O32) is the most reactive centre, but we will show that this is a minor product of the protonation. The energy analysis shows the higher stability of benzoxathiole II (Figure 3). This can be explained by the presence of conjugation between two N,N-aniline fragments and deactivation of second nitrogen upon protonation of the first one. But, the minimization in energy upon protonation of second nitrogen is reached by the redistribution of positive charge to central carbon and its combination with oxygen of neighbouring sulfo group through electrostatic attraction (Table 4).
Analysis of the total energy of different forms of PBV (see the list in the Supplementary Information) shows the presence of forms with quite close total energy but different geometry. That is why it may be assumed that in water solution, few forms for each state are possibly present. Using the Boltzmann’s distribution equation, the values of the most possible minor forms were calculated for states I, II and III (Figure 6). Thus, the state III exists in equilibrium between protonated nitrogen (III, Figure 3) and 2.7% of protonated sulfo group (IIIa, Figure 6). Non-benzoxathiole forms Ia (1.8%) and IIa (5.8%) exist as minor products in equilibrium with I and II, respectively (Figure 3).
The ion-molecular forms of the PBV in solution are in dynamic equilibrium depending on the pH value and may be represented in the form of distribution diagram (Figure 7).
The narrow pH ranges for the existence of III, IV and V forms (Figure 7) as well as the proximity of the corresponding spectral characteristics explain the ineffectiveness of classical instrumental methods of investigation as evidenced by the absence of information about the state of PBV in solutions in literature.
3.2 Spectrophotometric characteristics of Patented Blue V in solutions
The spectrophotometric characteristics of the electrically neutral form III, which exists in a range of pH 3-8 in water and organic solvents, are summarized in Table 5.

Chart of the acid-base forms of PBV distribution at different acidities in aqueous solution.
Analysis of the obtained results allows to conclude that the molar extinction coefficients of the dominant acid-base form of PBV in organic solvents increase almost linearly (\(\hbox {R}^{2}=0.87\)) with their dielectric permittivity (Figure 8a). The positions of the maximum of the absorption band correlate satisfactorily (\(\hbox {R}^{2} = 0.83\)) with the value of the Hansen parameter (Figure 8b) associated with the intermolecular interaction of PBV with the solvent due to the formation of hydrogen bonds.[33]

The influence of the dielectric permittivity of solvent on PBV molar absorptivity (a) and the value of Hansen parameter on absorption band maxima (b).
In addition, the ZINDO/S calculations were carried out for analysis of electronic transition in absorbance spectra.[34] It was shown that ZINDO/S method gives results compatible with long-range corrected DFT calculations.[35] In case of the hydrolyzed state VI, the value for the first excitation energy is 3.485 eV (356 nm) with the main contribution of \(\hbox {HOMO}\rightarrow \hbox {LUMO}+1\) (40%), \(\hbox {HOMO}\rightarrow \hbox {LUMO}+9\) (18%) and \(\hbox {HOMO}\rightarrow \hbox {LUMO}+10\) (28%) transitions. For anionic form V, the second calculated excited state of 2.360 eV (525 nm) have relatively high oscillator strength with the main contribution of HOMO-\(6\rightarrow \hbox {LUMO}\) (44%), HOMO-\(3\rightarrow \hbox {LUMO}\) (12%), and HOMO-\(2\rightarrow \hbox {LUMO}\) (24%) transitions. In case of state IV, the first excited state has the energy of 2.662 eV (466 nm) and the \(\hbox {HOMO}\rightarrow \hbox {LUMO}\) transition have the main contribution of 92% (Figure 9). Slightly different picture of excitation but with similar energy (2.680 eV –463 nm) was observed for the second excited state of form III. Transitions HOMO-\(3\rightarrow \hbox {LUMO}\) (42%), HOMO-\(4\rightarrow \hbox {LUMO}\) (22%), and HOMO-\(5\rightarrow \hbox {LUMO}\) (18%) have the main contributions to this excitation. The first excited state of form II has the energy of 4.190 eV (296 nm) and the main contribution have HOMO-\(5\rightarrow \hbox {LUMO}+4\) (15%), \(\hbox {HOMO}\rightarrow \hbox {LUMO}+4\) (8%) and HOMO-\(6\rightarrow \hbox {LUMO}+5\) (7%) transitions. For cationic form, I, the 5th excited state (4.701 eV; 264 nm) has relatively high oscillator strength with the main contributions of \(\hbox {HOMO}\rightarrow \hbox {LUMO}\) (34%), HOMO-\(1\rightarrow \hbox {LUMO}+4\) (11%) and \(\hbox {HOMO}\rightarrow \hbox {LUMO}+4\) (9%) transitions.
The significant blue shift in the case of form VI can be explained by the hydroxylation of the central atom and vanishing of conjugation between all three phenyl rings. Whereas the hypsochromic shift in the case of forms I and II computed with excluding of lone pair of one of the diethylamino groups from conjugation upon protonation.

HOMO and LUMO of form IV.
4 Conclusions
The acid-base properties of the Patented Blue V dye were studied by spectrophotometry and tristimulus colourimetry. The ionization constants of the Patented Blue V functional groups were determined. The theoretical investigations of the conformations and reactive sites of Patented Blue V helped to define the mechanism of protonation/deprotonation. Decrease in pH value leads to the protonation of the phenolic oxygen. Further protonation of diethylamino group transformed the dye to anilinium derivative. On the next protonation step, the dianilinium system is formed with simultaneous cyclization of a central carbon atom with neighboring \({\hbox {SO}_{3}}^{-}\) group. The protonation of sulfo group in ortho-position to phenolic OH is the final stage that we analyzed. The last two forms are characterized by the hypsochromic shift that is conducted with an exclusion of lone pair of electrons of one of the diethylamino groups from conjugation upon protonation. The DFT computations found the presence of forms of Patented Blue V with close values of total energy but different geometry. This leads us to confirm that forms for each state are possibly present in a real water solution. It should be noted that in the highly basic medium, the irreversible formation of triphenyl-substituted carbinol is expected. The absorbance spectra of this form of the dye is characterized by the significant blue shift that can be explained by the hydroxylation of central atom and vanishing of conjugation between all three phenyl rings. It has been shown that the dominant form of the Patented Blue V was the electroneutral form, and the molar absorptivity (\(\upvarepsilon _{639}=1.06\times 10^{5}\,\hbox {dm}^{3}\cdot \hbox {mol}^{-1}\cdot \hbox {cm}^{-1})\) increases with the increase of the dielectric permittivity of the solvent. The replacement of polar solvents by less polar ones is causing a bathochromic shift of the absorption band of the dye, the value of which is correlated with the value of the Hansen parameter.
References
Ceyhan B M, Gultekin F, Doguc D K and Kulac E 2013 Effects of maternally exposed coloring food additives on receptor expressions related to learning and memory in rats Food Chem. Toxicol. 56 145
Zevatskii Yu E, Samoilov D V and Mchedlov–Petrosyan N O 2009 Contemporary methods for the experimental determination of dissociation constants of organic acids in solutions Russ. J. Gen. Chem. 79 1859
Zevatski Yu E and Samoilov D V 2011 Modern methods for estimation of ionization constants of organic compounds in solution Russ. J. Org. Chem. 47 1445
Sovyn O R and Patsay I O 2012 Computer program “SpectroCalc-H5A” for spectrophotometric determination of acid dissociation constants Methods Objects Chem. Anal. 7 74 (in Ukrainian)
Ivanov V M, Monogarova O V and Oskolok K V 2015 Capabilities and prospects of the development of a chromaticity method in analytical chemistry J. Anal. Chem. 70 1165
Chebotarev A N and Snigur D V 2015 Study of the acid-base properties of quercetin in aqueous solutions by colour measurements J. Anal. Chem. 70 55
Chebotarev A N and Snigur D V 2016 Study of the acid-base properties of morin by tristimulus colorimetry Russ. J. Gen. Chem. 86 815
Chebotarev A N, Snigur D V, Zhukova Yu P, Bevziuk K V, Studenyak Ya I and Bazel Ya R 2017 Tristimulus colourimetric and spectrophotometric study of the state of 4-hydroxystyryl dyes in aqueous solutions Russ. J. Gen. Chem. 87 196
Stewart J J P 2013 Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters J. Mol. Model. 19 1
Bevziuk K, Chebotarev A, Snigur D, Bazel Ya, Fizer M and Sidey V 2017 Spectrophotometric and theoretical studies of the protonation of Allura Red AC and Ponceau 4R J. Mol. Struct. 1144 216
Fizer M, Sukharev S, Slivka M, Mariychuk R and Lendel V 2016 Preparation of bisthiourea and 5-Amino-4-benzoyl-1,2,4-triazol-3-thione complexes of Copper (II), Nickel and Zinc and their biological evolution J. Organomet. Chem. 804 6
Fizer M, Sidey V, Tupys A, Ostapiuk Y, Tymoshuk O and Bazel Ya 2017 On the structure of transition metals complexes with the new tridentate dye of thiazole series: theoretical and experimental studies J. Mol. Struct. 1149 669
Klamt A, Moya C and Palomar J 2015 A comprehensive comparison of the IEFPCM and SS(V)PE continuum solvation methods with the COSMO approach J. Chem. Theory Comput. 11 4220
Grimme S 2006 Semiempirical GGA-type density functional constructed with a long-range dispersion correction J. Comput. Chem. 27 1787
Grimme S, Antony J, Ehrlich S and Krieg H 2010 A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu J. Chem. Phys. 132 154104
Grimme S, Ehrlich S and Goerigk L 2011 Effect of the damping function in dispersion corrected density functional theory J. Comput. Chem. 32 1456
Burns L A, Vazquez-Mayagoitia A, Sumpter B G and Sherrill C D 2011 Density-functional approaches to noncovalent interactions: a comparison of dispersion corrections (DFT-D), exchange-hole dipole moment (XDM) theory, and specialized functionals J. Chem. Phys. 134 084107
Tao J, Perdew J P, Staroverov V N and Scuseria G E 2003 Climbing the density functional ladder: nonempirical meta–generalized gradient approximation designed for molecules and solids Phys. Rev. Lett. 91 146401
Medvedev M G, Bushmarinov I S, Sun J, Perdew J P and Lyssenko K A 2017 Density functional theory is straying from the path toward the exact functional Science 355 49
Riley K E, Tran K, Lane P, Murray J S and Politzer P 2016 Comparative analysis of electrostatic potential maxima and minima on molecular surfaces, as determined by three methods and a variety of basis sets J. Comput. Sci. 17 273
Cao J, Ren Q, Chen F and Lu T 2015 Comparative study on the methods for predicting the reactive site of nucleophilic reaction Sci. China Chem. 58 1845
Stewart J J P MOPAC 2016 Version 17.173W Stewart Computational Chemistry. Accessed on: http://OpenMOPAC.net (accessed on 12 June 2017)
Neese F 2012 The ORCA program system WIREs Comput. Mol. Sci. 2 73
Laikov D N 1997 Fast evaluation of density functional exchange-correlation terms using the expansion of the electron density in auxiliary basis sets Chem. Phys. Lett. 281 151
Johnson E R, Keinan S, Mori-Sanchez P, Contreras-Garcia J, Cohen A J and Yang W 2010 Revealing Noncovalent Interactions J. Am. Chem. Soc. 132 6498
Lu T and Chen F 2012 Multiwfn: A multifunctional wavefunction analyzer J. Comput. Chem. 33 580
Hanwell M D, Curtis D E, Lonie D C, Vandermeersch T, Zurek E and Hutchison G R 2012 Avogadro: an advanced semantic chemical editor, visualization, and analysis platform J. Cheminform. 4 1
Allouche A R 2011 Gabedit - A graphical user interface for computational chemistry softwares J. Comput. Chem. 32 174
Humphrey W, Dalke A and Schulten K 1996 VMD: Visual molecular dynamics J. Mol. Graph. 14 33
Lešková M, Bazel Ya, Torok M and Studenyak Ya 2013 Structure and properties of 2-[(E)-2-(4-dipropylaminophenyl)-1-ethenyl]-1,3,3-trimethyl-3H-indolium chloride Chem. Pap. 67 415
Billes F, Szabo A and Studenyak Ya 2011 Vibrational spectroscopic study on 2-[2-(4-dipropylamino-phenyl)-vinyl]-1,3,3-trimethyl-3H-indolium chloride Spectrochim. Acta A 78 967
Parr R G and Yang W 1989 In Density Functional Theory of Atoms and Molecules (New York, Oxford: Oxford University Press) p. 333
Hansen C M 2007 In Solubility Parameters: A User’s Handbook (Boca Raton: CRC Press Taylor & Francis Group) p. 521
Thompson M A and Zerner M C 1991 A Theoretical examination of the electronic structure and spectroscopy of the photosynthetic reaction center from rhodopseudomonas viridis J. Am. Chem. Soc. 113 8210
Brückner C and Engels B 2015 Benchmarking ground-state geometries and vertical excitation energies of a selection of p-type semiconducting molecules with different polarity J. Phys. Chem. A 119 12876
Acknowledgements
27 structures at different protonated states are shown with the difference in energy between the current form and the most stable form. Data for each state is available as Supplementary Information for this article at www.ias.ac.in/chemsci.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bevziuk, K., Chebotarev, A., Fizer, M. et al. Protonation of Patented Blue V in aqueous solutions: theoretical and experimental studies. J Chem Sci 130, 12 (2018). https://doi.org/10.1007/s12039-017-1411-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12039-017-1411-2