
Abstract
To develop an effective fluorescent chemosensor for relay recognition of Zn2+ and oxalate, a new fluorescent sensor based on binaphthol-quinoline Schiff base L 1 was designed and synthesized. In DMSO-H2O (1/1, v/v, HEPES 10 mM, pH = 7.4) solution, L 1 exhibits highly selective fluorescence turn on response to Zn2+ over other metal ions. The Zn2+ recognition event is barely interfered by other coexisting metal ions except Cu 2+, Co 2+ and Ni 2+. The in situ generated L 1 -Zn\(^{\mathrm {2+}}_{\mathrm {}}\)complex was further used as a chemosensing ensemble for oxalate detection. The complex L 1 -Zn2+ displays high selectivity to oxalate with significant fluorescence quenching through Zn2+ ion displacement approach. In addition, application of L 1 for imaging of Zn2+ and oxalate in living HeLa cells was also examined.

A new binaphthol-quinoline Schiff base-based fluorescent sensor L 1 exhibits highly selective relay recognition of Zn2+ and oxalate through fluorescence “off-on-off” functionality in DMSO-H2O (1/1, v/v, HEPES 10 mM, pH = 7.4) solution.
Similar content being viewed by others
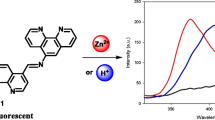
Avoid common mistakes on your manuscript.
1 Introduction
The development of chemosensors for highly selective detection of biologically and environmentally important metal ions and anions has received increasing attention owing to the vital roles they play in chemical, biological and environmental processes.[1] Zn2+, the second most abundant transition metal ion in human body, plays paramount roles in a wide range of biological processes such as gene transcription, signal transmission, and mammalian reproduction. Disruption of Zn2+ concentration in cells can result in a variety of pathological processes including Alzheimer’s disease, epilepsy and infantile diarrhea.[2] Therefore, there is a growing interest in developing selective and sensitive Zn2+ sensors to recognize the physiological significance of Zn2+ and its biomedical relevance.[3]
Among the biologically pivotal anions, oxalate is particularly important due to its important role especially in food chemistry and clinical analysis. Although various methods including enzyme electrode assay,[4] chromatography,[5]electrophoresis and electrochemistry[6] have been used for oxalate detection, fluorescence recognition of oxalate has received substantial attention due to its simplicity and waiver of sophisticated sample pretreatment.[7] Recent use of single sensor for relay recognition of metal ion and anion has aroused considerable interest of researchers due to the operational simplicity.[8] As far as we are aware, utilization of a single sensor for relay recognition of Zn2+ and oxalate has not been documented.
Herein, a fluorescence sensor based on Schiff base L 1 (Scheme 1) for sequential detection of Zn2+ and oxalate has been synthesized, which exhibits relay recognition behavior toward Zn2+ and oxalate in aqueous solution through fluorescence ‘off-on-off’ changes.

Synthesis of sensor L 1 and control compound L 2 .
2 Experimental
2.1 Instruments and reagents
Unless otherwise stated, solvents and reagents were purchased as analytical grade and were used without further purification. Double distilled water was used for spectral detection. 1H NMR and 13C NMR spectra were performed on an Agilent 400 MR spectrometer, and the chemical shifts (δ) were expressed in ppm and coupling constants (J) in Hertz. High-resolution mass spectroscopy (HRMS) was conducted on a Bruker micrOTOF-Q mass spectrometer. Fluorescence measurements were carried out on a Sanco 970-CRT spectrofluorometer (Shanghai Spectrum instruments Co., Ltd., China). UV-vis absorption spectra were recorded on a SP-1900 spectrophotometer (Shanghai Spectrum instruments Co., Ltd., China). The pH measurements were made with a Model PHS-25B meter (Shanghai, China).
2.2 Synthesis of chemosensor L 1 and control compound L 2
Chemosensor L 1 was synthesized as follows. A stirred solution of compounds (S)-1[9] (342 mg, 1 mmol) and 2a[10] (502.5 mg, 2.5 mmol) in absolute ethanol (50 mL) was heated to reflux for 2 h. After cooling to room temperature, the yellow precipitate formed was collected by filtration, which was washed three times with cold ethanol to give pure L 1 . Yield: 566 mg, 0.80%; M.p.: 207-208 ∘C;. 1H NMR (400 MHz, DMSO- d 6)δ: 12.70 (s, 2H, -OH), 10.58 (s, 2H, -NH), 9.01 (s, 2H, N =CH), 8.56 (d, J = 7.6 Hz, 2H, Ar-H), 8.49 (d, J = 3.2 Hz, 2H, Ar-H), 8.38 (s, 2H, Ar-H), 8.23 (d, J = 8.0 Hz, 2H, Ar-H), 8.07-8.05 (m, 2H, Ar-H), 7.59-7.50 (m, 5H, Ar-H), 7.43 (dd, J = 7.6, 4.0 Hz, 2H, Ar-H), 7.38-7.36 (m, 3H, Ar-H), 7.17-7.09 (m, 2H, Ar-H), 4.78 (dd, J = 16.4 Hz, 4H, -CH 2-); 13C NMR (100 MHz, CDCl 3)δ: 168.89, 166.71, 154.33, 148.48, 138.21, 135.90, 135.38, 134.86, 133.47, 129.17, 128.87, 127.66, 127.37, 126.78, 124.92, 123.59, 121.90, 121.29, 120.79, 117.09, 115.91, 64.05; HRMS-ESI(+): m/z= 709.2550 [M + 1] +.
Control compound L 2 was synthesized by a similar procedure was used for that of L 1 except that 2b[11] was used. Yield: 600 mg, 85%; M.p.: 159-160 ∘C; 1H NMR (400 MHz, DMSO- d 6)δ: 13.06 (s, 2H, -OH), 10.20 (s, 2H, NH), 8.96 (s, 2H, N =CH), 8.34 (s, 2H, Ar-H), 8.04 (t, J = 7.2 Hz, 4H, Ar-H), 7.91 (d, J = 7.6 Hz, 2H, Ar-H), 7.76 (d, J = 8.0 Hz, 2H, Ar-H), 7.68 (d, J = 7.2 Hz, 2H, Ar-H), 7.52-7.45 (m, 6H, Ar-H), 7.37-7.31 (m, 4H, Ar-H), 7.05 (d, J = 8.4 Hz, 2H, Ar-H), 4.72 (s, 4H, -CH 2-); 13C NMR (100 MHz, DMSO- d 6)δ:169.70, 168.07, 154.80, 135.30, 134.22, 134.13, 133.58, 129.57, 128.83, 128.56, 128.27, 127.55, 126.50, 126.33, 125.99, 125.17, 124.63, 123.68, 123.12, 122.33, 121.28, 116.61, 62.31; HRMS-ESI(+ ): m/z= 707.2653 [M + 1] +.
For 1H NMR spectra of L 1 and L 2 , please see Figures S1 and S2 (in Supplementary Information).
3 Results and Discussion
3.1 Fluorescence recognition of Zn2+ by L 1
Chemosensor L 1 was synthesized by condensation of (S)-1 and 2a in absolute ethanol, and the structure of L 1 was fully characterized by 1H NMR, 13C NMR and HRMS analysis. The fluorescence responses of L 1 (10 μM) to various metal ions were investigated in DMSO-H 2O (1/1, v/v, HEPES 10 mM, pH = 7.4) solution (Figure 1). L 1 itself displays almost negligible fluorescence emission on excitation at 359 nm, which may attributed to the combined effects of PET from phenolic moiety to quinoline fluorophore and the isomerization process of C =N bond.[12] Upon addition of 4.0 equiv. of Zn2+, a dramatic fluorescence enhancement at 512nm was observed, which could be attributed to the Zn2+ -binding induced suppression of PET and C =N isome rization. The Zn2+ induced green fluorescence color of L 1 solution can be observed by a naked eye under illumination at 365 nm with a portable UV lamp (Figure 1, inset). Whereas, addition of equal amounts of other metal ions such as K +, Na +, Ag +, Pb 2+, Sr 2+, Ba 2+, Cd 2+, Fe 2+, Fe 3+, Mn 2+, Mg 2+, Cr 3+, Al 3+, Ni +, Co 2+, Ca 2+, Cu 2+ and Hg 2+, promoted negligible fluorescence intensity changes of L 1 . The observed emission at 512 nm was supposed to come from the quinoline fluorophore,[13] and this hypothesis was confirmed by comparison of fluorescence responses of control compound L 2 and sensor L 1 to Zn2+ ion (Figure S3). Under identical conditions, L 2 does not show any noticeable fluorescence emission in the absence and presence of Zn2+, indicating that the quinoline moiety is responsible for the observed “turn-on” fluorescence of L 1 . These results indicate that L 1 has an excellent selectivity toward Zn2+ ion.

Changes in the fluorescence emission of L 1 (10 μM) in DMSO-H 2O (1/1, v/v, HEPES 10 mM, pH = 7.4) solution upon addition of 4 equiv. of various metal ion. Inset: naked eye observed fluorescence changes of L 1 solution in the presence of Zn2+ ions under a portable UV lamp at 365 nm.
The Zn2+ sensing behavior of L 1 to Zn2+ was then explored by fluorescence titration experiments. Incremental addition of Zn2+ to L 1 solution led to gradual increase in fluorescence intensity at 512 nm, which reached a plateau when 4.0 equiv. of Zn2+ was employed (Figure 2). Plotting of the normalized fluorescence intensity ((I- I min)/(I max- I min)) at 512 nm against Log[Zn2+] results in a neat linear relation (R = 0.9977) (Figure 3), and the detection limit of L 1 to Zn2+ was calculated to be 1.5 ×10 −6 M.[14] The association constant (K s) of L 1 and Zn2+ was estimated to be 2.15 ×10 6 M −1by non-linear least squares fitting of the titration data based on a 1:1 binding equation model (Figure 4). In addition, this 1:1 binding ratio was also supported by Job’s plot analysis (Figure S4). The UV-vis absorption spectrum of L 1 on incremental addition of Zn2+ shows ratiometric absorption changes (Figure S5). The absorption band at 394 nm of L 1 can be assigned to the absorption of Schiff base linked binol moiety. On gradual addition of Zn2+ to L 1 solution, the absorption intensity of L 1 at 394 nm decreased gradually, concomitantly, two new absorption bands centered at 357 nm and 443 nm emerged and gradually increased, and they can be attributed to the deprotonated amide-quinoline[13] and phenolic binol Schiff base,[15]respectively.

Fluorescence spectra changes of sensor L 1 (10 μM) in DMSO-H 2O (1/1, v/v, HEPES 10 mM, pH = 7.4) solution on step increasing in Zn2+ concentration (0 to 40 μM).

Normalized fluorescence intensity of L 1 solution (10 μM) to Log[Zn2+]; unit for [Zn2+] is M.

Nonlinear least-squares fitting of fluorescence intensity of L 1 (at 512 nm) employing a 1:1 binding mode equation. Unit for [Zn2+] is M.
To corroborate the high selectivity of L 1 to Zn2+, the competition experiments were subsequently carried out. As shown in Figure 5, on further addition of 4.0 equiv. of Zn2+ to the competitive metal ion containing L 1 solution, a dramatic fluorescence emission enhancement was observed except for Cu 2+, Ni 2+ and Co 2+. The influence of Cu 2+ is owing to its paramagnetic nature. On the other hand, the level of Ni 2+ and Co 2+ are very low in human body and they will have little influences on Zn2+ recognition in living cells. These results reveal that L 1 still has a potential applicability for Zn2+ detection.

Fluorescence intensity of L 1 (10 μM) in H 2O-DMSO (1/1, v/v, pH = 7.4) at 512 nm. The gray bars represent the emission intensity of L 1 in the presence of 4.0 equiv. of competing metal ion; the red bars represent the emission intensity of the above solution upon addition of 4.0 equiv. of Zn2+.
To obtain further insight into the binding mode of L 1 with Zn2+, 1H NMR spectra of L 1 in the absence and presence of Zn2+ were compared (Figure 6). The broad peak at 12.70 ppm in free L 1 is assigned to the hydroxyl proton (H a) (figure 6a), which is greatly decreased upon addition of 2.0 equiv. of Zn2+ (Figure 6b), indicating the Zn2+ binding induced de-protonation process. The imine protons (H b) in L 1 at 9.01 ppm (Figure 6a) shifted downfield to 10.76 ppm on interaction with Zn2+ (Figure 6b). The amide proton signal (H d) in L 1 at 10.58 ppm (Figure 6a) was greatly decreased on addition of Zn2+ (Figure 6b), indicative of the deprotonation of amide group. Moreover, the quinolone proton signals of H e (8.49 ppm) and H j (8.56 ppm) in free L 1 also shifted downfield to 8.98 ppm and 8.64 ppm, respectively. These results reveal that the phenolic oxygen, imine and amide nitrogen, and quinolone nitrogen atoms are all involved in the complex formation. Thus, a plausible binding mode of L 1 with Zn2+ ions was proposed and is illustrated in Scheme 2.

Partial 1H NMR spectrum (in DMSO- d 6) of L 1 in the presence of 0 (a), 2.0 (b) and 2.5 (c) equivalents of Zn2+.

Proposed binding mode between L 1 and Zn2+ ions.
3.2 Fluorescence recognition of oxalate by L 1 -Zn2+ complex
The in situ formed L 1 -Zn2+ complex (prepared by mixing L 1 with 4.0 equiv. of Zn2+) was envisioned to be a potential candidate for chemosensing ensemble of dicarboxylate recognition. Upon individual addition of 100 equiv. (relative to L 1 ) of different dicarboxylates to L 1 -Zn2+solution (10 μM based on L 1 ), oxalate elicited a dramatic fluorescence quenching, which is similar to the original emission state of free L 1 (Figure 7). Considering the low solubility of ZnC2O4 (K sp = 2.8×10−8), the observed fluorescence quenching is due to oxalate-induced de-complexation of L 1 -Zn2+ complex. Nevertheless, addition of other dicarboxylates including succinate, glutarate, adipate, phthalate, terephthalate, and isophthalate caused no significant fluorescence spectrum changes except that malonate promoted a slight decrease in fluorescence emission. These results demonstrate that the in situ generated L 1 -Zn2+ complex can act as an ensemble for oxalate recognition. Fluorescence titration with oxalate results in gradual emission quenching of Zn2+ -L 1 at 512 nm, and the quenching process terminated when 100 equiv. of oxalate was added (Figure 8). Based on the titration data, the detection limit of the ensemble to oxalate was estimated as 2.06 ×10 −5 M (Figure S6). Results of competition experiments reveal that co-existence of equal amount of other dicarboxylates did not cause any remarkable influence on oxalate detection (Figure 9).

Fluorescence spectra changes of L 1 -Zn2+(10 μM) in DMSO-H 2O (1/1, v/v, HEPES 10 mM, pH = 7.4) solution upon addition of 100 equiv. of various dicarboxylates.

Fluorescence spectra of sensor Zn2+ -L 1 (10 μM) in DMSO-H 2O (1/1, v/v, HEPES 10 mM, pH = 7.4) solution upon gradual increase in oxalate concentration (0 to 1 mM).

Change in fluorescence intensity of L 1 -Zn2+ (10 μM) in H 2O-DMSO (1/1, v/v, pH = 7.4) at 512 nm. The gray bars represent the emission intensity of Zn2+ -L 1 in the presence of 100 equiv. of competing dicarboxylates; the red bars represent the emission intensity of the above solution upon addition of 100 equiv. of oxalate.
3.3 Living cell imaging studies
To investigate the practical applicability of L 1 for sequential detection of Zn2+ and oxalate, we evaluated its potential utility for imaging Zn2+ and oxalate in living cells. The human cervical HeLa cancer cell lines incubated for 0.5 h at 37 ∘C with L 1 (5 μM) showed no fluorescence (Figure 10d). Further addition of Zn2+ (10 μM) to the pre-incubated cell for 1h exhibited a noticeable green fluorescence (Figure 10e). However, when oxalate (20 μM) was added to HeLa cell which was incubated with L 1 (10 μM) and Zn\(^{\mathrm {2+}}_{\mathrm {}}\)(10 μM) for 30 min at 37 ∘C, the previous observed green fluorescence was quenched greatly (Figure 10f). These results indicate that L 1 can be used as a sensor to probe the intracellular Zn2+ and oxalate.

Fluorescence images of Zn2+ and oxalate in HeLa cells. (d and a) Fluorescence image of HeLa cells with addition of L 1 and its bright field image. (e and b) Fluorescence image of HeLa cells incubated with L 1 (5 μM) for 30 min at 37 ∘C and then incubated with Zn2+(10 μM) for 30 min at 37 ∘C and its bright-field image. (f and c) Fluorescence images of HeLa cell incubated with L 1 (10 μM) and Zn2+(10 μM) for 30 min at 37 ∘C and then incubated with oxalate (20 μM) for 30 min at 37 ∘C and its bright field image.
4 Conclusions
In summary, we have developed a new fluorescence sensor based on Schiff base (L 1 ) for relay recognition of Zn2+ and oxalate in aqueous media. Sensor L 1 exhibits high selectivity and sensitivity for sequential detection of Zn2+ and oxalate with good anti-inferences ability. Imaging studies of living HeLa cells imaging studies indicate that this new fluorescent sensor holds great potential for biological applications for relay detection of Zn2+ and oxalate. Since the S enantiomer of binaphthol derivative was used in this work, we anticipate that the in situ formed chiral zinc complex could exert recognition behavior to a chiral analyte in a future work.
References
Berg J M and Shi Y 1996 Science 271 1081
Walker C F and Black R E 2004 Annu. Rev. Nutr. 24 255
(a) Chen Y, Bai Y, Han Z, He W and Guo Z 2015 Chem. Soc. Rev. 44 4517; (b) Jiang P and Guo Z 2004 Coord. Chem. Rev. 248 205; (c) Xu Z, Yoon J and Spring D R 2010 Chem. Soc. Rev. 39 1996; (d) Bhaumik C, Maity D, Das S and Baitalik S 2012 RSC Adv. 2 2581; (e) Mardanya S, Karmakar S, Das S and Baitalik S 2015 Sens. Actuators, B 206 701; (f) Mondal D, Bar M, Maity D and Baitalik S 2015 J. Phys. Chem. C 119 25429; (g) Ding A, Tang F, Wang T, Tao X and Yang J 2015 J. Chem. Sci. 127 375; (h) Chao D 2016 J. Chem. Sci. 128 133
Capra R H, Strumia M, Vadgama P M and Baruzzi A M 2005 Anal. Chimi. Acta 530 49
Li H, Chai X -S, DeMartini N, Zhan H and Fu S 2008 J. Chromatogr. A 1192 208
Liu Y, Huang J, Wang D, Hou H and You T 2010 Anal. Methods 2 855
(a) Tang L, Park J, Kim H-J, Kim Y, Kim S J, Chin J and Kim K M 2008 J. Am. Chem. Soc. 130 12606; (b) Hu M and Feng G 2012 Chem. Commun. 48 6951; (c) Wang G, Zhu H, Lin Y, Chen Y and Fu N 2015 Sens. Actuators, B 206 624; (d) He C, Qian X, Xu Y, Yang C, Yin L and Zhu W 2011 Dalton Trans. 40 1034
(a) Mummidivarapu V S, Nehra A, Hinge V K and Rao C P 2012 Org. Lett. 14 2968; (b) Yang Y, Yin C, Huo F, Chao J and Zhang Y 2014 Sens. Actuators, B 204 402; (c) Tang L, Zhou P, Huang Z, Zhao J and Cai M 2013 Tetrahedron Lett. 54 5948; (d) Tang L, Cai M, Zhou P, Zhao J, Zhong K, Hou S and Bian Y 2013 RSC Adv. 3 16802; (e) Peng Y, Dong Y-M, Dong M and Wang Y-W 2012 J. Org. Chem. 77 9072; (f) Tang L, Dai X, Cai M, Zhao J, Zhou P and Huang Z 2014 Spectrochim. Acta, Part A 122 656; (g) Kaur N and Alreja P 2015 J. Chem. Sci. 127 1253
Ye F, Zheng Z -J, Deng W -H, Zheng L -S, Deng Y, Xia C -G and Xu L -W 2013 Chem. - Eur. J. 8 2242
Zhu J -F, Yuan H, Chan W -H and Lee A W M 2010 Org. Biomol. Chem. 8 3957
Shi F, Shen J K, Chen D, Fog K, Thirstrup K, Hentzer M, Karlsson J -J, Menon V, Jones K A, Smith K E and Smith G 2011 ACS Med. Chem. Lett. 2 303
Sun Y -Q, Wang P, Liu J, Zhang J and Guo W 2012 Analyst 137 3430
Zhang Y, Guo X, Si W, Jia L and Qian X 2008 Org. Lett. 10 473
Lin W, Yuan L, Cao Z, Feng Y and Long L 2009 Chem. - Eur. J. 15 5096
Lu Q, Hou J, Wang J, Xu B, Zhang J and Yu X 2013 Chin. J. Chem. 31 641
Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 21176029, 21476029), Liaoning BaiQianWan Talents Program (No. 2012921057), and the Program for Liaoning Excellent Talents in University (LR2015001) for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information
Supplementary Information (Figures S1–S6) is available at www.ias.ac.in/chemsci.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
TANG, L., WU, D., HUANG, Z. et al. A fluorescent sensor based on binaphthol-quinoline Schiff base for relay recognition of Zn2+ and oxalate in aqueous media. J Chem Sci 128, 1337–1343 (2016). https://doi.org/10.1007/s12039-016-1124-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12039-016-1124-y