
Abstract
Hyperaccumulation of arsenic (As) by brake fern Pteris vittata has been described as an important genetic trait that provides an option for development of a sustainable phytoremediation process for As mitigation. Accumulation of very high concentration of arsenic in above-ground tissues may be the result of arsenic vacuole compartmentalization, but the mechanism(s) of arsenic uptake and transport by underground tissues are largely unknown. In this study, we made an attempt towards understanding the molecular mechanism of As hyperaccumulation in this plant. A time-dependent As accumulation study indicates an exponential accumulation of As from 7 to 30 days of arsenic exposure in fronds, and day 3–7 in roots. Root transcriptome analysis identified 554,973 transcripts. Further, subsets of 824 transcripts were differentially expressed between treated and control samples. Many of the genes of critical As-stress response, transcription factors and metal transporters, biosynthesis of chelating compounds involved in uptake and accumulation mechanisms were identified. The genes that were highly expressed such as cysteine-rich RLK, and ABC transporter G family member 26 needs further studies along with arsenite transmembrane transporter. The analysis of generated transcriptome dataset has provided valuable information and platform for further functional studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Arsenic (As) has geogenic and anthropogenic origin, widely present in the environment and poses health risk to all forms of life. People living in areas contaminated with As suffer from deformities and diseases occurring due to As toxicity (McCarty et al. 2011 and the references therein). Existing technologies for As removal relies on excavation of soil and treating with chemical, which makes it a costly method of remediating contaminated site (Wuana and Okieimen 2011). Phytoremediation is known to be an environment-friendly alternative clean-up method (Rungwa et al. 2013). However, it requires hyperaccumulator plants such as Brassica (Irtelli and Navari-Izzo 2008; Srivastava et al. 2009; Freitas-Silva et al. 2016; Rahman et al. 2016) and brake fern Pteris vittata (Ma et al. 2001; Wang et al. 2007; Sarangi and Chakrabarti 2008). The hyperaccumulator P. vittata can accumulate As to such a high level where other plants die (Gumaelius et al. 2004), or Pteris vittata has developed such mechanisms that it avoids or excludes the metal (Zhu et al. 2011). Whether the plant is resistant or accumulator depends on what happens at the root tissue (Meharg and Hartley-Whitaker 2002). Resistant plants reduce uptake of arsenate through suppression of high-affinity phosphate transporter (Meharg and MacNair 1992). So, Pteris must be having a mechanism that the other plants do not possess (Xie et al. 2009). Further, P. vittata root zone is found to be housed by specific endophytes with ability for As resistance and transformation. These endophytes are reported to play a significant role in promotion of plant growth, ameliorating soil conditions for bio-availability of metal to plants (Tiwari et al. 2016). Therefore, symbiotic interaction between plants and microbes for better improvement of metal uptake is receiving attention.
Arsenic gets into the plant root through the inorganic phosphate transport proteins in the form of arsenate (Meharg and Hartley-Whitaker 2002), whereas entry of arsenite in rice plant was reported through some root aquaporins such as nodulin 26-like intrinsic membrane proteins (NIPs) (Ma et al. 2008) and plasma membrane intrinsic proteins (PIPs) (Mosa et al. 2012). Once in the root cell, arsenate is converted into the arsenite in the root through arsenate reductase (Xu et al. 2007). This arsenite then gets complexed with glutathione and some phytochelators (Raab et al. 2004). These complexes stored in vacuole (Lombi et al. 2002). Thus As is sequestered in the vacuole, and its toxic effects are minimized. Target gene identification and expression studies related to As sequestration and detoxification was carried out in plants such as O. sativa, P. vittata and Holcus lanatus (as cited above). Here, the aim of the present study was to look at the molecular level in terms of global transcriptome and proteins that are involved in As hyperaccumulation in Indian ecotype of P. vittata. However, the molecular mechanism underlying As hyperaccumulation have not yet been completely understood and requires further elucidation at the gene and protein level, especially the full set of genes that are up-regulated or down-regulated in order to deal with the As stress and accumulation and the transcription factors regulating these genes.
The present study specifically aimed to (1) develop reference transcriptomes of an Indian eco-type of P. vittata, and (2) identify root-specific differentially expressed As-stress-related genes (including transcription factors and metal transporters) that are inherent to hyperaccumulator plants.
2 Materials and methods
2.1 Plant materials and arsenic accumulation analysis
Spores of P. vittata were germinated in plastic pots with sand and soil at a ratio of 1:1 and were maintained in greenhouse condition. Sporophytes of 6-month-olds were transplanted in As-contaminated (10 mg/kg) soil (further indicated as treated plant sample) and without As soil (control). The plants were allowed to grow for 3, 7, 15, 30 and 45 days under greenhouse condition. The plants were harvested at each time interval and washed thoroughly with Milli-Q water. The plants were separated into two parts, i.e. above-ground (fronds) and below-ground (roots including rhizomes); these were separately oven-dried at 60°C for 48 h. Dried samples (0.1 g) were acid-digested with concentrated nitric acid (Cai et al. 2000) and samples were prepared for total As analysis using ICP-OES iCAP 6300 DUO (Thermo Scientific, USA). Elemental As analyses of the samples were performed under optimized conditions according to U.S. Environmental Protection Agency (US EPA) method (US EPA 1991).
2.2 RNA isolation and Illumina sequencing
The below-ground tissues (roots including rhizome) were sampled separately from plants grown in As-treated and control soil. Tissues were thoroughly rinsed in chilled Milli-Q water, excess water wiped with tissue paper, tissues were weighed to 1 gm and immediately ground to fine powder in presence of 50 mg of polyvinylpyrrolidone (PVPP) with liquid nitrogen. Further, total RNA was extracted using extraction buffer (100mM Tris-Cl (pH-8.0), 10 mM EDTA (pH-8.0), 100 mM LiCl, 2 % SDS (v/v), 5 % β-mercaptoethanol) and treated with water saturated Phenol:Chloroform (1:1). RNA was precipitated with isopropanol and 3 M sodium acetate. Extracted total RNA preparation was purified by silica based on column DNA digestion using DNase I and removal of colouring compounds associated with below-ground tissues (humic acid, phenolic compounds) using Zymo-Spin™ IV-HRC spin filters (ZR plant RNA MiniPrep, Zymo Research, USA). Quantity and purity of extracted total RNA were determined using NanoDrop (Thermo Fisher Scientific Inc, USA), Qubit 2.0 Fluorometer with Qubit RNA BR Assay kit (Life Technologies, USA) and Agilent 2100 bioanalyzer (Agilent Technologies, USA), respectively. The isolated total RNA was used to enrich mRNA and cDNA library construction. Paired-end sequencing of cDNA libraries was constructed using TruSeq RNA Library Preparation Kit as per the manufacturer’s instructions (Illumina Inc., CA) and sequencing with an Illumina HiSeq 2500 sequencing platform (Kukurba and Montgomery 2015).
2.3 Sequence analysis, annotation and differential gene expression
RNA-sequencing and de novo assembly was accomplished with short reads assembling program – Trinity (version- trinityrnaseq_r20140717) (Grabherr et al. 2011). The transcripts of length ≥ 200 bp were only used to determine the transcript expression using Bowtie2 program (ver. 2.2.2.6). Transcripts with ≥1 FPKM (fragments per kilobase of exon model per million fragments mapped) were used for further downstream annotations. BLASTX search was performed with obtained transcript sequences against several databases, including NCBI Nr, UniProt, Gene Ontology, using a cut-off E-value of 10−5 and % identity cut-off of 40%. Further, these transcripts were classified based on GO annotation for molecular function, biological process and cellular component (Conesa et al. 2005). KEGG (Kyoto encyclopaedia of genes and genomes) pathway analysis was based on the comparative results between annotated transcripts and the current KEGG database (Kanehisa et al. 2016). Differential gene expression analysis between treated and control libraries were performed using DEseq program (ver. 1.16.0) (Anders and Huber 2010).
2.4 Reverse transcriptase-PCR (RT-PCR) validation of RNA-sequencing analysis
To validate the mRNA abundance of arsenic related genes, we randomly selected 6 genes identified in RNA-seq analysis to perform semi-quantitative RT-PCR analysis. Total RNA (1 μg) from control and treated samples were used to synthesize cDNAs using SuperScript IV first-strand synthesis kit (Thermo Inc., USA) according to manufacturers guidelines. The specifics of genes and their respective primers for RT-PCR are listed in (supplementary table 4). We used elongation factor (EF-1α) as an internal control for normalization.
3 Results
3.1 Time-dependent As accumulation
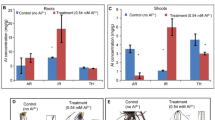
The below-ground tissue samples (roots including rhizome) were analysed to study the time of maximum influx of As into P. vittata upon As exposure vis-à-vis induction of expression of As-related genes. Total As concentration in both above-ground (fronds) and below-ground tissues were increased with increasing exposure time from 0 to 30 day. Time-dependent As accumulation indicated that the exponential accumulation of As starts from day 7 to day 30 in fronds, while in roots, sudden increase of As from day 3 to 7 was observed. However, there were no much drastic change in the As uptake rate from day 7 onwards was detected in roots. After 30 days, decrease in As content was detected both in leaf and root tissues as measured on 45 day, while 50 days onwards plants did not survived. Highest As uptake in fronds (2571.07 µg gm−1 dry wt.) and in roots (1885.93 µg gm−1 dry wt.) was detected on 30 day of As exposure (figure 1). Based on time-dependent As accumulation study, below-ground tissues were harvested and pooled from 3 and 7 day for isolation of total RNA.

Arsenic accumulation in foliar (above-ground) and root (below-ground) samples.
3.2 RNA sequencing and de novo assembly
In the present study, tissue-specific libraries from below-ground tissues of P. vittata were constructed for understanding the molecular mechanism underlying As hyperaccumulation. To capture the maximum transcript diversity expressed in below-ground tissues upon As exposure, pooled tissues from roots and rhizome were used to isolate total RNA. This total RNA from control and treated samples were used to construct cDNA libraries for sequencing. These libraries were subjected to sequencing on Illumina HiSeq 2500 platform that generated a total of 4666.6 Mb and 3256.28 Mb raw reads from the control and treated library, respectively. The obtained fastq files comprising raw reads were trimmed for adapters and low-quality reads prior to performing de novo assembly to avoid sequence-specific biasness. The clean reads so obtained had average GC content for control and treated library sequences of 54.72% and 56.93%, respectively, indicating marginal GC richness of the transcripts. The trimmed reads were aligned to the assembled transcriptome (length ≥ 200bp) that resulted in 140,480 unique transcripts for control and 1,52,573 for treated library with expression ≥1.0 FPKM. Overall, a total of 5,61,740 transcripts were assembled using Trinity with default options, whereas 5,54,973 transcripts obtained after cd-hit clustering with 56.4 mean GC % (table 1). The raw reads and the assembly data for treated library were deposited at the SRA section of NCBI under the accession SRX2354811.
3.3 Transcripts annotation and pathway analysis
The filtered, assembled transcripts were primarily compared with NCBI non-redundant (Nr) protein database using BLASTX program. Matches with E-value cut-off of 10−5 and % identity cut-off of 40% were retained for further annotation using UniProt, Gene Ontology, KEGG databases. The BLASTX search result indicated that 217,344 transcripts have similarity of ≥ 60% at protein level. Out of these, a total of 67,596 (31.1 %) were annotated against Nr, 46,438 (21.36 %) against UniProt, 10,324 (4.75 %) against GO, and 5,665 (2.6 %) against KEGG database. Based on NR database annotation, the top BLASTX hit of each transcript return with most frequent organisms were Ricinus communis (7,020), Dorcoceras hygrometricum (5,847), Physcomitrella patens subsp. patens (3,716), Marchantia polymorpha subsp. polymorpha (2,870), and Selaginella moellendorffii (2,313).
The Gene Ontology (GO) terms were assigned to 10,324 transcripts and the terms were summarized into the following GO categories; biological processes, molecular functions and cellular components. The top 20 terms of each category are shown in figure 2A, B and C. Among the biological processes category, highest transcripts belong to translation (1,247); in molecular function category, maximum transcripts belong to ATP binding (2,480), whereas in cellular component category, maximum number of transcripts belong to integral component of membrane and structural constituent of ribosome (1,541 and 1,205, respectively).

Gene Ontology annotation of transcriptome. Top 20 GO terms with number of transcripts are summarized for each category as follows: (A) Biological process, (B) Molecular function and (C) Cellular components.
To better understand the function of sequenced transcripts, these were searched against the KEGG database. KEGG pathway analysis showed that 5,665 transcripts were assigned to 96 pathways, including metabolic pathways with 611 (10.78%) transcripts, biosynthesis of secondary metabolites 295 (5.2%), microbial metabolism in diverse environments 135 (2.3%), and oxidative phosphorylation 60 (1%), (supplementary table 1). Further exploration of stress-related pathway, such as oxidative phosphorylation (KEGG pathway ID: ko00190), revealed that most genes including proton channel and cytochrome redox complexes were prominently identified.
3.4 Identification of abiotic stress-related transcription factors
Regulation of gene expression upon As exposure was of interest in the present study. Alignment and analysis of differential gene expression of annotated transcripts of treated and control libraries resulted in the identification of transcripts belonging to 21 transcription factor families. The members of MYB, Orphan, AP2, Dof, WRKY, bHLH, HOMEOBOX, NAC, Trihelix, MADS, C3HDZ and Tubby-like F-box protein were dominant (supplementary table 2).
3.5 Transporter proteins
Large number of metal transporter families including transmembrane transporter, ABC-type, aquaporins, NRAMP and ZIP family proteins were reported to be involved in heavy metal (HM) uptake, transport and distribution. Significant number of transporters including ABC transporter family proteins, sulphate transpoters, inorganic phosphate transmembrane transporter, efflux family proteins, HM-associated domain containing proteins, and aquaporins were identified as differentially expressed genes (DEGs) upon As-stress. Among the differentially expressed metal transporters, the major portion belongs to inorganic phosphate transmembrane transporter; ABC-type, aquaporins, NRAMPs, ZIPs, and arsenite transporters were identified in the present study (supplementary table 3). Further, the genes encoding MT-like proteins 2 and phytochelatins (PCs)-related genes were also identified.
3.6 Differential gene expression analysis
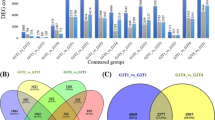
By comparing the transcripts from As-treated and control libraries, 5,54,973 transcripts were subjected to DEseq program for identification of DEGs. Relatively large numbers of differentially expressed transcripts were identified. Among all the identified DEGs, 420 were identified to be up-regulated and 404 were down-regulated (figure 3). Two of the unique transcript c110929_g1_i1 and c215021_g1_i1 were highly up-regulated (log2FC: 13.2995 and 13.0142, respectively), whereas the unique transcript c113888_g1_i1 was found to be highly down-regulated (log2FC: -14.2293).

Bar diagram depicting differential gene expression between As-treated and control libraries.
Further, semi-quantitative RT-PCR analysis of six arsenic/metal-related genes showed significant level of differential expression. Among these six genes, only the gene RACS did not show any difference in expression, whereas the other genes were significantly up-regulated (figure 4).

Semi-quantitative RT-PCR assays of arsenic-responsive genes in arsenic treated (T) and control (C) tissues. Glutaredoxin like protein 4 (GTR4), arsenite transporter like protein (AT), arsenate reductase (AR), response to arsenic containing substance (RACS), Metal tolerance protein 10 (MTP10), phytochelatin (PC) and elongation factor 1 alpha (EF1α).
4 Discussion
Arsenic is ubiquitous in the earth’s crust in the form of arsenopyrite (Zhao et al. 2010). Atmospheric flux of As is due to volcanic action, erosion of rocks and forest fires, or through anthropogenic activities (Duker et al. 2005; Neumann et al. 2010). Compared to physic-chemical and mechanical As remediation method, phytoremediation is being evaluated as an eco-friendly, cost-effective, sustainable method. The findings of As-hyperaccumulating fern species (Ma et al. 2001) and investigation of the molecular mechanisms essential to effectively control HM uptake, translocation and accumulation by plant genetic manipulation and genomic research (Wang et al. 2013) has received considerable attention.
4.1 Arsenic accumulation in below-ground tissue
In the tissue-specific As accumulation study, majority of As accumulated in the above-ground biomass (Potdukhe et al. 2015), which supports the findings of Ma et al. (2001) that 93% of the total As accumulated in the above-ground biomass was concentrated in the fronds. Low As was recorded in below-ground tissue, but rapid uptake of As was higher in below-ground tissue within short time (3 to 7 day) of As exposure compared to above-ground tissue (figure 1). It was observed that As concentrations increased with time up to 30 days in fronds, while there was rapid uptake up to day 7 and there was not much change observed from day 7 onwards in below-ground tissues. The decrease in As concentration in both above- and below-ground tissues after 30 days of As exposure could be attributed to the dilution factor resulting from an increase in the plant biomass, which was also noted by Cai et al. (2004).
4.2 Root transcripts associated with As stress and regulatory network
Based on time-dependent As accumulation study and pooling the below-ground tissues (roots and rhizome) from two different time frames (day 3 and 7) resulted in 1,40,480 unique transcripts for control and 1,52,573 for treated library with expression ≥1.0 FPKM. Further, overall 5,54,973 transcripts were obtained after cd-hit clustering and about 2,17,344 transcripts were shown similarity of more than 60% at protein level in BLASTX search, these results indicating the representation of high genes diversity in constructed libraries. In this study, we found that a broad set of unique and novel genes was significantly up- or down-regulated in response to As exposure. A global analysis of transcriptome was reported to assists in identification of critical genes, expression and understanding the regulatory mechanisms in response to abiotic stresses in plant such as high-salinity, drought and cold stresses (Shan et al. 2013) and HM stress (Li et al. 2014). Arsenate and phosphate are reported to have striking similarities and share the same transport pathway in higher plants (Ullrich-Eberius et al. 1989). The uptake of As in the form of arsanate involves inorganic phosphate transport system as shown in P. vittata (Wang et al. 2002) and Arabidopsis thaliana (Shin et al. 2004; Gonzalez et al. 2005), whereas arsenite can be actively taken up by plant roots through the subfamilies of aquaporins (Meharg and Jardine 2003; Bienert et al. 2008a). In the present study, the transcripts of inorganic phosphate transporter (PiT) and a great number of transcripts for different subfamilies of aquaporins, such as plasma membrane intrinsic proteins (PIPs), nodulin 26-like intrinsic proteins (NIPs), tonoplast intrinsic proteins (TIPs) and major intrinsic proteins (MIPs), were identified (supplementary table 3). Along with Pi transporters, higher transcripts of aquaporins as identified in the present study might be responsible for direct influx of arsenite (Abedin et al. 2002; Meharg and Jardine 2003) and higher accumulation of As in the form of arsenite. Entry of As in root cells activates stress including oxidative stress responsive signalling molecules via protein kinase cascades (Rao et al. 2011), and oxidative phosphorylation system.
In the present investigation, cysteine-rich RLK (receptor-like protein kinase) 8 was found to be highly up-regulated. Interestingly, the up-regulation of CRK (cysteine-rich RLK) were also reported for Cr stress as was elucidated from microarray data of expression of signalling genes in rice roots (Trinh et al. 2014). This indicates that changes in cysteine-rich RLK expression might be related to the response of metals/metalloids stress. High up-regulation of cysteine-rich RLK 8, which is reported to be induced by reactive oxygen species (ROS) (Wrzaczek et al. 2010; Burdiak et al. 2015) and ubiquinol-cytochrome c reductase cytochrome b subunit (table 2), might be induced by As-stress. Further, these cascades induce different transcription factors (TFs) such as WRKY, bZIP, MYB families, in the nucleus to regulate the expression of some functional genes (Huang et al. 2012; Thapa et al. 2012). These genes might encode transporters (arsenite transporters, ABC-type, aquaporins, ZIP and Nramps) and metal chelators such as MTs and PCs, which would detoxify and accumulate As in plant cells. In the present study, about 67 transcripts belonging to different TFs families and 54 transcripts encoding HM transporter proteins including arsenite transporter were identified (supplementary tables 2 and 3). Among the major transporters, ABCCs transporter are full size and contain forward-oriented nucleotide binding domain (NBD) and a trans-membrane domain (TMD). Members of this family generally play some role in detoxification (Verrier et al. 2008). Proteomic data obtained for Arabidopsis vacuoles suggest that most ABCCs of Arabidopsis reside in the tonoplast, and in addition to AtABCC1/2, other ABCCs, may contribute to the overall glutathione conjugate transport activity (Jaquinod et al. 2007). Expression of ABCC-type multidrug resistance-associated proteins (MRPs) in the present study indicates high activity localization near the tonoplast of the vacuole. Song et al. (2010) have shown extreme sensitive of A. thaliana to arsenic and arsenic-based herbicides in the absence of two ABCC-type transporters, AtABCC1 and AtABCC2. In differential gene expression analysis, some critical genes were prominently up-regulated, which includes cysteine-rich receptor-like protein kinase 8 (CRK8), ubiquinol-cytochrome c reductase cytochrome b subunit, ABC transporter G family member 26 and F-type H+-transporting ATPase subunit (table 2). The down-regulated genes were identified as iron complex outer-membrane receptor protein, mechanosensitive ion channel protein 1, and tartrate-resistant acid phosphatase type 5 (table 3). The genes that are directly related to As uptake, transport and accumulation were moderately up-regulated (table 4). Moreover, up-regulation of GTR4, AT, AR, MTP10 and PC genes as observed in RT-PCR assay corroborate the reported literature. Based on the DEGs enrichments in the present study and reported literature (Shin et al. 2004; Raab et al. 2004; Ma et al. 2008; Ellis et al. 2006; Bienert et al. 2008b; Thapa et al. 2012; Cesaro et al. 2015; Tiwari et al. 2016), a possible regulatory network of As uptake, transport and accumulation was put forward in the below-ground tissue of P. vittata (figure 5). The present investigation reports root transcriptome data and provides useful information about the root-specific As-stress-related genes that may pave the way for future functional studies.

Schematic diagram of root uptake, accumulation and vascular transport of arsenic.
References
Abedin MJ, Feldmann J and Meharg AA 2002 Uptake kinetics of arsenic species in rice plants. Plant Physiol. 128 1120–1128
Anders S and Huber W 2010 Differential expression analysis for sequence count data. Genome boil. 11 R106
Bienert GP, Schussler MD and Jahn TP 2008a Metalloids: Essential, beneficial or toxic? Major intrinsic proteins sort it out. Trends Biochem. Sci. 33 20–26
Bienert GP, Thorsen M, Schüssler MD, Nilsson HR, Wagner A, Tamas MJ and Jahn TP 2008b A subgroup of plant aquaporins facilitate the bidirectional diffusion of As(OH)3 and Sb(OH)3 across membranes. BMC Biol. 6 26
Burdiak P, Rusaczonek A, Witon D, Glow D and Karpinski S 2015 Cysteine-rich receptor-like kinase CRK5 as a regulator of growth, development, and ultraviolet radiation responses in Arabidopsis thaliana. J. Exp. Bot. 66 3325–3337
Cai Y, Georgiadis M and Fourqurean JW 2000 Determination of arsenic in seagrass using inductively coupled plasma mass spectrometry. Spectrochimica Acta Part B B55 1411–1422
Cai Y, Su J and Ma LQ. 2004 Low molecular weight thiols in arsenic hyperaccumulator Pteris vittata upon exposure to arsenic and other trace elements. Environ. Pollut. 129 69–78
Cesaro P, Cattaneo C, Bona E, Berta G and Cavaletto M 2015 The arsenic hyperaccumulating Pteris vittata expresses two arsenate reductases. Sci. Rep. 5 14525
Conesa A, Gotz S, Garrcia-Gomez M, Terol J, Talon M and Robles M 2005 Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21 3674–3676
Duker AA, Carranza EJM, Hale M (2005) Arsenic geochemistry and health. Environ. Int. 31 631–641
Ellis DR, Gumaelius L, Indriolo E, Pickering IJ, Banks JA and Salt DE (2006) A novel arsenate reductase from the arsenic hyperaccumulating fern Pteris vittata. Plant Physiol. 141 1544–1554
Freitas-Silva L, Araujo TO, Silva LC, Oliveira JA and Araujo JM 2016 Arsenic accumulation in Brassicaceae seedlings and its effects on growth and plant anatomy. Ecotoxicol. Environ. Saf. 124 1–9
Gonzalez E., Solano R., Rubio V., Leyva A and Paz-Ares J 2005 PHOSPHATE TRANSPORTER TRAFFIC FACILITATOR1 is a plant-specific SEC12-related protein that enables the endoplasmic reticulum exit of a high-affinity phosphate transporter in Arabidopsis. Plant Cell 17 3500–3512
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N et al. 2011 Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotech. 29 644–652
Gumaelius L, Lahner B, Salt D and Banks JA 2004 Arsenic hyperaccumulation in gametophytes of P. vittata: a new model system for analysis of arsenic hyperaccumulation. Plant Physiol. 136 3198–3208
Huang TL, Nguyen QT, Fu SF, Lin CY, Chen YC and Huang HJ 2012 Transcriptomic changes and signalling pathways induced by arsenic stress in rice roots. Plant Mol. Biol. 80 587–608
Irtelli B and Navari-Izzo F 2008 Uptake kinetics of different arsenic species by Brassica carinata. Plant Soil 303 105–113
Jaquinod M, Villiers F, Kieffer-Jaquinod S, Hugouvieux V, Bruley C, Garin J and Bourguignon J 2007 A proteomics dissection of Arabidopsis thaliana vacuoles isolated from cell culture. Mol .Cell. Proteomics 6 394–412
Kanehisa M, Sato Y, Kawashima M, Furumichi M and Tanabe M 2016 KEGG as a reference resource for gene and protein annotation. Nucl. Acids Res. 44 D457–D462
Kukurba KR and Montgomery SB 2015 RNA sequencing and analysis. Cold Spring Harb. Protoc. 11 951–969
Li JM, Liu B, Cheng F, Wang XW, Aarts MGM and Wu J 2014 Expression profiling reveals functionally redundant multiple-copy genes related to zinc, iron and cadmium responses in Brassica rapa. New Phytol. 203 182–194
Lombi E, Zhao F-J, Fuhrmann M, Ma LQ and McGrath SP 2002 Arsenic distribution and speciation in the fronds of the hyperaccumulator Pteris vittata. New Phytol. 156 195–203
Ma JF, Yamaji N, Mitani N, Xu X-Y, Su Y-H, McGrath SP and Zhao F-J 2008 Transporters of arsenite in rice and their role in arsenic accumulation in rice grain. Proc. Natl. Acad. Sci. USA 105 9931–9935
Ma LQ, Komar KM, Tu C, Zhang W, Cai Y and Kennelley ED 2001 A fern that hyperaccumulates arsenic. Nature 409 579
McCarty KM, Hanh HT and Kim K-W 2011 Arsenic geochemistry and human health in South East Asia. Rev. Environ. Health 26 71–78
Meharg AA and Hartley-Whitaker J 2002 Arsenic uptake and metabolism in arsenic resistant and nonresistant plant species. New Phytol. 154 29–43
Meharg AA and Jardine L 2003 Arsenite transport into paddy rice (Oryza sativa) roots. New Phytol. 157 39–44
Meharg, AA and MacNair MR 1992 Suppression of the high affinity phosphate uptake system: a mechanism of arsenate tolerance in Holcus lanatus L. J. Exp. Bot. 43 519–524
Mosa KA, Kumar K, Chhikara S, Mcdermott J, Liu Z, Musante C, White JC and Dhankher OP 2012 Members of rice plasma membrane intrinsic proteins subfamily are involved in arsenite permeability and tolerance in plants. Transgenic Res. 21 1265–1277
Neumann RB, Ashfaque KN, Badruzzaman ABM, Ali MA, Shoemaker JK and Harvey CF 2010 Anthropogenic influences on groundwater arsenic concentrations in Bangladesh. Nat. Geosci. 3 46–52
Potdukhe R, Sarangi BK, Pandey RA and Thul ST 2015 “Arsenic hyperaccumulation efficiency depends on time and tissue in Pteris vittata” presented in International Symposium on Biodiversity, Agriculture, Environment and Forestry. 11-12, Dec. 2015, Book of Abstract, pp 194
Raab A, Feldmann J and Meharg AA 2004 The Nature of arsenic-phytochelatin complexes in Holcus lanatus and Pteris cretica. Plant Physiol. 134 1113–1122
Rahman M, Haq N and Williams ID 2016 Phytoaccumulation of arsenic, cadmium and lead by Brassica juncea parents and their f1 hybrids. J. Environ. Prot. 7 613–622
Rao KP, Vani G, Kumar K, Wankhede DP, Misra M, Gupta M and Sinha AK 2011 Arsenic stress activates MAP kinase in rice roots and leaves. Arch. Biochem. Biophys. 506 73–82
Rungwa S, Arpa G, Sakulas H, Harakuwe A and Timi D 2013 Phytoremediation – An eco-friendly and sustainable method of heavy metal removal from closed mine environments in Papua New Guinea, Procedia. Earth Planet Sci. 6 269–277
Sarangi BK and Chakrabarti T 2008 Characterization of an ecotype of brake-fern, Pteris vittata, for arsenic tolerance and accumulation in plant biomass. Cytol. Genet. 42 16–31
Shan XH, Li YD, Jiang Y, Jiang ZL, Hao WY and Yuan YP 2013 Transcriptome profile analysis of maize seedlings in response to high-salinity, drought and cold stresses by deep sequencing. Plant Mol. Biol. Rep. 31 1485–1491
Shin H, Shin HS, Dewbre GR and Harrison MJ 2004 Phosphate transport in Arabidopsis: Pht1;1 and Pht1;4 play a major role in phosphate acquisition from both low- and high-phosphate environments. Plant J. 39 629–642
Song WY, Park J, Mendoza-Cózatl DG, Suter-Grotemeyer M, Shim D, Hörtensteiner S, Geisler M, Weder B, Rea PA, Rentsch D, Schroeder JI, Lee Y and Martinoia E 2010 Arsenic tolerance in Arabidopsis is mediated by two ABCC-type phytochelatin transporters. Proc. Natl. Acad. Sci. USA 107 21187–2192
Srivastava S, Srivastava AK, Suprasanna P and D’souza SF 2009 Comparative biochemical and transcriptional profiling of two contrasting varieties of Brassica juncea L. in response to arsenic exposure reveals mechanisms of stress perception and tolerance. J. Exp. Bot. 60 3419–3431
Thapa G, Sadhukhan A, Panda SK and Sahoo L 2012 Molecular mechanistic model of plant heavy metal tolerance. Biometals 25 489–505
Tiwari S, Sarangi BK and Thul ST 2016 Identification of arsenic resistant endophytic bacteria from Pteris vittata roots and characterization for arsenic remediation application. J. Environ. Manage. 180 359–365
Trinh NN, Huang TL, Chi WC, Fu SF, Chen CC and Huang HZ 2014 Chromium stress response effect on signal transduction and expression of signaling genes in rice. Physiol. Plant 150 205–224
US EPA 1991 Office of Research and Development. Washington, D.C.: Determination of metals and trace elements in water and wastes by inductively coupled plasma-atomic emission spectrometry; EPA (series 200). EPA/600/4-91/010 pp 31–83
Ullrich-Eberius CI, Sanz A and Novacky AJ 1989 Evaluation of arsenate- and vanadate-associated changes of electrical membrane potential and phosphate transport in Lemna gibba G1. J. Exp. Bot. 40 119–128
Verrier PJ, Bird D, Burla B, Dassa E, Forestier C, Geisler M, et al. 2008 Plant ABC proteins–a unified nomenclature and updated inventory. Trends Plant Sci. 13 151–159
Wang H, Wong MH, Lan C, Baker AJM, Qin Y, Chen G, et al. 2007 Uptake and accumulation of arsenic by 11 Pteris taxa from southern China. Environ. Pollut. 145 225–233
Wang JR, Zhao FJ, Meharg AA, Raab A, Feldmann J and McGrath SP 2002 Mechanisms of arsenic hyperaccumulation in Pteris vittata. Uptake kinetics, interactions with phosphate, and arsenic speciation. Plant Physiol. 130 1552–1561
Wang Y, Xu L, Chen YL, Shen H, Gong YQ, Limera C and Liu L 2013 Transcriptome profiling of radish (Raphanus sativus L.) root and identification of genes involved in response to lead (Pb) stress with next generation sequencing. PLoS ONE 8 e66539
Wrzaczek M, Brosche M, Salojärvi J, Kangasjärvi S, Idänheimo N, Mersmann S, et al. (2010) Transcriptional regulation of the CRK/DUF26 group of receptor-like protein kinases by ozone and plant hormones in Arabidopsis. BMC Plant Biol. 10 95
Wuana RA and Okieimen FE 2011 Heavy metals in contaminated soils: a review of sources, chemistry, risks and best available strategies for remediation. ISRN Ecology Article ID 402647, https://doi.org/10.5402/2011/402647.
Xie Q-E, Yan X-L, Liao X-Y and Li X 2009 The arsenic hyperaccumulator fern Pteris vittata L. Environ. Sci. Technol. 43 8488–8495
Xu XY, McGrath SP and Zhao FJ 2007 Rapid reduction of arsenate in the medium mediated by plant roots. New Phytol. 176 590–599
Zhao FJ, McGrath SP and Meharg AA 2010 Arsenic as a food chain contaminant: mechanisms of plant uptake and metabolism and mitigation strategies. Annu. Rev. Plant Biol. 61 535–59
Zhu XF, Zheng C, Hu YT, Jiang T, Liu Y, Dong NY, Yang JL and Zheng SJ 2011 Cadmium-induced oxalate secretion from root apex is associated with cadmium exclusion and resistance in Lycopersicon esulentum. Plant Cell Environ. 34 1055–1064
Acknowledgements
This work was funded by Department of Biotechnology (DBT), New Delhi India, under Grant ID: BT/PR-5523/BCE/08/909/2012. The authors acknowledge the kind support and encouragement extended by Director, NEERI, Nagpur. The authors are also wish to acknowledge SciGenom Labs, India for the generation of sequencing data. This has CSIR-NEERI Communication No.: CSIR-NEERI/KRC/2017/JULY/EBGD/8.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Gagandeep Kang.
Corresponding editor: Gagandeep Kang
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Potdukhe, R.M., Bedi, P., Sarangi, B.K. et al. Root transcripts associated with arsenic accumulation in hyperaccumulator Pteris vittata. J Biosci 43, 105–115 (2018). https://doi.org/10.1007/s12038-018-9735-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12038-018-9735-8