
Abstract
The efficient and prolonged neurotransmission is reliant on the coordinated action of numerous synaptic proteins in the presynaptic compartment that remodels synaptic vesicles for neurotransmitter packaging and facilitates their exocytosis. Once a cycle of neurotransmission is completed, membranes and associated proteins are endocytosed into the cytoplasm for recycling or degradation. Both exocytosis and endocytosis are closely regulated in a timely and spatially constrained manner. Recent research demonstrated the impact of dysfunctional synaptic vesicle retrieval in causing retrograde degeneration of midbrain neurons and has highlighted the importance of such endocytic proteins, including auxilin, synaptojanin1 (SJ1), and endophilin A (EndoA) in neurodegenerative diseases. Additionally, the role of other associated proteins, including leucine-rich repeat kinase 2 (LRRK2), adaptor proteins, and retromer proteins, is being investigated for their roles in regulating synaptic vesicle recycling. Research suggests that the degradation of defective vesicles via presynaptic autophagy, followed by their recycling, not only revitalizes them in the active zone but also contributes to strengthening synaptic plasticity. The presynaptic autophagy rejuvenating terminals and maintaining neuroplasticity is unique in autophagosome formation. It involves several synaptic proteins to support autophagosome construction in tiny compartments and their retrograde trafficking toward the cell bodies. Despite having a comprehensive understanding of ATG proteins in autophagy, we still lack a framework to explain how autophagy is triggered and potentiated in compact presynaptic compartments. Here, we reviewed synaptic proteins’ involvement in forming presynaptic autophagosomes and in retrograde trafficking of terminal cargos. The review also discusses the status of endocytic proteins and endocytosis-regulating proteins in neurodegenerative diseases and strategies to combat neurodegeneration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurons are post-mitotic cells with intricate cellular architecture. Unlike non-polar cells, neuronal morphology is differentiated into compartments—dendrites, cell body (soma), axon, and arborized terminals [1]. The polarized morphology and increased surface area amplify the energy budget required to maintain ion gradients, resting potential, action potential firing, and efficient synaptic transmission [2, 3]; together, increasing the metabolic rate and making them prone to oxidative stress, organelles dysfunction, protein aberrations, and neurodegeneration [3]. Furthermore, the neuronal compartments (soma and axon terminals) differ in morphology, chemical composition, and biochemistry [4]. While the soma of neurons is almost like non-polar cells, the extended axon and terminals are devoid of the nucleus and lack major synthetic machinery to synthesize biomolecules [5]; therefore, axon terminals primarily rely on biosynthetic processes occurring in the cell body for the urgent supply of organelles, proteins, and other biomolecules required for neurotransmission. However, during synaptic maintenance and plasticity, presynaptic compartments abruptly adapt autonomy to synthesize local proteins following the distribution of ribosomes, mRNAs, and other synthetic machinery in the compartments [6].
Apart from the reliance on cell body, axon terminals and synaptic boutons are shrunken-cytoplasmic zones with increased bioenergetics due to high mitochondrial density [1], which altogether bows them down in the state of being likely to be affected. Research in many neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD), showed that aberrant proteins and organelles accumulate in the axon terminals much earlier than degeneration events [7,8,9]. It is often impossible for neurons to distribute or eliminate the redundancy due to their post-mitotic nature; mainly, the anomalies are unevenly segregated more in presynaptic terminals than in the neuronal soma [10]. It is probably because of the higher density and significant volume occupancy of terminals and synapses, which uniquely makes them predominantly vulnerable [11, 12], putting them at the degeneration edge. Therefore, presynaptic terminals in neurons deteriorate before their soma degenerates [13,14,15,16].
Autophagy is a lysosome-dependent evolutionarily conserved clearance process. Typically, every mammalian cell relies upon autophagy to digest its anomalies. It rejuvenates the cell by catabolizing and recycling several subcellular constituents, including redundant proteins, lipids, and dysfunctional organelles. Also, the increased catabolic process delivers nutrition during starvation by digesting damaged proteins and organelles when no longer required [17]. The process involves the phagophore formation that sequesters ubiquitin-tagged superfluous proteins and organelles and subsequently matures until its open edges fuse to create an enclosed autophagosome vesicle. The mature autophagosome carries the sequestered cytoplasmic debris to the lysosome, which digests and recycles the components back into the cytosol [18]. Neurons depend on efficient clearance mechanisms like autophagy to regularly digest and recycle damaged organelles and aberrant proteins and sustain neuronal health and function [19, 20]. Research showed that in many neurological disorders, including AD, PD, HD, and amyotrophic lateral sclerosis (ALS), defective autophagy leads to proteotoxic and oxidative stress due to augmented protein misfolding and mitochondrial dysfunction, respectively [21,22,23,24,25,26,27].
One of the current strategies to combat neurodegeneration in such diseases is to protect deteriorating neurons by enhancing neuronal autophagy to boost the clearance of cellular debris. However, the therapeutic opportunities to treat neurodegenerative diseases by targeting abnormal neuronal autophagy in deteriorating neurons are minimal. One reason for unsatisfying results could be the ignorance of neuronal cytoarchitecture while studying autophagy in neurons. Researchers study neuronal autophagy as they study it in other non-polar cells. But, due to the peerless cytomorphology of the neuron and intra-neuronal difference in the chemical composition of the cell body and axon terminals, they possess compartment-specific autophagy [4, 10]. That is why boosting autophagy in neurons often digests the junks but fails to protect crucial presynaptic terminals in the absence/breakdown of local (presynaptic) clearance. Hoffmann et al. demonstrated that presynaptic boutons possess a robust, highly, and locally regulated distinct autophagy that responds only to local damages and temporally digest misfolded synaptic proteins therein to maintain the synaptic health, integrity, and function [28]. Thus, understanding local quality control mechanisms (presynaptic autophagy), other than general autophagy in neurons, in arborized terminals and presynaptic compartments, is timely, urgent, and needed to ameliorate the early degeneration of synaptic terminals in neurodegenerative diseases.
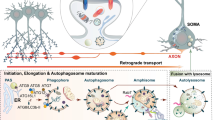
Endophilin A (EndoA) Remodels Vesicular Structure: The Docking Site for the ATG Proteins
Endophilin A (EndoA) is a presynaptic protein that regulates multiple membrane remodeling through several mechanisms, such as scaffolding, inserting amphipathic α-helices into the lipid bilayer, and protein crowding. By scaffolding and protein crowding mechanisms, the EndoA forcibly changes the conformation of intrinsic proteins and recruits its binding partners, such as dynamin-1 and synaptojanin 1 (SJ1), respectively [29, 30]. Inserting amphipathic α-helices into the lipid bilayer generates wedge force [29]. Structurally, EndoA consists of two major domains: an N-terminal BIN/amphiphysin/Rvs (BAR) domain—for the induction, stabilization, and sensing membrane curvature of the clathrin-coated vesicle (CCV)–and a C-terminal src homology 3 (SH3) domain that facilitates proteins’ interaction and recruitment to the membrane curvature site (Fig. 1) [29, 31]. Typically, the BAR domain signals the protein to induce only the internal curvature of the membrane through the scaffolding mechanism. But, on binding with the curved membrane (like the synaptic membrane), the EndoA inserts its amphipathic α-helices (residue 60–87) into the bilipid layer. The protein’s BAR domain forms tight contact with the membrane, and its amphipathic α-helices penetrate deeply into the bilipid layer, causing the membrane to shape itself tubular [32]. However, the surface ingression of amphipathic α-helices—where the scaffolding effect is significantly weakened due to the distantly placed BAR domain—remodels the membrane into a vesicle [29, 32,33,34]. Interestingly, PD-linked leucine-rich repeat kinase 2 (LRRK2) phosphorylates the EndoA at S75, residing in the protein’s amphipathic α-helix [29]. The corresponding phosphorylation toggles a conformational switch from the tubular to the vesicular structure [29, 35, 36]. The phosphorylated EndoA associates loosely with the membrane to promote a highly curved membrane compared to the non-phosphorylated form. Research showed that autophagic proteins (ATGs) use the increased membrane curvature as a docking site, and LRRK2-mediated phosphorylated EndoA promotes ATG3 recruitment to the vesicles [36]. The site-specific (S75) mutation in EndoA blocks autophagy in the terminal compartments, which implies the direct role of membrane-bending protein EndoA in presynaptic autophagy, apart from its vital role in endocytosis [36, 37]. Murdoch et al. showed that reduced expression levels of EndoA in mammalian models result in impaired autophagosome formation, decreased autophagy, and aggregation of ubiquitinated and dysfunctional proteins over time, which eventually lead to impaired movement, age-dependent ataxia, and neurodegeneration [38].

Structure of AP4, AP2, EndoA, SJ1, and SNX protein family showing roles of their subunits and domains
Interestingly, Endophilin B1 (EndoB1), an endophilin protein family member, also regulates autophagy in neurons [39]. Like EndoA, EndoB1 has BAR and SH3 domains at N-and C-terminal positions, respectively [40]. The originally discovered Bax-interacting protein, EndoB1, is a versatile protein with involvement in various cellular events, such as apoptosis, autophagy, and mitochondrial health [41]. EndoB1 interacts with the Beclin1 via ultraviolet irradiation resistant–associated gene (UVRAG) [39]. Beclin1 is a nucleation-promoting protein that forms the EndoB1-Beclin1 complex and activates the PI3KC3 to promote autophagy [39]. Contrary to LRRK2-mediated phosphorylation of EndoA, Cdk5 activity phosphorylates EndoB1 at the T145-position in its BAR domain [42]. Research reported significantly decreased EndoB1 protein expression in AD patients, where the cause of pathogenesis is the aggregation of amyloid-β and its reduced clearance in hippocampal neurons [41]. According to Wang et al., the increased load of amyloid-β is secondary to loss of EndoB1-mediated autophagy [41]. Thus, the phosphorylation of both EndoA and EndoB1 induces autophagy for synaptic health [16, 36, 37], but through a different mechanism.
Synaptojanin 1 Removes PI3P to Promote Autophagosome Biogenesis
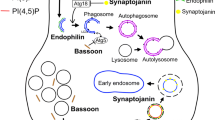
SJ1 is a poly-phosphoinositide phosphatase synaptic protein having dual but parallel roles in endocytic recycling and presynaptic autophagy. The protein comprises an N-terminal Sac1 domain to dephosphorylate PI4P and a central 5-phosphatase domain to remove phosphate from PI(4,5)P2 and PI(3,4,5)P3. It also consists of RNA recognizing motif (RRM), and an unstructured C-terminal proline-rich domain (PRD); the PRD interacts with SH3 domain–containing endocytic proteins, primarily EndoA, during clathrin uncoating (Fig. 1) [43,44,45]. The binding of SJ1 to curvature-generating EndoA—near the neck of an endocytic pit—couples to dephosphorylation of synaptic vesicle–enriched PI(4,5)P2 at 5- and 4-phosphosite by SJ1’s 5-phosphatase and the Sac1 domain, respectively. 5-phosphatase domain’s preferential hydrolysis of PI(4,5)P2 at 5-position [44, 46] lowers the affinity of clathrin adaptors from the nascent synaptic vesicular membrane. Thus, the detachment of clathrin adaptors from the synaptic vesicle promotes the downstream proteins, including auxilin, recruitment to disassemble the clathrin lattice. Once the clathrin disperses in the cytosol, the Sac1 domain’s 4-phosphatase activity helps to release the auxilin for the next cycle of uncoating [43].
In addition to its activity as a PI4P phosphatase, the Sac1 domain can dephosphorylate PI3P and PI(3,5)P2, both of which are non-synaptic membrane-enriched phosphoinositide phosphates [44]. The differently phosphorylated phosphoinositide lipids act as molecular tags for translocating specific proteins to the membrane to confer the membrane’s identity [47,48,49,50] For example, PI(4,5)P2 is abundantly found on the synaptic membrane and functions as tags to bind clathrin adaptors and other endocytic proteins. Similarly, PI3P and PI(3,5)P2 are critical phosphoinositide phosphates on the phagophore that recognize ATG18 to promote autophagosome biogenesis [51]. The ATG18 on the phagophore membrane encourages the recruitment of ATG16, a binding partner of the ATG12-ATG5 conjugate [52]. The ATG12-ATG5 conjugate converts LC3 I (un-lipidated form) to LC3 II (lipidated form). At the same time, ATG16 of the ATG12-ATG5-ATG16 complex determines the position of LC3 lipidation [52,53,54]—the conversion of LC3 I to LC3 II results in membrane elongation and formation of autophagosomes.
Sac1 domain–mediated hydrolysis of PI3P and PI(3,5)P2 from the membrane removes early-autophagosome markers, including ATG18, thereby promoting the elongation protein, ATG8/LC3, recruitment to the autophagosome [44, 55]. The PD-associated R258Q mutation in the Sac1 domain of SJ1 aggregates PI3P/PI(3,5)P2-binding protein ATG18 on premature autophagosomes that fail to mature at Drosophila synapses and in human neurites [44]. Pan et al. also reported that Parkinsonian mutation (R258Q and R839C) of SJ1 augments autophagosome formation but decreases autophagy [56].
While SJ1 regulates presynaptic autophagy through ATG18, research showed that the active zone constituting cytoskeleton matrix protein Bassoon regulates the process through ATG5. Bassoon consists of an ATG5-binding peptide motif (CC2v1) in its structure to bind and inactivate ATG5’s role as an E3-like ligase. The binding of ATG5 with Bassoon disrupts ATG5/12 interaction with ATG16. Thus, it inhibits the catabolic events in the presynaptic compartments [57].
The Adaptor Protein (AP)2 Complex Mediates Retrograde Trafficking of Autophagosomes
The adaptor protein (AP)2 complex is the membrane-derived protein complex that participates in the clathrin-mediated coating. In AD-linked hippocampal neurons, the AP2 complex prevents amyloidogenesis by regulating amyloid precursor protein (APP) processing and amyloid-β formation [58, 59]. The endocytic adaptor interacts with the two crucial proteins of HD, i.e., Huntingtin and Huntingtin-interacting protein 1 (HIP1); however, the physiological role of AP2 binding with these proteins is elusive [60]. The AP2 complex primarily anchors the clathrin to the membrane’s PI(4,5)P2, at 5-phosphosite present on synaptic vesicles [61]. The clathrin-AP2 complex is a crucial center for synaptic proteins’ interactions, which regulate clathrin uncoating of clathrin-coated pits [61]. Once the AP2 complex is recruited to the completely formed CCV, the EndoA translocates dynamin-2 to the collar of the CCV to pinch off the CCV from the sites into presynaptic axoplasm [62, 63]. Moreover, the AP2 complex is also responsible for the arborization of dendrites via its upstream kinase. The AP2-associated kinase1 is an upstream kinase that activates the AP2 complex protein to promote dendritic branching [64].
The AP2 is a heterotetramer protein comprising four subunits: α, β, µ, and σ subunits (Fig. 1). The first two subunits participate in the retrograde trafficking of the autophagosomes: the α-subunit binds to LC3 protein on the late-stage autophagosome (marked by LC3 and Rab7). At the same time, the β-subunit anchors the p150Glued subunit of dynactin [65, 66]. The dynactin is a cofactor of a retrograde motor protein dynein [66]. Thus, the LC3-AP2-p150Glued complex is especially crucial in neurons because the spatially segregated autophagosomes in distal terminals must travel a considerable distance retrograde (Fig. 3) with high precision and rate to accurately reach the soma for ultimate clearance [65]. The AP2 complex also participates in ATG9A (mammalian homolog of ATG9) trafficking; it translocates the protein from the presynaptic membrane to the endosome [40, 67].
The AP3/AP4 Complex Anterograde Transports ATG9 into Presynaptic Sites for Exocytosis
Reports show many other AP complexes that have their role in presynaptic autophagy [11, 68, 69]. In C. elegans, one such protein is the AP3 complex that buds off the vesicles from the trans-Golgi network (TGN) and delivers them to the presynaptic compartments. Interestingly, these TGN-derived vesicles released from the juxtanuclear regions are ATG9-positive [11]. In general, ATG proteins are cytosolic proteins, but ATG-9 is the sole evolutionary-conserved transmembrane protein in the family of autophagy proteins and is localized to various subcellular loci. In neurons, the protein is localized to multiple compartments, including TGN-derived vesicles, plasma membranes, early endosomes, recycling endosomes, and phagophores [70]. ATG9 traffics between these sites to create its pool ready to contribute to the autophagosome biogenesis upon autophagy induction [71,72,73]. The protein works with ATG2 to promote nucleation of the nascent phagophore [74, 75]. The ATG2 plays a vital role in lipid transportation to support membrane elongation [76]. While membrane-bound ATG9 randomly traffics in axoplasm, the AP3 complex binds the cytosolic tail of ATG9 and mediates its anterograde transportation to the plasma membrane of the presynaptic site [11]. The mammalian homolog of AP3 complex is a AP4 complex that drives the ATG9A to the presynaptic membrane [11, 68, 69]. It also interacts with the cytosolic tail of AD-linked amyloid precursor protein (APP) [77]. The AP4 complex is a heterotetrameric adaptor protein that consists of ε, β4, µ4, and σ4 subunits; the ε-subunit binding to ATG9A translocates it to the terminal compartments (Figs. 1 and 2) [68, 69]. In mammalian neurons, AP4-ε KO leads to ATG9A aggregation in TGN and depletion in the peripheral cytoplasm [69]. Intriguingly, the AP2 complex also participates in ATG9 trafficking; it translocates the protein from presynaptic membrane to the endosome (Fig. 2). Both AP2 and AP4 complex bind tyrosine- and di-leucine-enriched sorting motif in the cytosolic tail of ATG9—to traffic the autophagic protein [68].

Anterograde trafficking and delivery of ATG9 to the autophagosome in the presynaptic compartment: The AP4 complex binds the ATG9 protein inserted in the TGN-derived vesicle and anterograde transports it to the presynaptic compartment for exocytosis. The ATG9 protein translocated to the presynaptic plasma membrane is endocytosed into the synaptic vesicle, recycled into the endosome, and then to the phagophore. The phagophore matures into the autophagosome
TBC1D5 and AP2 Complex Together Create an ATG9 Pool for Autophagosome Biogenesis
Popovic and Dikic reported that the deletion of AP2 blocks the presynaptic (intra-compartmental) trafficking of ATG9 to the late endosome under the basal or upon autophagy induction [46, 62]. Under the basal conditions, the deletion enhanced ATG9 localization on the plasma membrane of presynaptic compartments, which infers failed endocytosis of ATG9-positive membrane in the absence of AP2. Upon autophagy induction, the ATG9-positive TGN-derived vesicles accumulate in the juxtanuclear region. Also, the deletion hinders the binding of ATG9 with its intra-compartmental trafficking-carrier TBC1D5 [62]. The TBC1D5 is a crucial Rab-GTPase (RabGAP) protein that interacts with the retromer protein complex to regulate axonal retrograde trafficking [78, 79]. Research showed that TBC1D5 regulates the growth and function of synapses, and their loss leads to aberration in the presynaptic terminals, including abnormal development, change in morphology, synaptic vesicle density, and excessive satellite buttons and branch formation [80].
In a functional neuron, upon autophagy induction, the ATG9-positive TGN-vesicle pool is driven to the presynaptic compartment for exocytosis. At the same time, the plasma membrane–associated ATG9 is endocytosed to the clathrin-AP2-complex positive CCVs [62, 72]. The shedding off clathrin from the vesicle membrane by SJ1 and auxilin provides an opportunity for TBC1D5 to interact with the AP2 bound to the uncoated early endosome [62]. Instantly, the TBC1D5 recruits to the endosomal membrane containing AP2 complex and ATG9; the membrane, along with the proteins, is transferred to the recycling endosome and then to the forming pre-autophagosomal structure to support the supply of membrane, ATG9, and AP2. The ATG9 is an autophagy initiation protein, while the other two proteins, AP2 and TBC1D5, bind to the autophagosome elongation protein LC3. Here, the TBC1D5 interacts with LC3, with its LC3 interacting region 1 (LIR1), and promotes protein internalization and sequesters cytoplasmic materials [62, 81, 82], while the AP2 complex directs retrograde trafficking of the autophagosome (Fig. 3) [59, 62]. Interestingly, TBC1D5 depletion leads to the hang-up of clathrin-mediated endocytosis and missorting of ATG9, suggesting a dual role of TBC1D5 in endocytosis and autophagy [62]. However, it is yet to be determined whether the retrograde trafficking facilitator AP2 complex directly binds to LC3 on the mature autophagosome or indirectly through interacting with the LC3-bound TBC1D5. Also, how the AP2 switches its function—from endocytosis to retrograde trafficking, and vice-versa—in the neuronal terminal is still obscure.

Formation of the precursor-autophagosome structure and retrograde transport of autophagosome: The plasma membrane–localized ATG9 and ATG16 are endocytosed differently to form a separate pool of vesicles. ATG9 recycles through the conventional route (via early endosome to the recycling endosome), while the ATG16 is directly delivered to the recycling endosome. SNX18 and the dynamin-2 buds off the precursor-autophagosome structure containing ATG9, ATG16, AP2 complex, and TBC1D5. The precursor structure is supplied to the phagophore to form an autophagosome. The LC3-bound AP2 complex protein interacts with the dynactin cofactor of dynein, a retrograde motor protein, to promote retrograde trafficking of the autophagosome
Role of Retromer Complex and Sorting Nexin (SNX) Proteins
The retromer complex and membrane-associated sorting nexin (SNX) proteins are vital in regulating the sorting of endosomal proteins [83, 84]. Notably, numerous neurodegenerative disorders, including PD and ALS, are related to retromer and SNX protein dysfunction [26, 83, 85,86,87,88,89]. In mammals, two major subcomplexes of the retromer complex are a vacuolar protein sorting–associated protein 26 (VPS26)-VPS29-VPS35 trimeric subcomplex and membrane-associated SNX dimers. VPS26 and VPS29 subunits of the VPS26-VPS29-VPS35 complex bind to a scaffold of the core protein VPS35 at its C- and N-terminal, respectively [90, 91]. The SNX dimers are commonly a collective composition of SNX1, SNX2, SNX5, and SNX6 [92]. While the trimeric assembly of VPS26, VPS29, and VPS35 directly recognizes the cargo and binds to it, the proteins of the SNX family consist of a phox-homology (PX) domain and a C-terminal BAR domain to induce membrane remodeling [93,94,95]. Members of SNX are reportedly involved in clathrin-dependent endocytosis and in preserving endosome-emanating tubules. SNX proteins are essential for mental health, and their reduced expression is implicated in several neurological disorders, including AD and PD [96]. Most SNX proteins are associated with the membrane’s PI3P; therefore, PI3P-enriched early endosomes are a great reservoir of SNXs [97].
A. The retromer complex regulates autophagosome processivity
In neurons, the VPS26-VPS29-VPS35 retromer complex recycles proteins linked to the endolysosomal pathway; it is widely involved in aging, PD, and AD [98]. The VPS29 subunit of the VPS26-VPS29-VPS35 retromer complex interacts with the TBC1D5, a trafficking-carrier protein for ATG9 [62, 81]. As clathrin proteins disperse from the endocytic vesicles, the TBC1D5 leaves the interacting partner VPS29 and translocates to the early endosome to bind the AP2 complex. Further, the two proteins (AP2 complex and TBC1D5) work collectively to facilitate autophagosome formation, maturation, and retrograde transportation. Thus, the retromer subunit VPS29 regulates the switching of the presynaptic autophagy [62]. Moreover, VPS29 plays a pivotal role in brain health in aging [98]. Thereunto VPS29, VPS35, the cargo-binding biggest co-protein of the retromer complex, also participates in presynaptic autophagy by regulating LRRK2 kinase activity, and the PD-linked mutation (D620N) dramatically increases LRRK2-mediated phosphorylation of Rab8, Rab10, and Rab12 [99]. The elevated phosphorylation of Rab proteins by LRRK2 leads to decreased processivity of trafficking autophagosomes [100] and impaired mitophagy in neurons [101]. A study revealed the reduced expression of VPS35 and VPS26 proteins in the entorhinal cortex of AD patients [102].
B. SNX18 and dynamin-2 together generate phagophore’s precursor structures
Research on SNX18 showed that the protein generates tubular buds containing ATG9 and ATG16 from the recycling endosome [71]. ATG9 and ATG16 are trafficked from the presynaptic plasma membrane to the phagophore’s precursor structures in endosomes via different routes to form two pools of vesicles (ATG9- or ATG16-positive) that coalescence at recycling endosome [103]. While the delivery of ATG9 to the recycling endosome is via a conventional path through early endosomes [103], the ATG16 traffics through a distinct undeciphered way (Fig. 3). Interestingly, the PX domain of SNX18 binds to the PI3P of the recycling endosome membrane, while BAR domain scaffolds the membrane to induce internal curvature and tubule formation [40, 71]. These ATG9- and ATG16-positive tubules originating from the recycling endosomes are precursor of phagophores/autophagosomes [40, 103]. These precursor autophagosomal tubular structures bud off from the endosome following the interaction of dynamin-2 with the SNX18. The ligation of PRD of dynamin-2 to the SH3 domain of SNX18 mediates their interaction [71]. The ATG9-positive phagophore precursor vesicles, which are budded off from recycling endosomes, are phosphorylated (by ULK1) and redistributed to pre-autophagosomal structure upon autophagy induction [104,105,106].
Future investigation might show if the SNX18-dynamin-2 protein complex works alone (or the only complex) to bud off the precursor tubule for autophagosome or if some other SH3 domain–containing endocytic proteins are also involved or could compensate for the SNX18 function. This investigation must clarify the story because several SH3 domain–containing membrane remodeling proteins interacting with PRD of dynamin-2 reside in the presynaptic compartments, including EndoA.
LRRK2: A Kinase Switch to Toggle Presynaptic Autophagy
The LRRK2 is a PD-linked multidomain protein that accounts for ~ 1% of sporadic and ~ 5% of genetic cases [107,108,109,110]. Piccoli et al. investigated LRRK2’s localization in the synaptosomal compartment; it interacts with several synaptic proteins, modulates their trafficking and distribution, and regulates their dynamics [111]. The protein regulates the synaptic vesicle endocytosis and membrane remodeling by phosphorylating the BAR domain, specifically to S75 of the H1 helix in the BAR domain of EndoA protein [29, 36]. The LRRK2-mediated phosphorylation is implicated in switching the EndoA function; the non-phosphorylated EndoA deforms the membrane to tubules, while the phosphorylation induces the protein to create a highly curved membrane. The S75-phosphorylation in the H1 helix prevents the EndoA from penetrating deep into the membrane and associating tightly with it, thus inducing a highly curved membrane, as those found at the growing phagophore’s edge [36]. Interestingly, the phosphorylated EndoA-created curved membrane serves as the docking site for an autophagy protein, ATG3 [36], as does the curvature of growing phagophore for the recruitment of autophagic factors, including ATG3, ATG14, and ATG1 [112,113,114,115,116].
The LRRK2 also modulates vesicular trafficking by phosphorylating the Rab proteins. The report shows that increased kinase activity of LRRK2 due to PD-causing G2019S mutation decreases the processivity of trafficking autophagosome, leading to defective axonal transport, acidification, and maturation of autophagosome [100, 109]. Moreover, the mutant LRRK2 protein disrupts the synaptic vesicle endocytosis in PD-linked dopaminergic neurons by abruptly phosphorylating the auxilin in its clathrin-binding domain at S627 [117]. The hyperactivated mutant LRRK2 abnormally recruits the motor adaptor JNK-interacting protein 4 (JIP4) to the autophagosome, promotes binding of JIP4 to LRRK2-phosphorylated Rab proteins, and induces the anterograde protein kinesin to bind with the activated JIP4 on the autophagosome, thereby promoting the anterograde trafficking and reversing the migrating autophagosomes back into the presynaptic compartments [100, 109]. Thus, the G2019S mutation stuck autophagosomes to an unproductive decision between the anterograde and retrograde motors [100]. Moreover, the overexpression of Rab29—one of the several substrates of LRRK2—also induces the hyperactivation of LRRK2 kinase activity and decreases the processivity of autophagosome trafficking [100].
Wauters et al. [101] showed that the LRRK2 kinase activity is essential for the PTEN-induced kinase 1 (PINK1)– and parkin RBR E3 ubiquitin–protein ligase (PRKN)–dependent elimination of depolarized mitochondria through mitophagy. The two usual mutations in LRRK2—G2019S and R1441C—that cause PD led to the aggregation of depolarized mitochondria due to mitophagy failure [101, 118]. PINK-PRKN-mediated clearance of dysfunctional mitochondria requires Rab10 clustering on depolarized mitochondria to recruit and bind the autophagy receptor optineurin (OPTN) on dysfunctional mitochondria to facilitate mitophagy [118]. The mutation-induced hyperactivation of LRRK2 phosphorylation activity enhanced the Rab10 phosphorylation at T73, resulting in decreased interaction of Rab10 and OPTN on dysfunctional mitochondria and impaired mitochondrial clearance [101].
Discussion
The intriguing question is how the presynaptic autophagy is executed in the axon terminals to eliminate aberrant proteins and organelles redundancy. Studies showed that autophagosomes are primarily formed in the presynaptic compartments, where the protein redundancy is relatively higher due to a high metabolic rate [12, 20, 51, 119]. Autophagosome formation in the distantly located presynaptic compartments is a conserved mechanism that rejuvenates axon terminals, enhances their neuroplasticity, and maintains synaptic homeostasis [20, 44, 119,120,121,122]. In a highly selective manner, newly developing autophagosomes engulf the ubiquitinated presynaptic junk and travel to the somatodendritic compartment to digest the molecular garbage through lysosomal fusion [123, 124]. During their journey, autophagosomes mature into autolysosomes and digest the redundant synaptic proteins and dysfunctional organelles while reaching the soma [10, 12, 125, 126]. Defective axonal transport significantly induces neurodegeneration in AD, PD, ALS, and HD [127, 128]. The unsolved question is, “Are presynaptic autophagosomes distinct from somal autophagosomes?” Maday and coworkers demonstrated that autophagosomes arising in the axon fiber of hippocampal neurons are morphologically different from those derived in the somatodendritic zone [123, 124]. While autophagosomes formed in the soma and dendritic regions are less developed and mobile, the axonal autophagosomes arriving in the somatodendritic compartment are LAMP1-positive (late endosome/lysosome marker), mature enough, and dynamic in nature [124, 129]. Another research by Hoffmann et al. demonstrated that the terminal compartments possess separate autophagy machinery that gets engaged within minutes following a local insult. They developed a vector system that generated a spatiotemporally controlled ROS-induced damage of local proteins within presynaptic compartments. To their surprise, the insult induces rapid accumulation of LC3-positive vesicles in presynaptic compartments within 5 min, which spread into axons over time (1–2 h) [28]. Thus, autophagosomes formed in presynaptic compartments differ drastically from their counterparts originating in the neuronal cell body or non-polar cells [10, 28, 124, 129, 130]. In the terminal compartments, a few synaptic proteins are parallelly involved in ATG assembly, substrate internalization, membrane distortion/remodeling, and lipid metabolism to support autophagosome formation [29, 38, 44, 131]. At the same time, few proteins escort the autophagosomes retrograde to the cell body [62, 71, 101]. Failure of presynaptic autophagy increases nonfunctional protein accumulation in presynaptic compartments, leading to compromised synaptic transmission. Thus, we reviewed the critical studies and recent findings highlighting the involvement of endocytic/synaptic proteins in the formation and retrograde trafficking of presynaptic autophagosome and examined the altered expression of synaptic proteins in disease onset/progression Table 1.
The present review appeals to study neuronal autophagy differently from that in non-polar cells. Autophagy in neurons might seem the same as in other cells because of the communal site of lysosomal degradation of autophagosomes in the soma. However, the autophagosome formation in neurons is compartmentalized, which makes neuronal autophagy peerless. In axonal/presynaptic autophagosomes, several synaptic proteins support and regulate their formation. However, it will be necessary for researchers to determine in the future whether axonal autophagosomes are molecularly extraordinary and whether they respond to synaptic regulatory signals. Synaptic loss is the earliest hallmark of many neurodegenerative diseases. Therefore, identifying biomarkers of presynaptic autophagy dysfunctions could facilitate early diagnosis that could be extrapolated to monitor disease. Secondly, the involvement of synaptic proteins in autophagosome formation, distribution, and trafficking needs further validation. Future research will validate whether their role in presynaptic autophagy is uniquely obligatory or facultative addition in the classical autophagic process. It will also be compulsory to research the autophagic function of other endocytic and non-endocytic proteins residing in the presynaptic compartments and how autophagy collaborates with other degradation mechanisms to impact neurotransmission. In brief, future investigations on presynaptic autophagy will present significant potential in elucidating the etiology of neurodegenerative disorders and formulating successful treatments to mitigate neurodegeneration. Thus, it is crucial and timely to investigate synaptic autophagy as a field.
Data Availability
No datasets were generated or analysed during the current study.
References
Cheng X-T, Huang N, Sheng Z-H (2022) Programming axonal mitochondrial maintenance and bioenergetics in neurodegeneration and regeneration. Neuron 110:1899–1923. https://doi.org/10.1016/j.neuron.2022.03.015
DiRocco RJ, Hall WG (1981) Metabolic neural mapping in neonatal rats. J Neurosci Res 6:13–19. https://doi.org/10.1002/JNR.490060103
Camandola S, Mattson MP (2017) Brain metabolism in health, aging, and neurodegeneration. EMBO J 36:1474–1492. https://doi.org/10.15252/embj.201695810
Birdsall V, Waites CL (2019) Autophagy at the synapse. Neurosci Lett 697:24–28. https://doi.org/10.1016/J.NEULET.2018.05.033
Guedes-Dias P, Holzbaur ELF (2019) Axonal transport: driving synaptic function. Science (1979) 366:eaaw9997. https://doi.org/10.1126/science.aaw9997
Sun C, Nold A, Fusco CM et al (2021) The prevalence and specificity of local protein synthesis during neuronal synaptic plasticity. Sci Adv 7:eabj0790. https://doi.org/10.1126/SCIADV.ABJ0790/SUPPL_FILE/SCIADV.ABJ0790_SM.PDF
Burke WJ, Chung HD, Huang JS et al (1988) Evidence for retrograde degeneration of epinephrine neurons in Alzheimer’s disease. Ann Neurol 24:532–536. https://doi.org/10.1002/ANA.410240409
Burke RE, O’Malley K (2013) Axon degeneration in Parkinson’s disease. Exp Neurol 246:72–83
Li H, Li SH, Yu ZX et al (2001) Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J Neurosci 21:8473–8481. https://doi.org/10.1523/JNEUROSCI.21-21-08473.2001
Mishra AK, Dixit A (2022) Dopaminergic axons: key recitalists in Parkinson’s disease. Neurochem Res 47:234–248. https://doi.org/10.1007/s11064-021-03464-1
Yang S, Park D, Manning L et al (2022) Presynaptic autophagy is coupled to the synaptic vesicle cycle via ATG-9. Neuron 110:824-840.e10. https://doi.org/10.1016/j.neuron.2021.12.031
Lüningschrör P, Sendtner M (2018) Autophagy in the presynaptic compartment. Curr Opin Neurobiol 51:80–85. https://doi.org/10.1016/J.CONB.2018.02.023
Shen W, Ganetzky B (2009) Autophagy promotes synapse development in Drosophila. J Cell Biol 187:71–79. https://doi.org/10.1083/jcb.200907109
Bowling H, Klann E (2014) Shaping dendritic spines in autism spectrum disorder: mTORC1-dependent macroautophagy. Neuron 83:994–996. https://doi.org/10.1016/j.neuron.2014.08.021
Tang G, Gudsnuk K, Kuo SH et al (2014) Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83:1131–1143. https://doi.org/10.1016/J.NEURON.2014.07.040
Hernandez-Diaz S, Ghimire S, Sanchez-Mirasierra I et al (2022) Endophilin-B regulates autophagy during synapse development and neurodegeneration. Neurobiol Dis 163:105595. https://doi.org/10.1016/J.NBD.2021.105595
Negrete-Hurtado A, Overhoff M, Bera S et al (2020) Autophagy lipidation machinery regulates axonal microtubule dynamics but is dispensable for survival of mammalian neurons. Nat Commun 11:1535. https://doi.org/10.1038/S41467-020-15287-9
Mishra AK, Mishra S, Rajput C et al (2018) Cypermethrin activates autophagosome formation albeit inhibits autophagy owing to poor lysosome quality: relevance to Parkinson’s disease. Neurotox Res 33:377–387. https://doi.org/10.1007/S12640-017-9800-3
Maday S (2016) Mechanisms of neuronal homeostasis: autophagy in the axon. Brain Res 1649:143–150. https://doi.org/10.1016/J.BRAINRES.2016.03.047
Stavoe AKH, Holzbaur ELF (2019) Autophagy in neurons. Annu Rev Cell Dev Biol 35:477–500. https://doi.org/10.1146/ANNUREV-CELLBIO-100818-125242
Xie W, Wan OW, Chung KKK (2010) New insights into the role of mitochondrial dysfunction and protein aggregation in Parkinson’s disease. Biochim Biophys Acta (BBA) - Mol Basis Dis 1802:935–941. https://doi.org/10.1016/j.bbadis.2010.07.014
Wang X, Wang W, Li L et al (2014) Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta (BBA) - Mol Basis Dis 1842:1240–1247. https://doi.org/10.1016/j.bbadis.2013.10.015
Hollis F, Kanellopoulos AK, Bagni C (2017) Mitochondrial dysfunction in autism spectrum disorder: clinical features and perspectives. Curr Opin Neurobiol 45:178–187. https://doi.org/10.1016/j.conb.2017.05.018
Shacham T, Sharma N, Lederkremer GZ (2019) Protein misfolding and ER stress in Huntington’s disease. Front Mol Biosci 6:20. https://doi.org/10.3389/fmolb.2019.00020
Nakamura T, Lipton SA (2010) Redox regulation of mitochondrial fission, protein misfolding, synaptic damage, and neuronal cell death: potential implications for Alzheimer’s and Parkinson’s diseases. Apoptosis 15:1354–1363. https://doi.org/10.1007/s10495-010-0476-x
Pérez-Torres EJ, Utkina-Sosunova I, Mishra V et al (2022) Retromer dysfunction in amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 119:e2118755119. https://doi.org/10.1073/pnas.2118755119
Wong YC, Holzbaur ELF (2014) The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci 34:1293–1305. https://doi.org/10.1523/JNEUROSCI.1870-13.2014
Hoffmann S, Orlando M, Andrzejak E et al (2019) Light-activated ROS production induces synaptic autophagy. J Neurosci 39:2163–2183. https://doi.org/10.1523/JNEUROSCI.1317-18.2019
Ambroso MR, Hegde BG, Langen R (2014) Endophilin A1 induces different membrane shapes using a conformational switch that is regulated by phosphorylation. Proc Natl Acad Sci USA 111:6982–6987. https://doi.org/10.1073/pnas.1402233111
Watanabe S, Mamer LE, Raychaudhuri S et al (2018) Synaptojanin and endophilin mediate neck formation during ultrafast endocytosis. Neuron 98:1184-1197.e6. https://doi.org/10.1016/j.neuron.2018.06.005
Wu T, Baumgart T (2014) BIN1 membrane curvature sensing and generation show autoinhibition regulated by downstream ligands and PI(4,5)P2. Biochemistry 53:7297–7309. https://doi.org/10.1021/BI501082R
Isas JM, Ambroso MR, Hegde PB et al (2015) Tubulation by amphiphysin requires concentration-dependent switching from wedging to scaffolding. Structure 23:873–881. https://doi.org/10.1016/J.STR.2015.02.014
Poudel KR, Dong Y, Yu H et al (2016) A time course of orchestrated endophilin action in sensing, bending, and stabilizing curved membranes. Mol Biol Cell 27:2119–2132. https://doi.org/10.1091/mbc.E16-04-0264
Mizuno N, Jao CC, Langen R, Steven AC (2010) Multiple modes of endophilin-mediated conversion of lipid vesicles into coated tubes: implications for synaptic endocytosis. J Biol Chem 285:23351–23358. https://doi.org/10.1074/jbc.M110.143776
Matta S, van Kolen K, da Cunha R et al (2012) LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 75:1008–1021. https://doi.org/10.1016/j.neuron.2012.08.022
Soukup SF, Kuenen S, Vanhauwaert R et al (2016) A LRRK2-dependent EndophilinA phosphoswitch is critical for macroautophagy at presynaptic terminals. Neuron 92:829–844. https://doi.org/10.1016/j.neuron.2016.09.037
Soukup SF, Verstreken P (2017) EndoA/Endophilin-A creates docking stations for autophagic proteins at synapses. Autophagy 13:971–972. https://doi.org/10.1080/15548627.2017.1286440
Murdoch JD, Rostosky CM, Gowrisankaran S et al (2016) Endophilin-A deficiency induces the Foxo3a-Fbxo32 Network in the brain and causes dysregulation of autophagy and the ubiquitin-proteasome system. Cell Rep 17:1071–1086. https://doi.org/10.1016/j.celrep.2016.09.058
Takahashi Y, Meyerkord CL, Wang HG (2009) Bif-1/Endophilin B1: a candidate for crescent driving force in autophagy. Cell Death Differ 16:947–955
De Tito S, Hervás JH, van Vliet AR, Tooze SA (2020) The Golgi as an assembly line to the autophagosome. Trends Biochem Sci 45:484–496. https://doi.org/10.1016/J.TIBS.2020.03.010
Wang DB, Kinoshita Y, Kinoshita C et al (2015) Loss of endophilin-B1 exacerbates Alzheimer’s disease pathology. Brain 138:2005–2019. https://doi.org/10.1093/brain/awv128
Wong ASL, Lee RHK, Cheung AY et al (2011) Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson’s disease. Nat Cell Biol 13:568–579. https://doi.org/10.1038/ncb2217
Cao M, Wu Y, Ashrafi G et al (2017) Parkinson sac domain mutation in synaptojanin 1 impairs clathrin uncoating at synapses and triggers dystrophic changes in dopaminergic axons. Neuron 93:882-896.e5. https://doi.org/10.1016/j.neuron.2017.01.019
Vanhauwaert R, Kuenen S, Masius R et al (2017) The SAC 1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J 36:1392–1411. https://doi.org/10.15252/embj.201695773
Inoshita T, Cui C, Hattori N, Imai Y (2018) Regulation of membrane dynamics by Parkinson’s disease-associated genes. J Genet 97:715–725
Backues SK, Orban DP, Bernard A et al (2015) Atg23 and Atg27 act at the early stages of Atg9 trafficking in S. cerevisiae. Traffic 16:172–190. https://doi.org/10.1111/TRA.12240
Schink KO, Raiborg C, Stenmark H (2013) Phosphatidylinositol 3-phosphate, a lipid that regulates membrane dynamics, protein sorting and cell signalling. BioEssays 35:900–912. https://doi.org/10.1002/bies.201300064
Wallroth A, Haucke V (2018) Phosphoinositide conversion in endocytosis and the endolysosomal system. J Biol Chem 293:1526–1535. https://doi.org/10.1074/jbc.R117.000629
Ketel K, Krauss M, Nicot AS et al (2016) A phosphoinositide conversion mechanism for exit from endosomes. Nature 529:408–412. https://doi.org/10.1038/nature16516
Marat AL, Haucke V (2016) Phosphatidylinositol 3-phosphates—at the interface between cell signalling and membrane traffic. EMBO J 35:561–579. https://doi.org/10.15252/embj.201593564
Hill SE, Colón-Ramos DA (2020) The journey of the synaptic autophagosome: a cell biological perspective. Neuron 105:961–973. https://doi.org/10.1016/j.neuron.2020.01.018
Sawa-Makarska J, Baumann V, Coudevylle N et al (1979) (2020) Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science 369:eaaz7714. https://doi.org/10.1126/SCIENCE.AAZ7714
Martens S, Fracchiolla D (2020) Activation and targeting of ATG8 protein lipidation. Cell Discov 6:23. https://doi.org/10.1038/S41421-020-0155-1
Nishimura T, Tooze SA (2020) Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov 6:32
Søreng K, Neufeld TP, Simonsen A (2018) Membrane trafficking in autophagy. Int Rev Cell Mol Biol 336:1–92. https://doi.org/10.1016/BS.IRCMB.2017.07.001
Pan P-Y, Zhu J, Rizvi A et al (2021) Synaptojanin1 deficiency upregulates basal autophagosome formation in astrocytes. J Biol Chem 297:100873. https://doi.org/10.1016/j.jbc.2021.100873
Reimer RJ, Schneider K, Leal-Ortiz S et al (2017) Bassoon controls presynaptic autophagy through Atg5. Neuron 93:897-913.e7. https://doi.org/10.1016/J.NEURON.2017.01.026
Bera S, Camblor-Perujo S, Calleja Barca E et al (2020) AP2 reduces amyloidogenesis by promoting BACE1 trafficking and degradation in neurons. EMBO Rep 21:e47954. https://doi.org/10.15252/embr.201947954
Tian Y, Chang JC, Fan EY et al (2013) Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci USA 110:17071–17076. https://doi.org/10.1073/pnas.1315110110
Legendre-Guillemin V, Metzler M, Charbonneau M et al (2002) HIP1 and HIP12 display differential binding to F-actin, AP2, and clathrin. Identification of a novel interaction with clathrin light chain. J Biol Chem 277:19897–19904. https://doi.org/10.1074/jbc.M112310200
Höning S, Ricotta D, Krauss M et al (2005) Phosphatidylinositol-(4,5)-bisphosphate regulates sorting signal recognition by the clathrin-associated adaptor complex AP2. Mol Cell 18:519–531. https://doi.org/10.1016/j.molcel.2005.04.019
Popovic D, Dikic I (2014) TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep 15:392–401. https://doi.org/10.1002/embr.201337995
Redpath GMI, Betzler VM, Rossatti P, Rossy J (2020) Membrane heterogeneity controls cellular endocytic trafficking. Front Cell Dev Biol 8:757. https://doi.org/10.3389/FCELL.2020.00757
Ultanir SK, Hertz NT, Li G et al (2012) Chemical genetic identification of NDR1/2 kinase substrates AAK1 and Rabin8 uncovers their roles in dendrite arborization and spine development. Neuron 73:1127–1142. https://doi.org/10.1016/j.neuron.2012.01.019
Kononenko NL, Claßen GA, Kuijpers M et al (2017) Retrograde transport of TrkB-containing autophagosomes via the adaptor AP-2 mediates neuronal complexity and prevents neurodegeneration. Nat Commun 8:14819. https://doi.org/10.1038/ncomms14819
Fu MM, Nirschl JJ, Holzbaur EL (2014) LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev Cell 29:577–590. https://doi.org/10.1016/J.DEVCEL.2014.04.015
Zhou C, Ma K, Gao R et al (2017) Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res 27:184–201. https://doi.org/10.1038/cr.2016.146
Mattera R, Park SY, De Pace R et al (2017) AP-4 mediates export of ATG9A from the trans-Golgi network to promote autophagosome formation. Proc Natl Acad Sci USA 114:E10697–E10706. https://doi.org/10.1073/pnas.1717327114
de Pace R, Skirzewski M, Damme M et al (2018) Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genet 14:e1007363. https://doi.org/10.1371/journal.pgen.1007363
Yang S, Colón-Ramos DA (2022) Transmembrane protein ATG-9 links presynaptic autophagy with the synaptic vesicle cycle. Autophagy 18:1746–1747. https://doi.org/10.1080/15548627.2022.2049151
Søreng K, Munson MJ, Lamb CA et al (2018) SNX 18 regulates ATG 9A trafficking from recycling endosomes by recruiting Dynamin-2. EMBO Rep 19:e44837. https://doi.org/10.15252/embr.201744837
Imai K, Hao F, Fujita N et al (2016) Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J Cell Sci 129:3781–3791. https://doi.org/10.1242/jcs.196196
Stavoe AKH, Hill SE, Hall DH, Colón-Ramos DA (2016) KIF1A/UNC-104 transports ATG-9 to regulate neurodevelopment and autophagy at synapses. Dev Cell 38:171–185. https://doi.org/10.1016/J.DEVCEL.2016.06.012
Chowdhury S, Otomo C, Leitner A et al (2018) Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex. Proc Natl Acad Sci USA 115:E9792–E9801. https://doi.org/10.1073/pnas.1811874115
Maeda S, Otomo C, Otomo T (2019) The autophagic membrane tether ATG2A transfers lipids between membranes. Elife 8:e45777. https://doi.org/10.7554/eLife.45777
Hernandez-Diaz S, Soukup S-F (2020) The role of lipids in autophagy and its implication in neurodegeneration. Cell Stress 4:167–186. https://doi.org/10.15698/cst2020.07.225
Burgos PV, Mardones GA, Rojas AL et al (2010) Sorting of the Alzheimer’s disease amyloid precursor protein mediated by the AP-4 complex. Dev Cell 18:425–436. https://doi.org/10.1016/J.DEVCEL.2010.01.015
Deinhardt K, Salinas S, Verastegui C et al (2006) Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 52:293–305. https://doi.org/10.1016/j.neuron.2006.08.018
Seaman MNJ, Mukadam AS, Breusegem SY (2018) Inhibition of TBC1D5 activates Rab7a and can enhance the function of the retromer cargo-selective complex. J Cell Sci 131:jcs217398. https://doi.org/10.1242/JCS.217398
Zhou X, Gan G, Sun Y et al (2023) GTPase-activating protein TBC1D5 coordinates with retromer to constrain synaptic growth by inhibiting BMP signaling. J Genet Genom 50:163–177. https://doi.org/10.1016/j.jgg.2022.11.009
Popovic D, Akutsu M, Novak I et al (2012) Rab GTPase-activating proteins in autophagy: regulation of endocytic and autophagy pathways by direct binding to human ATG8 modifiers. Mol Cell Biol 32:1733–1744. https://doi.org/10.1128/MCB.06717-11
Roy S, Leidal AM, Ye J et al (2017) Autophagy-dependent shuttling of TBC1D5 controls plasma membrane translocation of GLUT1 and glucose uptake. Mol Cell 67:84-95.e5. https://doi.org/10.1016/J.MOLCEL.2017.05.020
Zhang H, Huang T, Hong Y et al (2018) The retromer complex and sorting nexins in neurodegenerative diseases. Front Aging Neurosci 10:79. https://doi.org/10.3389/fnagi.2018.00079
McNally KE, Cullen PJ (2018) Endosomal retrieval of cargo: retromer is not alone. Trends Cell Biol 28:807–822. https://doi.org/10.1016/j.tcb.2018.06.005
Reitz C (2018) Retromer dysfunction and neurodegenerative disease. Curr Genom 19:279–288. https://doi.org/10.2174/1389202919666171024122809
Chen X, Kordich JK, Williams ET et al (2019) Parkinson’s disease-linked D620N VPS35 knockin mice manifest tau neuropathology and dopaminergic neurodegeneration. Proc Natl Acad Sci USA 116:5765–5774. https://doi.org/10.1073/pnas.1814909116
Seaman MNJ (2021) The retromer complex: from genesis to revelations. Trends Biochem Sci 46:608–620. https://doi.org/10.1016/j.tibs.2020.12.009
Vazquez-Sanchez S, Bobeldijk S, Dekker MP et al (2018) VPS35 depletion does not impair presynaptic structure and function. Sci Rep 8:2996. https://doi.org/10.1038/s41598-018-20448-4
Lin G, Wang L, Marcogliese PC, Bellen HJ (2019) Sphingolipids in the pathogenesis of Parkinson’s disease and parkinsonism. Trends Endocrinol Metab 30:106–117. https://doi.org/10.1016/j.tem.2018.11.003
Cui Y, Yang Z, Flores-Rodriguez N et al (2021) Formation of retromer transport carriers is disrupted by the Parkinson disease-linked Vps35 D620N variant. Traffic 22:123–136. https://doi.org/10.1111/tra.12779
Simoes S, Neufeld JL, Triana-Baltzer G et al (2020) Tau and other proteins found in Alzheimer’s disease spinal fluid are linked to retromer-mediated endosomal traffic in mice and humans. Sci Transl Med 12:eaba6334. https://doi.org/10.1126/SCITRANSLMED.ABA6334
Yong X, Hu W, Zhou X et al (2018) Expression and purification of the SNX1/SNX6 complex. Protein Expr Purif 151:93–98. https://doi.org/10.1016/J.PEP.2018.06.010
Kvainickas A, Jimenez-Orgaz A, Nägele H et al (2017) Cargo-selective SNX-BAR proteins mediate retromer trimer independent retrograde transport. J Cell Biol 216:3677–3693. https://doi.org/10.1083/jcb.201702137
Shortill SP, Frier MS, Conibear E (2022) You can go your own way: SNX-BAR coat complexes direct traffic at late endosomes. Curr Opin Cell Biol 76:102087. https://doi.org/10.1016/j.ceb.2022.102087
Simonetti B, Paul B, Chaudhari K et al (2019) Molecular identification of a BAR domain-containing coat complex for endosomal recycling of transmembrane proteins. Nat Cell Biol 21:1219–1233. https://doi.org/10.1038/S41556-019-0393-3
Vieira N, Rito T, Correia-Neves M, Sousa N (2021) Sorting out sorting nexins functions in the nervous system in health and disease. Mol Neurobiol 58:4070–4106. https://doi.org/10.1007/s12035-021-02388-9
Rodgers SJ, Jones EI, Arumugam S et al (2022) Endosome maturation links PI3Kα signaling to lysosome repopulation during basal autophagy. EMBO J 41:e110398. https://doi.org/10.15252/embj.2021110398
Ye H, Ojelade SA, Li-Kroeger D et al (2020) Retromer subunit, VPS29, regulates synaptic transmission and is required for endolysosomal function in the aging brain. Elife 9:e51977. https://doi.org/10.7554/ELIFE.51977
Mir R, Tonelli F, Lis P et al (2018) The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem J 475:1861–1883. https://doi.org/10.1042/BCJ20180248
Boecker CA, Holzbaur ELF (2021) Hyperactive LRRK2 kinase impairs the trafficking of axonal autophagosomes. Autophagy 17:2043–2045. https://doi.org/10.1080/15548627.2021.1936933
Wauters F, Cornelissen T, Imberechts D et al (2020) LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 16:203–222. https://doi.org/10.1080/15548627.2019.1603548
Small SA, Kent K, Pierce A et al (2005) Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann Neurol 58:909–919. https://doi.org/10.1002/ana.20667
Puri C, Renna M, Bento CF et al (2014) ATG16L1 meets ATG9 in recycling endosomes additional roles for the plasma membrane and endocytosis in autophagosome biogenesis. Autophagy 10:182–184. https://doi.org/10.4161/auto.27174
Papinski D, Kraft C (2014) Atg1 kinase organizes autophagosome formation by phosphorylating Atg9. Autophagy 10:1338–1340. https://doi.org/10.4161/AUTO.28971
Feng Y, Klionsky DJ (2017) Autophagic membrane delivery through ATG9. Cell Res 27:161–162. https://doi.org/10.1038/CR.2017.4
Mack HID, Zheng B, Asara JM, Thomas SM (2012) AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 8:1197–1214. https://doi.org/10.4161/AUTO.20586
Li J-Q, Tan L, Yu J-T (2014) The role of the LRRK2 gene in Parkinsonism. Mol Neurodegener 9:47. https://doi.org/10.1186/1750-1326-9-47
Bardien S, Lesage S, Brice A, Carr J (2011) Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson’s disease. Parkinsonism Relat Disord 17:501–508. https://doi.org/10.1016/j.parkreldis.2010.11.008
Boecker CA, Goldsmith J, Dou D et al (2021) Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Curr Biol 31:2140-2154.e6. https://doi.org/10.1016/j.cub.2021.02.061
Pang SYY, Lo RCN, Ho PWL et al (2022) LRRK2, GBA and their interaction in the regulation of autophagy: implications on therapeutics in Parkinson’s disease. Transl Neurodegener 11:5. https://doi.org/10.1186/s40035-022-00281-6
Piccoli G, Condliffe SB, Bauer M et al (2011) LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J Neurosci 31:2225–2237. https://doi.org/10.1523/JNEUROSCI.3730-10.2011
Brier LW, Ge L, Stjepanovic G et al (2019) Regulation of LC3 lipidation by the autophagyspecific class III phosphatidylinositol-3 kinase complex. Mol Biol Cell 30:1098–1107. https://doi.org/10.1091/MBC.E18-11-0743
Hervás JH, Landajuela A, Antón Z et al (2017) Human ATG3 binding to lipid bilayers: role of lipid geometry, and electric charge. Sci Rep 7:15614. https://doi.org/10.1038/S41598-017-15057-6
Hurley JH, Schulman BA (2014) Atomistic autophagy: the structures of cellular self-digestion. Cell 157:300–311. https://doi.org/10.1016/J.CELL.2014.01.070
Nath S, Dancourt J, Shteyn V et al (2014) Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat Cell Biol 16:415–424. https://doi.org/10.1038/ncb2940
Ragusa MJ, Stanley RE, Hurley JH (2012) Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell 151:1501–1512. https://doi.org/10.1016/J.CELL.2012.11.028
Nguyen M, Krainc D (2018) LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc Natl Acad Sci USA 115:5576–5581. https://doi.org/10.1073/pnas.1717590115
Li W, Fu YH, Halliday GM, Sue CM (2021) PARK genes link mitochondrial dysfunction and alpha-synuclein pathology in sporadic Parkinson’s disease. Front Cell Dev Biol 9:612476. https://doi.org/10.3389/FCELL.2021.612476
Stavoe AKH, Holzbaur ELF (2019) Axonal autophagy: mini-review for autophagy in the CNS. Neurosci Lett 697:17–23. https://doi.org/10.1016/J.NEULET.2018.03.025
Cai Q, Ganesan D (2022) Regulation of neuronal autophagy and the implications in neurodegenerative diseases. Neurobiol Dis 162:105582. https://doi.org/10.1016/j.nbd.2021.105582
Liang YT (2019) Emerging concepts and functions of autophagy as a regulator of synaptic components and plasticity. Cells 8:34
Coughlan ML, Maday S (2023) Beyond housekeeping: autophagy regulates PKA signaling at synapses. Trends Neurosci 46:167–169. https://doi.org/10.1016/J.TINS.2023.01.002
Maday S, Holzbaur ELF (2014) Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell 30:71–85. https://doi.org/10.1016/J.DEVCEL.2014.06.001
Maday S, Holzbaur ELF (2016) Compartment-specific regulation of autophagy in primary neurons. J Neurosci 36:5933–5945. https://doi.org/10.1523/JNEUROSCI.4401-15.2016
Watson ET, Pauers MM, Seibert MJ et al (2023) Synaptic vesicle proteins are selectively delivered to axons in mammalian neurons. Elife 12:e82568. https://doi.org/10.7554/ELIFE.82568
Yagensky O, Dehaghi TK, Chua JJE (2016) the roles of microtubule-based transport at presynaptic nerve terminals. Front Synaptic Neurosci 8:3. https://doi.org/10.3389/FNSYN.2016.00003
Perlson E, Maday S, Fu MM et al (2010) Retrograde axonal transport: pathways to cell death? Trends Neurosci 33:335–344. https://doi.org/10.1016/J.TINS.2010.03.006
Abeliovich A, Gitler AD (2016) Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539:207–216. https://doi.org/10.1038/NATURE20414
Dong A, Kulkarni VV, Maday S (2019) Methods for imaging autophagosome dynamics in primary neurons. Methods Mol Biol 1880:243–256. https://doi.org/10.1007/978-1-4939-8873-0_16
Kulkarni VV, Stempel MH, Anand A et al (2022) Retrograde axonal autophagy and endocytic pathways are parallel and separate in neurons. J Neurosci 42:8524–8541. https://doi.org/10.1523/JNEUROSCI.1292-22.2022
Kuijpers M, Haucke V (2016) Autophagosome formation by endophilin keeps synapses in shape. Neuron 92:675–677. https://doi.org/10.1016/j.neuron.2016.11.016
Yu Q, Wang Y, Du F et al (2018) Overexpression of endophilin A1 exacerbates synaptic alterations in a mouse model of Alzheimer’s disease. Nat Commun 9:2968. https://doi.org/10.1038/s41467-018-04389-0
Ando K, Yilmaz Z, Suain V et al (2020) The lipid phosphatase Synaptojanin 1 undergoes a significant alteration in expression and solubility and is associated with brain lesions in Alzheimer’s disease. Acta Neuropathol Commun 8:79. https://doi.org/10.1186/S40478-020-00954-1
Miranda AM, Herman M, Cheng R et al (2018) Excess Synaptojanin 1 contributes to place cell dysfunction and memory deficits in the aging hippocampus in three types of Alzheimer’s disease. Cell Rep 23:2967–2975. https://doi.org/10.1016/J.CELREP.2018.05.011
Drouet V, Lesage S (2014) Synaptojanin 1 mutation in Parkinson’s disease brings further insight into the neuropathological mechanisms. Biomed Res Int 2014:289728. https://doi.org/10.1155/2014/289728
Gundelfinger ED, Karpova A, Pielot R et al (2022) Organization of presynaptic autophagy-related processes. Front Synaptic Neurosci 14:829354. https://doi.org/10.3389/FNSYN.2022.829354
Acknowledgements
Government Shaheed Gendsingh College, Charama, Chhattisgarh and Munger University, Munger, Bihar, India are sincerely acknowledged for providing facilities to Abhishek Kumar Mishra and Dipak Kumar, respectively.
Author information
Authors and Affiliations
Contributions
A.K.M. contributed to the literature search and drafted the initial version of the manuscript. A.K.M., M.K.T., and D.K. contributed to designing, preparing, and editing the manuscript. S.P.G. participated in preparing the final version of the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Research Involving Human Participants and/or Animals
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manish Kumar Tripathi and Dipak Kumar contributed equally.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mishra, A.K., Tripathi, M.K., Kumar, D. et al. Neurons Specialize in Presynaptic Autophagy: A Perspective to Ameliorate Neurodegeneration. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04399-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04399-8