
Abstract
Proliferation of specific nucleotide sequences within the coding and non-coding regions of numerous genes has been implicated in approximately 40 neurodegenerative disorders. Cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS), a neurodegenerative disorder, is distinguished by a pathological triad of sensory neuropathy, bilateral vestibular areflexia and cerebellar impairments. It manifests in adults gradually and is autosomal recessive and multi-system ataxia. Predominantly, CANVAS is associated with biallelic AAGGG repeat expansions in intron 2 of the RFC1 gene. Although various motifs have been identified, only a subset induces pathological consequences, by forming stable secondary structures that disrupt gene functions both in vitro and in vivo. The pathogenesis of CANVAS remains a subject of intensive research, yet its precise mechanisms remain elusive. Herein, we aim to comprehensively review the epidemiology, clinical ramifications, molecular mechanisms, genetics, and potential therapeutics in light of the current findings, extending an overview of the most significant research on CANVAS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The global prevalence of neurodegenerative disorders presents a significant and escalating health concern, thereby constituting a formidable challenge for contemporary medicine. Characterized by a gradual onset and a progressive trajectory, these disorders demonstrate an increasing propensity with advancing age, leading to a projected rise in the observable symptoms of these conditions over time. The predominance of neurological conditions remains consistent across nations, regardless of the economic stratification. The etiopathogenesis of such neurodegenerative diseases can be simply attributed to multifarious mechanisms.
A third of the genome (33%) approximately consists of repetitive DNA sequences, believed to play important roles in the process of species differentiation. Some of these repeat expansions could be largely pathogenic in nature. Such pathogenic repeat expansions (REs) have been identified in association with over 30 hereditary human diseases, predominantly those affecting the nervous system [1]. These disorders encompass a diverse spectrum, each distinguished by the expansion of particular genetic sequences. The approach of long-read technologies represents a recent and distinctive avenue for methodically probing the role of tandem and expanded repeats in shaping the genetic landscape of human disorders [2]. Cerebellar Ataxia, Neuropathy, and Vestibular Areflexia Syndrome (CANVAS) are the co-occurrence of cerebellar ataxia with neuropathy and vestibular areflexia, which is a late onset, gradual, autosomal recessive, multi-system ataxia marked by simultaneous dysfunctioning of sensory neurons, the vestibular apparatus, and the cerebellum [3]. The emergence of CANVAS, as a distinct disease, has been a gradual phenomenon unfolding over the past three decades. Nonetheless, the genesis of this disease is posited to date back to around 23,000 B.C. in Europe. The etiology of CANVAS encompasses acquired, hereditary, and non-hereditary factors [4].
In clinical terms, CANVAS was introduced as a novel identity in 2011 [5]. It comprehends a wide range of clinical traits such as imbalance, sensory peripheral symptoms, gait impairment, oscillopsia, dry cough, autonomic dysfunction, dysarthria and dysphagia [4, 6]. CANVAS may encompass a polygenic basis, implying the involvement of multiple causative genes. This condition also exhibits substantial phenotypic heterogeneity and has the potential to present phenocopies, further complicating the diagnosis. Studies have found complete CANVAS instances to be those in which the vestibular system, cerebellum, and sensory peripheral nerves are all involved simultaneously with a mean onset age of above 50 years. Recent research has also enhanced our understanding of the condition, revealing additional associated motor and non-motor symptoms, including oscillopsia, sweat gland denervation, cough and autonomic dysfunctions to be associated with CANVAS [4]. Research into unexplained genetic ataxias, including CANVAS, is currently focused on investigating their phenotypic similarities to established conditions associated with repeat expansions (REs). For CANVAS particularly, a primary etiological factor is the presence of a biallelic intronic recessive AAGGG repeat expansion, ranging from 250 to over 2000 repeats, in the replication factor complex subunit 1 (RFC1) gene. The RFC1 gene, serves an integral role in both DNA replication and repair, constituting replication factor C (RFC), which contributes majorly in DNA replication and repair by loading the PCNA (proliferating cell nuclear antigen) on the DNA [4, 6,7,8]. Predictions suggest the presence of different repeat motifs to be associated with the disease, a few non-pathogenic and a few distinct pathogenic extended repeat motifs, where (AAGGG)n•(CCCTT)n is the most prevalent pathogenic repeat, whereas (AAAAG)n•(CTTTT)n is the most prevalent non-pathogenic repeat [7]. The pathogenic allele differs from the non-pathogenic allele repeats in size as well as in nucleotide composition, setting it apart from the majority of other repeat expansion diseases [6]. So far, the precise route to the pathogenicity of CANVAS remains elusive, specifically about why only certain expanded motifs lead to pathology, while others do not. It is predicted that all pathogenic motifs in RFC1 form extremely stable G quadruplexes (a secondary DNA structure), which have been proven to influence gene transcription in other similar conditions and appear to have evolved from a single haplotype [9]. Recent findings further affirm this idea by demonstrating that only pathogenic patterns persist in the RFC1 transcript [7]. This review, thus, amalgamates the genetic basis with the syndromic clinical features of CANVAS to enable more precise clinical diagnosis and future research directions regarding molecular mechanisms behind the pathophysiology of the disease.
Epidemiology of CANVAS
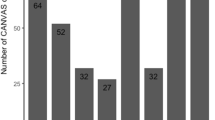
While clinical descriptions of CANVAS are uncommon, it is possible that its prevalence could exceed initial expectations. Quite a few population-based assessments reported elevated carrier frequencies of the pathogenic (AAGGG)n motif coupled with dwindled biallelic rates (Fig. 1). The majority of confirmed patients so far, both clinically and genetically, have come from communities in Europe [10, 11]. At the outset, 92% of instances having complete CANVAS, 54% of cases with cerebellar ataxia and sensory neuropathy, and 22% with late-onset ataxias showed biallelic pathological RFC1 expansions [11]. Contemporary studies have independently revealed the presence of the pathological biallelic RFC1 expansion in ataxia cohorts; however, the reports indicated higher prevalence in full-blown CANVAS cases and lower prevalence in cases involving late-onset ataxia and incomplete CANVAS. In line with diverse studies, individuals from northern European origin have shown up with carrier frequency of RFC1 heterozygous (AAGGG)exp [1] to range from 0.7 to 4%, with an approximate prevalence of RFC1-related diseases of 1/20,000, i.e. 1:625. The Chinese population has also shown an analogous allele frequency of 2.24%. Whereas, 1.8% of the Japanese population (1/55) have shown heterozygous RFC1 (AAGGG)exp [12]. The occurrence of harmful RFC1 gene expansions (AAGGG)n has been demonstrated to differ notably among various groups of individuals with late-onset ataxia. This ranges from 1.1% in a study involving Canadian and Brazilian participants to as high as 28.9% in a British cohort. In other studies, the prevalence was found to be 3.2% in a North American group, 6.5% in a Greek cohort, 5.2–10.8% in Japanese cohorts, 14% in a Turkish cohort, 14.5% in an Italian cohort, 15% in a French cohort and around 20.2% in a German cohort (Table 1) [13,14,15]. Variations in the criteria used for participant selection can be assumed to contribute significantly to the range of prevalence rates observed across numerous cohort studies focused on late-onset ataxias. Another potential factor could be the presence of population-specific variables that may exert an influence on these figures. The AAGGG expansion manifests at a comparable allele frequency in Asian populations, though the impact of RFC1 on disease within these populations is significantly less pronounced in comparison to European populations. Cortese et al. reported the non-pathogenic allele frequency to be 13% for (AAAAG)exp, 7.9% for (AAAGG)exp and 2.1% for (AAGAG)exp [11]. The allelic distribution for biallelic (AAGGG)exp in association with CANVAS has been reported to be around 0.7–6.8% [16]. These collective findings emphasize that late-onset ataxia patients, especially those with accompanying sensory neuropathy, frequently exhibit genetically confirmed CANVAS. Hence, it is crucial to conduct screenings for biallelic RFC1 expansions in individuals presenting with complete CANVAS, partial CANVAS, or unexplained sensory ataxia. This proactive approach can ensure comprehensive care for these patients.

Percentage prevalence of biallelic expanded (AAGGG)n repeat. Diverse cohort-based studies have revealed the occurrence of biallelic (AAGGG)n repeat extension in CANVAS across different geographical regions worldwide
Clinical Considerations Linked to the Syndrome
In 2011, Szmulewicz et al. introduced the acronym “CANVAS” to describe a condition associated with the late emergence of symptoms including cerebellar ataxia, sensory neuropathy and vestibular areflexia [17]. Concurrently, these patients exhibited cerebellar atrophy and persistent chronic cough. The chronological course of CANVAS demonstrates an escalating profile and amplification of its overall symptoms or characteristics (Fig. 2). Migliaccio et al. initially documented four cases featuring gradual sporadic cerebellar ataxia, coupled with a decline in vestibulo-ocular reflex [18]. In a study involving 80 individuals experiencing late-onset cerebellar ataxia, the findings revealed that 33% of the patients had an expression of multiple system atrophy (MSA), another 33% were associated with acquired causes, and the remaining cases were diagnosed as idiopathic late-onset cerebellar ataxia (ILOCA), linked to CANVAS [19]. Derived from an examination of 150 cases, where 22 familial cases were diagnosed with CANVAS alongside ILOCA, the study emphasizes that CANVAS contributes to 20% of the cases involving ILOCA [11]. But in the most current scenario, the central characteristics of the clinically possible CANVAS have evolved. There have been significant efforts to understand the clinical aspects of the disease and it has been observed that the emergence of the last element in the diagnostic triad may extend beyond a decade. Therefore, a patient initially presenting with cerebellar ataxia and bilateral vestibulopathy (CABV), or any other combination of two of the three primary features of CANVAS, should undergo initial assessments. Consequently, it is recommended that these individuals undergo regular reassessment to ascertain whether they have begun to develop a progressive form of CANVAS [20]. According to Cortese et al. the disease turns to transition gradually from initial occurrence of sensory neuron deficits to later manifestations of vestibular and cerebellar dysfunctions [4].

An illustration portraying the developing profile of CANVAS symptoms. This figure provides a comprehensive depiction of the advancing trajectory of symptoms associated with CANVAS
Research involving five Turkish families with a high incidence of consanguineous marriages revealed the presence of gait ataxia accompanied by sensory and autonomic disturbances. The study further detailed lightheadedness and cold feet as prevalent autonomic symptoms associated with CANVAS [21]. Thus, as per observations, patients exhibit a vast range of behavioural signs, from pure cerebellar ataxia to more complex clinical characteristics, contributing to the continuously fostering clinical spectrum. These may also appear in conjunction with additional features like pyramidal tract disorder, muscle fasciculation, autonomic and cognitive impairments, chronic cough, parkinsonism, involuntary movement and elevated creatine kinase levels (hyperCKemia). Individuals may also have noise-induced hearing loss or presbycusis, two unrelated instances of hearing loss [22]. Noteworthy is the revelation that a significant majority, exceeding 60%, of patients may experience an unexplained dry cough [3]. In CANVAS, the ongoing neurodegenerative process leads to a gradual disruption of cerebellar neurons that are likely implicated in the regulation of the cough reflex. This implies that as the condition advances, the neurological deterioration specifically affects the neurons in the cerebellum responsible for controlling the cough response [23]. Chronic cough thus has now been recognized as a fundamental characteristic of genetically confirmed CANVAS. Marked by hypersensitivity, chronic cough is known to precede the fundamental symptoms by a span of 30 years or more. This finding highlights the evolving understanding of the condition, emphasizing that it encompasses not only the previously established symptoms but also the presence of persistent coughing. Ninety-two percent of the patients with CANVAS-endorsed history of chronic cough that predated gait instability by a median duration of 16 years had their history genetically validated [24]. This discovery emphasizes the significance of recognizing this symptom in the evaluation of individuals with RFC1 REs. Recent research thus has illuminated a wider clinical spectrum associated with RFC1 REs, surpassing prior understanding [25]. This newfound depth of insight promises more precise diagnoses and potentially transformative treatment approaches for affected individuals.
Furthermore, sensory neuropathy, coupled with both central and peripheral axonopathy, notably in cutaneous sensory nerve action potentials (SNAP), frequently showcase as the primary symptom in genetically confirmed cases. Sensory impairment in CANVAS is caused by a prominent dorsal root ganglionopathy, which results in compromised proprioception. It is also characterized by reduced sensitivity to pinprick, referred to as pinprick hypoesthesia, as well as diminished sensations in joint position and vibration. These sensory impairments follow a length-dependent distribution. Rather than being a “neuropathy”, the somatic sensory impairment identified in CANVAS is a “neuronopathy” [22]. The cerebellar symptoms are most likely linked to a loss of Purkinje cells [26, 27].
In a 2018 study that involved the analysis of five patients exhibiting gait imbalance and cough, intact motor reflexes, as indicated by tendon jerks, were observed, and no discernible hearing loss was detected [28]. The research further indicated well-functioning peripheral motor fibres and muscle afferent fibres. Nonetheless, a pronounced loss in sensory nerve action potential was documented. This observation aligns with the understanding that, during ganglionopathy and axonopathy, the corticospinal tracts remain unaffected, as seen in the H reflex where A alpha fibres remain intact. In another case, as noted in a study on two families from the Asia–Pacific region, motor neurons were found to be affected, showcasing key CANVAS symptoms, muscle weakness in extremities, and signs of motor neuron denervation in areas such as the hypoglossal nuclei and spinal cord [29]. Unsteadiness, therefore, is a prevalent clinical characteristic of CANVAS, attributed to bilateral vestibular dysfunction. This results in somatosensory deficits, yet hearing remains unaffected. The bilateral vestibular dysfunction is also associated with oscillopsia, where objects seem to oscillate during head movements. Additionally, a study involving two siblings linked sweat gland denervation to CANVAS [30].
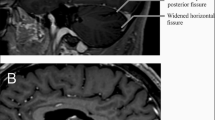
Therefore, in cases of late-onset ataxia, especially when coinciding with sensory neuropathy, it is advisable to conduct screening for biallelic RFC1 expansions [14]. Cerebellar atrophy, which is defined clearly as a diminution of vermian Purkinje cells, is the most prevalent MRI result; however, there may be other, less frequent, related abnormalities as well [17, 31, 32]. In addition to the cerebellum and its connections, the basal ganglia dopaminergic circuitry is also affected by neurodegeneration in RFC1/CANVAS. It is recently discovered that RFC1/CANVAS frequently exhibits nigrostriatal dysfunction [33, 34]. Subsequent symptoms may also include the development of orthostatic hypotension, neuropathic pain, dysphasia, dysarthria, challenges with urinary erectile, retention functions and dryness in eyes and mouth [22, 32, 35]. A recent study by El Houjeiry et al. described the first case of CANVAS syndrome which was initially presented with isolated spinal cord lesion which mimicked dysimmune myelitis [36]. Very recently, another study reported two cases of RFC1-associated CANVAS with the brain MRI illustrating the (pseudo-)eye-of-the-tiger sign [31]. In fact, this study highlights that RFC1-associated CANVAS should be considered as an alternative imaging diagnosis in cases exhibiting the (pseudo-) eye-of-the-tiger sign. This finding expands our understanding of potential differential diagnoses in imaging studies. Thus, the prevailing features now indulge more than just the classical triad of symptoms; they also encompass an enduring chronic cough, along with discernible signs of dysautonomia and neurogenic pain. These revelations contribute to a more profound comprehension of this condition and its diverse expressions, offering a path towards more focused and efficacious medical interventions.
Pathology of the Disease
From a neuroscientific perspective, CANVAS can be characterized as a neuronopathy implicating the dorsal root ganglia, multiple cranial nerve pathways and concomitant cerebellar atrophy. Figure 3 clearly defines the three primarily associated clinical features along with their distinct characteristics. Dorsal root ganglionopathy is now considered to be responsible for sensory impairment in CANVAS, which leads to degeneration of neuron cell bodies [32, 37]. The sensory dysfunction in CANVAS does not adhere to the typical length-dependent paradigm often seen in neuropathies. Distinctively, CANVAS demonstrates a non-length-dependent sensory deficit, clinically described as sensory ganglionopathy or neuronopathy, marking its unique pathophysiological profile [38]. In a study conducted by Szmulewicz et al., which explored the neuropathology of the brain and spinal cord in two individuals diagnosed with CANVAS, it was observed that the sensory deficit results from dorsal root ganglionopathy accompanied by secondary tract degeneration [17]. Scarpa ganglion cells also exhibited reduction [39]. The sensory deficit’s pathology stems from the neuronal loss in cranial nerve V, VII and VIII ganglia, as well as the dorsal root ganglion in the spinal cord. Neuronal loss in the dorsal root ganglion leads to axonal degeneration, subsequent removal of the myelin sheath, and T2 hyperintensity in the posterior columns of the spinal cord [17]. Post-mortem temporal bone histology revealed ganglionopathy in facial, vestibular and trigeminal nerves [37]. In the investigative examination of cerebellar sections, a notable reduction of Purkinje cells and vermal atrophy was observed, associated with the formation of torpedo bodies. Additionally, gliosis of the Bergmann layer was discerned [37]. The research team identified neuronal loss in the inferior olivary nuclei of the cerebellum, which correlates with the sensory deficits. The axonal degeneration coexisted with nerve thinning [40]. Consequently, the cross-sectional area of peripheral nerves in these subjects was significantly reduced in comparison to healthy controls. Further, degenerative changes within the nuclei of the pons and evident neuronal loss characterized the CANVAS patients [41]. The potential degeneration of the mesencephalic nucleus has also been accounted for by the observed masseter areflexia. Vermal involvement pattern causes Crus 1 to act functionally analogous to the oculomotor region of the cerebellum [42]. Vestibular system pathology is linked to the loss of vestibular ganglion, termed vestibular neuronopathy, observed in all five temporal bones, along with atrophy of vestibular nerves in axons and dendrites [39]. Notably, the vestibular nuclei and receptor cells remain unaffected, and there is no evidence of trans-synaptic degeneration [42]. The maintenance of the vestibular nuclei, despite the degeneration of axons, is due to the varied afferent nerve inputs they receive, including those from the visual system and the midbrain [39]. Additionally, sensory nerve damage in the geniculate and trigeminal ganglia, as observed in two temporal bones, contributes to the profound vestibular impairment in CANVAS patients, attributing it to vestibular ganglionopathy [39]. Auditory functions remain intact, given that neither degeneration nor neuronal loss is observed in the cochlear ganglia and auditory nerves. The front and side sections of the spinal cord, along with the thoracic columns, have known to stay intact or unaffected. Additionally, fossa decompression emerges as a potential complication associated with bilateral vestibulopathy [22].

Defining clinical features of the CANVAS syndrome. The diagram highlights the key characteristics associated with cerebellar dysfunctions, vestibular dysfunctions and sensory neuropathy that collectively form the cause underlying the RFC1-CANVAS disease
In patients diagnosed with CANVAS, oscillopsia is often expressed as a persistent symptom primarily linked to cerebellar ataxia, a consequence of downbeat nystagmus [43]. The pathology of CANVAS also encompasses the involvement of unmyelinated C fibres and A (delta) fibres, as evidenced by clinical revelations such as pinprick anaesthesia and spasmodic coughing episodes. Predominantly, the spasmodic cough is attributed to the engagement of C fibres, which oversee sensory innervation of the upper respiratory and oesophageal pathways. Intriguingly, these C fibres are associated with nociceptors situated within the larynx. The informational relay by these fibres converges at specific regions of the brain stem, controlling involuntary cough reflexes through efferent pathways linked to associated nucleus governing muscular responses. Thus, the pathophysiology of CANVAS involves a complex interplay of neuronal degeneration across various cranial nerve pathways, dorsal root ganglia and cerebellar structures, leading to diverse clinical manifestations.
Genetic Foundations
CANVAS was first described in 2004 and further outlined in 2011, with a suspected hereditary aetiology [17, 44]. Ever since, it has been believed to have an extensive genetic basis. However, the underlying molecular and genetic basis of CANVAS poses a mystifying enigma [14]. Recently CANVAS has been recognised as the consequence of a biallelic intronic repeat expansion in the gene encoding the replication factor C subunit 1 (RFC1) on chromosome 4 [4, 45]. RFC1 gene contains the genetic information for the largest subunit of Replication Factor C, a pentameric complex which functions as a clamp loader and facilitates the attachment of polymerases, aiding in extension of nucleic acid chains. It orchestrates the activities of both DNA replication and repair. RFC1-ataxia is known to be triggered by a catastrophic extension of the AAGGG pentanucleotide in the poly(A) tail of an AluSx3 element in RFC1 intron 2, which substitutes the wild-type sequence of 11 AAAAG repeats, present 2952 base pairs upstream of exon 3 and 2863 base pairs downstream of exon 2. REs, particularly more than 100 repeat modifications, modify the 3-D structure of the RFC1 protein. Whilst the first dissemination described an AAGGG pathogenic expansion with 400 to 2000 repeats (AAGGG400-2000), lesser extensions (AAGGG100-160) have also been documented [46]. Although the reference genome has (AAAAG)11 pure repetitions, recent studies have revealed astounding genetic heterogeneity. It thus, is also susceptible to other repeat sequences such as AAAGGG, AACGG, AAGGC, AACGG, AAAGG, AAGAG, AGAGG, and ACAGG in addition to AAAAG and AAGGG (Table 2) [47]. AAGGG can be preceded by non-pathogenic repeats of configuration (AAAGG), with repeats up to 51–53. Thus, it is very clear that the pentanucleotide expansion possesses vibrant character and diverse configurations. In relation to the increased heterogeneity at the RFC1 locus, a total of seven distinct expanded alleles have been laid out, three of which have been linked to the disease: AAGGG, ACAGG and the Māori allele [(AAAGG)15–25 (AAGGG)exp (AAAGG)10] and the rest four are believed to be of benign or unclear pathogenicity: AAAAG, AAAGG, AAGAG and AGAGG [48]. The expansion of the pathogenic sequence is hypothesized to take place via replication slippage, indicating instability within the A and G-rich motif. The robust base stacking interactions associated with the A and G-rich motifs (AAGGG and AAAGG) contribute to expansion eventually. The occurrence of pseudo-dominance may be a result of the high frequency of heterozygotes among carriers in various populations. The length of repeats varies between 15 and 200 for (AAAAG)exp and 40–1000 for (AAAGG)exp, both of which belong to non-pathogenic alleles. Whereas, for (AAGGG)exp, which is pathogenic in nature, the repeat size ranges from 400 to 2000 repeats [11]. According to Dominik et al., all the pathogenic variants shared a common region of around 66 kb, indicating a recent recombination event [9]. In fact, all the pathogenic alleles along with carriers AAGGG and AAAGG have also shown a larger shared region indicating the derivation of these expanded variants from an ancestral haplotype (dating 56–100 years back).
In an investigation where biallelic expansion was detected, the patients exhibited a congruent core haplotype, encompassing 27 single nucleotide polymorphisms. This suggests a shared origin for the point mutations within the RFC1 gene. This core haplotype for RFC1 spans 0.36 Mb and comprises four genes: (a) TMEM156, (b) KLHL5, (c) WDR19 and (d) RFC1. Utilization of bioinformatics localized the pathogenic repeat expansion to a specific locus on chromosome four (chr4:38887351–40463592, hg19), which consistently showed association with CANVAS across all family samples examined [11, 49].
In a combined analysis, 537 samples were examined, leading to the identification of 23 heterozygous and one homozygous individual, resulting in an allele frequency of 0.023. In two CANVAS-affected individuals, the motif AAAAG on chromosome 4 (chr4:39350045–39350095, hg19) was substituted by AAAGG [49]. It highlighted another investigative insight, that the most recent common ancestor (MRCA) of CANVAS existed approximately 25,880 years ago, originating in Europe, and the divergence of this MRCA was attributed to a distinct founder effect. From this MRCA, four descendant subgroups were discerned. Group “A” has its origins tracing back 5600 years, while Group “B” bifurcated into “B1” and “B2” with a MRCA around 4180 years ago. Group “C” showcased an MRCA dating back 1860 years. The final Group “N” was characterized only by the shared core haplotype. Notably, while CANVAS overexpression is predominant in those of European descent, instances in non-European lineages such as Japanese, Lebanese and Native American have also been documented. Heterozygous individuals for the RFC1 gene repeat expansion remain asymptomatic but are classified as carriers. Carrier frequencies across populations were variable, with Europe at approximately 0.7%, China ranging from 1 to 2.2%, and Canada at 4%. In various studies, this frequency oscillated between 0.7 and 6.5% [12]. The motif (AAAAG)12–200 has an allele frequency of 0.13, while the regular sequence (AAAAG)11 stands at 0.75 [4]. Intriguingly, such expansions in the poly(A) stretch of the Alu element have parallels in other neurodegenerative ailments, including FRDA and several types of SCA associated with late-onset ataxia. This pattern intimates a potential shared pathogenetic mechanism rooted in Alu region polymorphisms across these neurodegenerative conditions. The motif sequences also suggest the potential existence of intricate nucleic acid structures. It is evident that pathogenic motifs may exhibit a propensity for forming structures (G-quadruplexes and triplexes) not observed in non-pathogenic motifs.
There have been a few reports that have pointed towards the frameshift or nonsense variants of RFC1 to be linked to the condition. Arteche-López et al. reported the presence of the nonsense c.724C > T p.(Arg242*) mutants along with the pathological AAGGG expansion in the RFC1 gene in two compound heterozygous patients [50], whereas another investigation by Benkirane et al. reported p.Arg388* and c.575delA as the RFC1 variants in patients with compound heterozygosity of pathogenic AAGGG expansion [51]. Nevertheless, the exact etiological basis remains yet to be elucidated [7, 9].
Molecular Mechanisms
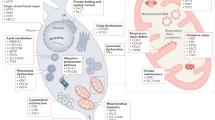
Presently, the specific pathogenic mechanisms in CANVAS remain unidentified. Nonetheless, it is possible to hypothesize potential mechanisms based on observations in CANVAS as well as in other similar disorders involving REs. Various associated pathogenic mechanisms include RNA loss-of-function, protein gain-of-function, RNA gain-of-function, repeat-associated non-AUG (RAN) translation, interaction with RNA-binding proteins resulting in sequestration, formation of R-loops, or a combination of these mechanisms (Fig. 4). The mechanisms underlying the selective sensory neuronopathy and damage to Purkinje cells in CANVAS are also currently unknown. However, the following molecular mechanisms are known to be largely associated with the following:
-
a)
DNA repair mechanism: The RFC1 gene encodes a DNA-dependent ATPase that plays a crucial role in loading the DNA clamp onto DNA. This clamp recruit’s polymerase enzymes for replication, and the resulting complex catalyses the reaction that opens the PCNA of the DNA-clamp protein, allowing it to encircle DNA [52]. Additionally, this complex is involved in pathways related to DNA repair, participating in processes such as mismatch repair and excision repair as responses to DNA damage. It is noteworthy that mutations in genes associated with DNA damage repair, including PCNA, ATM and ATR have been linked to ataxia. This suggests that if the DNA repair system in the cerebellum is not working properly due to mutations, it could make the cerebellum more prone to damage, possibly leading to the development of ataxia. Nonetheless, the precise mechanism by which the RFC1 repeat expansion induces ataxia remains to be cleared. One hypothesis proposes that alterations in the polymorphic zone may modify the gene expression of proteins involved in DNA repair, ultimately resulting in damage to the cerebellum and peripheral nerves, particularly the small fibres associated with sensation [53]. Furthermore, cells with elevated energy demands are susceptible to oxidative stress, potentially causing DNA damage and impairment. Although the RFC1 complex is a crucial isoform known to interact with ligase, various transcriptional factors and ASF1 27 (Anti-silencing factor 1, a H3-H4 histone chaperone), there is currently no supporting evidence for this hypothesis [54]. Mutations in genes related to DNA repair can also contribute to mitochondrial dysfunction. This dysfunction serves as a potential final pathway for ataxia due to a combination of factors, including the vulnerability of mitochondrial DNA to damage by oxidative reactive oxygen species (ROS) and the substantial energy demand of Purkinje cells, resulting in high ROS production and consequent mitochondrial damage and dysfunction. However, the specific reasons for this process occurring in particular cells remain unknown.
-
b)
Transcriptional silencing: It is induced by hypermethylation of heterochromatin initiated due to REs. The mechanism by which the REs mediates transcriptional silencing remains undefined. According to a hypothesis, the transcription of RNA from DNA with repeat expansion results in the formation of R-loop that halts the transcription process by causing polymerase stalling. This, in turn, triggers the recruitment of the PRC2 complex, responsible for methylation, leading to stable silencing of the expanded DNA. A potential mechanism through which R-loop formation may occur is via the DNA Damage Response (DDR) system. The amplification of the expansion can enhance R-loop formation, subsequently triggering the recruitment of the DDR system. This can precipitate mitochondrial dysfunction and apoptosis, a phenomenon evident in the hexanucleotide expansion disorder associated with ALS. Diseases linked to G-rich motifs have been observed to correlate with R-loop formation, as evidenced by the in vivo studies. Supporting evidence for this concept comes from a study demonstrating the silencing of FMR1 in the later stages of embryo development [55]. A clear example of this silencing mechanism can also be seen in C9 amyotrophic lateral sclerosis (ALS), which results from the expansion of GGGGCC repeats in C9ORF72 [56]. However, no evidence was found in a study involving CANVAS-derived cell lines, of transcriptional reduction of RFC1. Analysis by Rafehi et al. of GTEx RNA-seq data showed that pathogenic allele did not inhibit normal expression of RFC1 as compared to reference sequence [49]. The somatic instability evident in disorders such as spinocerebellar ataxia type 10 (ATTCT) and familial adult myoclonic epilepsy 1 and 3 (TTTCA) may offer additional insights into the molecular foundations of CANVAS [57].
-
c)
RNA toxicity: A very recent investigation reported formation of toxic RNA foci in two Japanese women (83 and 85 years old, respectively) at the time of their death. These two CANVAS patients, identified with compound heterozygosity (biallelic ACAGG-exp and AAGGG-exp), point towards the RFC1 loss of function to be associated with CANVAS. Additionally, the same two patients also revealed the formation of RNA foci as per the analysis by RNA FISH (Fluorescence in situ hybridization), indicating the presence of RNA toxicity in the neuronal tissues to also have partial association in the CANVAS pathogenesis [58]. Another report by Benkirane et al. also assisted the loss-of-function of the gene hypothesis by showing a considerable reduction in the RFC1 mRNA levels in the blood of patients [51]. RNA foci (gain of toxic function) and loss of function of the gene have also been linked to many similar repeat expansion disorders.
-
d)
Altered RNA splicing: RNA splicing, a critical component of mRNA processing, was first identified in 1977, describing the existence of exons and introns. However, aberrance in RNA splicing can result in the retention of intronic sequences within the final protein product. This misincorporation can alter the protein’s secondary and tertiary structures, potentially leading to protein aggregation or nuclear sequestration, with downstream pathway implications. Nevertheless, post-mortem analyses have not identified such protein aggregates in the affected brain regions. Supporting this mechanistic perspective, Traschutz et al. observed a modest increase in the retention of intron 2 from the RFC1 gene in its precursor mRNA from muscle biopsy samples [59]. The meaning and broader impact of this finding remain under-explored in current literature and necessitate further investigation.
-
e)
G-quadruplex formation: The pathogenic mechanism of CANVAS is unusual in two ways: the repeat expansion of RFC1 falls inside the AluSx3 poly(A) tail, which leads to plausible expansion by retrotransposons. Secondly, repetition also does not affect the expression level of RFC1 gene products. Thus, CANVAS is yet to be understood and is a unique, intriguing disease. The association of particular repeat motifs to cause the condition is contributed by the unusual nucleic acid structures that are formed by the pathogenic forms. The pathogenic AAGGG in its expanded form facilitates the formation of G-quadruplex (non-B-form) structures that are more stabilized [60]. The tendency to form non-B form structures can be attributed to the self or interassociation of different homopurine or homopyrimidine sequences. A recent report suggests that (AAGGG)n in both DNA and RNA forms resulted in the formation of the four-stranded G-quadruplex structures in the potassium solution [7]. These G-quadruplexes are built of subunits called G-quartets that in turn are built through the self-association of four guanine residues through Hoogsteen hydrogen bonds arranged in a planar form around a monovalent cation. The formed G-quadruplex is known to regulate transcription by either interrupting the movement of RNA polymerase or by tethering onto duplex-DNA. These pathogenic repeats are also observed to attain triplex (triple-stranded) forms. Both triplexes and quadruplexes have not been observed with non-pathogenic forms. “Dominik et al. also showed the G4 scores (a bioinformatics-based analysis) to be high for most of the pathogenic repeat configurations compared to the non-pathogenic repeat ones by using the G4Hunter and QGRS-Mapper, confirming the expected propensity of G-quadruplex formation in CANVAS [9].” However, one pathogenic repeat (ACAGG), prevalent in the Asian population, seems to diverge from this pattern. Very recent research by Kudo et al. reveals that ACAGG does not exhibit a propensity to form G-quadruplex structures; instead, the ACAGG RNA tend to adopt a unique slipped hairpin configuration. These findings clearly suggests that while pathogenic repeats generally develop rigid secondary formations, none of the non-pathogenic repeats exhibit identifiable secondary structures in nucleic acids, highlighting a clear contrast between the secondary structure dynamics of pathogenic and non-pathogenic repeats [61].

Possible mechanisms associated with the pathogenesis of CANVAS. Expanded pentanucleotide repeats in the AluSx3 element in the intron 2 of replication factor C subunit 1 (RFC1) gene lead to the formation of toxic RNA, that leads to formation of secondary structures as well as RNA foci that sequesters RBPs (RNA binding proteins). This leads to the formation of mutant proteins either through translation or RAN (Non-AUG) translation, both of which cause aggregation of proteins
Prospective Diagnostic and Therapeutic Strategies
Due to the insufficient comprehension of the bilateral vestibular areflexia and sensory disorders as the cues to the right diagnosis, the diagnosis of CANVAS may be missing. The disorder is highly probable to remain underdiagnosed. The possibility of repeat expansion in RFC1 should be taken into account when encountering cases involving sensory ataxia neuropathy. This consideration is especially relevant, though not exclusively limited to, situations where there is a concurrent presence of cerebellar dysfunction, vestibular involvement, and cough [4]. Patients with genetically inexplicable, gradually worsening adult-onset ataxia and sensory or sensorimotor axonal neuropathy should be assessed for the possibility of (AAGGG)n REs in the RFC1 gene in order to strengthen the existing diagnostic practices [25]. When compared with individuals only diagnosed with cerebellar ataxia, the inclusion of neuropathy as a symptom boosts the positive effect of testing by 20.1% (with a confidence interval of 9.7% to 30.6%). Furthermore, the combination of both neuropathy and vestibulopathy elevates the benefit to 70.6% (with a confidence interval of 44.2 to 97.0%) [13]. In patients who exhibit a notable delay in the emergence of all three cardinal characteristics, diagnosis may be harder to arrive at. Thus, identification of the condition poses greater challenges when the characteristic features develop over an extended time period. Expanded (AAAGG) REs have at times appeared to mimic the (AAGGG) expansion in cases where the evaluation is purely based on the RP-PCR results. In such conditions, the pure characteristic of the expansion is largely misapprehended [62]. Therefore, to combat this, the diagnostic criteria could be staged according to the pathology found in CANVAS [22].
Upon tracking down the genetic anomaly, it may prove beneficial to assess these alternative phenotypes for a more comprehensive outlining of the phenotype, and potentially for understanding the presence of multiple distinct pathologies. Next-generation sequencing (NGS) and other advanced genomic technologies are revolutionizing the landscape of molecular screening and clinical healthcare. These cutting-edge tools have ushered in a new era, allowing for more precise and comprehensive analysis of genetic material. Multimodal RFC1 repeat screening (Southern blot, PCR, whole-exome/genome sequencing-based approaches) along with longitudinal and cross-sectional deep phenotyping, vestibulo-ocular reflex quantification by the video head impulse test and optical genome mapping has been largely useful as of now [25, 59, 63]. To screen for the RFC1 expansions associated with CANVAS, a systematic process is followed that is depicted by a workflow here (Fig. 5). This initially includes RFC1-flanking PCR to identify if amplification in the RFC1 region is present or not, followed by RP-PCR (Repeat Primed PCR) to confirm the presence of biallelic (AAGGG)n [13]. This step ensures the specificity of the expansion associated with CANVAS. Subsequently, the repeat lengths are further assessed using Southern blotting [3]. This technique allows for the precise determination of the repeat lengths and distinguishes between homozygous, heterozygous, and non-expanded alleles associated with CANVAS. Thus, accurate screening for timely detection of the disease is done following a concrete workflow, that is a composite of different reliable techniques.

Comprehensive screening workflow for RFC1 repeats expansion. The screening process involves RFC1 flanking PCR to identify the presence of amplification in the RFC1 region, RP-PCR to identify the presence of biallelic (AAGGG)n, southern blotting to assess the repeat length, long-range PCR to confirm the structural integrity, sequencing for nucleotide-level resolution and optical genome mapping for high-throughput analysis
A recent investigation has revealed a substantial rise in the serum neurofilament levels in individuals with RFC1 disease. The observed increase in neurofilament levels serves as a compelling indicator, pointing towards its potential utility as a biomarker in advancing the accuracy of diagnosis and enhancing treatment strategies for RFC1 disease [64]. Another imperative approach could be the conduction of comprehensive assessment of familial medical background and facilitating access to genetic counselling for all individuals. Taking this into consideration, there is a significant prospective for strengthening the diagnostic accuracy to a significant degree. Precise diagnosis of CANVAS is a crucial therapeutic endeavour that shall influence the management, prognosis, ease for patients and most importantly, the prospect of future therapy. A recent study by Ghorbani et al. successfully advocated the incorporation of RFC1 screening to the genetic assessment workflow by employing novel strategies that yields extensive fragments such as the use of optical genome mapping over the southern blotting, which has been considered a gold standard for determining the repeat expansion lengths till now, to succumb the labour intensiveness and time-consumption [25].
CANVAS-associated pathogenesis can be addressed through therapeutic approaches targeting both upstream and downstream mechanisms. These are largely aimed at reducing the toxic effects caused by the expanded repeats. A range of potential therapeutic nucleic acid-targeting strategies having considerable importance in tackling other similar RE neurodegenerative disorders can be of immense importance in this disease too. Since small molecules have been of potential value as a therapeutic approach in other similar diseases, the G-quadruplex structures created by extended CANVAS repetitions containing transcripts can also be targeted using small molecules to neutralize the toxicity and thereby for the treatment of the disorder [65,66,67]. Targeting the repeat expanded DNA/RNA using Antisense oligonucleotides and RNA interference (RNAi strategy) represents other attractive therapeutic modalities that can be made to target the upstream portion of the genetic cause, as well as regulating the downstream effects. Both active and passive immunization have been investigated as potential strategies for targeting toxic proteins, and this avenue has also shown promise in addressing other neurodegenerative conditions. CRISPR/Cas9 technology can also be engineered to precisely cleave the specific REs responsible for CANVAS, facilitating gene correction via nonhomologous end joining, suppressing the transcription of repeat-containing RNAs and triggering certain downstream modifications, such as hindering the export of hazardous repeat-containing RNA to the cytosol [68]. These approaches, either individually or collectively can largely target the upstream mechanisms of the disease progression such as; releasing of sequestered RNA Binding proteins (RBPs), repressing RAN (non-AUG mediated) translation, altering of splicing defects, reduction of toxic foci formation and prevention of DNA/RNA hybrid formation, all of which forms the firm basis behind the cause of the disease. Additionally, these strategies can also significantly intervene in various downstream mechanisms, by targeting specific proteins involved, correcting downstream cellular pathways, targeting different nuclear export factors to reduce the nuclear export and targeting SINE sequences from which repeat expansions are derived (Fig. 6). Thus, it is crucial to persistently drive forward research endeavors in this domain, given that the diverse and multifaceted nature of these strategies offers significant potential in effectively preventing the advancement of the disease.

Exploring therapeutics avenues for CANVAS: targeting upstream and downstream mechanisms. The diagram depicts the possible therapeutic strategies with a high potential for targeting the upstream and downstream mechanisms associated with CANVAS. Small molecules, ASO (antisense oligonucleotides), RNAi and CRISPR Cas9 can be used to target different upstream and downstream regulatory functions
Conclusions
CANVAS, a RFC1 mediated autosomal recessive disease, has been fully elucidated only in the last decade or so. It has a polygenic basis and diverse clinical symptoms. Due to the expanded repetitions in the RFC1 gene, CANVAS has emerged as one of the prevailing instances of autosomal recessive ataxias. Elucidating the exact pathogenic mechanism is currently the focus of comprehensive scientific research; however, a few studies point towards stable G quadruplexes formation by specific motifs in RFC1 may influence gene transcription, offering insights into its genetic basis. Initially more common in European communities, recent studies have identified the pathological RFC1 expansion in ataxia cases worldwide, albeit at lower rates. The broad clinical spectrum ranges from pure ataxia to complex features like pyramidal tract issues and cognitive impairment. Additionally, CANVAS presents an enduring chronic cough, dysautonomia, and neurogenic pain. Sensory neuropathy and chronic cough often precede gait instability, broadening our understanding and paving the way for targeted therapies. The aetiology of CANVAS implicates a biallelic intronic repeat expansion within the RFC1 gene on chromosome 4, primarily involving the AAGGG pentanucleotide sequence, leading to genetic disruption. The genetic heterogeneity observed, stemming from variations in repeat sequences, adds complexity to the understanding of this condition. While specific expanded alleles are linked to disease, the intricate molecular mechanisms underlying CANVAS necessitate deeper research. The underdiagnosis of CANVAS, attributed to challenges in recognizing its key clinical indicators, demands a heightened awareness within the medical community. Screening for RFC1 REs, particularly in cases featuring sensory ataxia neuropathy, holds potential for enhancing the diagnostic accuracy. Integrating neuropathy and vestibulopathy as diagnostic criteria shall substantially improve the efficacy of testing by many folds. With advanced genomic technologies being pivotal in enabling comprehensive genetic analysis, targeting G-quadruplex structures through small molecules, RNAi, ASOs, CRISPR/Cas9 technology and conducting thorough familial medical assessments coupled with genetic counselling may further refine diagnostic approaches and pave the way for more effective therapeutic interventions.
Data Availability
The datasets supporting the findings of this study are included in this article.
References
Tankard RM, Bennett MF, Degorski P, Delatycki MB, Lockhart PJ, Bahlo M (2018) Detecting expansions of tandem repeats in cohorts sequenced with short-read sequencing data. Am J Hum Genet 103(6):858–873. https://doi.org/10.1016/j.ajhg.2018.10.015
Depienne C, Mandel JL (2021) 30 years of repeat expansion disorders: what have we learned and what are the remaining challenges? Am J Hum Genet 108(5):764–785. https://doi.org/10.1016/j.ajhg.2021.03.011
Dominik N, Galassi Deforie V, Cortese A, Houlden H (2021) CANVAS: a late onset ataxia due to biallelic intronic AAGGG expansions. J Neurol 268(3):1119–1126. https://doi.org/10.1007/s00415-020-10183-0
Cortese A et al (2020) Cerebellar ataxia, neuropathy, vestibular areflexia syndrome due to RFC1 repeat expansion. Brain 143(2):489–490. https://doi.org/10.1093/brain/awz418
Szmulewicz DJ, Waterston JA, Macdougall HG, Mossman S, Chancellor AM, Mclean CA, Merchant S, Patrikios P et al (2011) Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS): a review of the clinical features and video-oculographic diagnosis. Ann N Y Acad Sci 1233(1):139–147. https://doi.org/10.1111/j.1749-6632.2011.06158.x
Hisey JA, Radchenko EA, Ceschi S, Rastokina A, Nicholas H, Mcginty RJ, Matos-rodrigues G, Hernandez A et al (2023). Pathogenic CANVAS (AAGGG). https://doi.org/10.1101/2023.07.25.550509
Abdi MH, Zamiri B, Pazuki G, Sardari S, Pearson CE (2023) Pathogenic CANVAS-causing but not nonpathogenic RFC1 DNA/RNA repeat motifs form quadruplex or triplex structures. J Biol Chem 299(10):105202. https://doi.org/10.1016/j.jbc.2023.105202
Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, Nakazawa Y, Niimi A et al (2010) Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell 37(5):714–727. https://doi.org/10.1016/j.molcel.2010.02.009
Dominik N, Magri S, Currò R, Abati E, Facchini S, Corbetta M, Macpherson H, Di Bella D et al (2023) Normal and pathogenic variation of RFC1 repeat expansions: implications for clinical diagnosis. Brain. https://doi.org/10.1093/brain/awad240
Tsuchiya M, Nan H, Koh K, Ichinose Y, Gao L, Shimozono K, Hata T, Kim Y-J et al (2020) RFC1 repeat expansion in Japanese patients with late-onset cerebellar ataxia. J Hum Genet 65(12):1143–1147. https://doi.org/10.1038/s10038-020-0807-x
Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yan YW, Humphrey J, Jaunmuktane Z et al (2019) Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Gen 51(4), 649–658. https://doi.org/10.1038/s41588-019-0372-4
Ando M et al (2022) Genetic and clinical features of cerebellar ataxia with RFC1 biallelic repeat expansions in Japan. Front Neurol 13. https://doi.org/10.3389/fneur.2022.952493.
Aboud Syriani D et al (2020) ‘Prevalence of RFC1-mediated spinocerebellar ataxia in a North American ataxia cohort. Neurology: Gene 6(3). https://doi.org/10.1212/NXG.0000000000000440.
Kontogeorgiou Z, Kartanou C, Tsirligkani C, Anagnostou E, Rentzos M, Stefanis L, Karadima G, Koutsis G (2021) Biallelic RFC1 pentanucleotide repeat expansions in Greek patients with late-onset ataxia. Clin Genet 100(1):90–94. https://doi.org/10.1111/cge.13960
Scriba CK et al (2023) BRAIN COMMUNICATIONS RFC1 in an Australasian neurological disease cohort : extending the genetic heterogeneity and implications for diagnostics. Brain Comm 5(4):1–14. https://doi.org/10.1093/braincomms/fcad208
Thieme A, Depienne C, Timmann D (2021) Cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS): from clinical diagnosis towards genetic testing. Med Gen 33(4):301–310. https://doi.org/10.1515/medgen-2021-2098
Szmulewicz DJ, Waterston JA, Halmagyi GM, Mossman S, Chancellor AM, McLean CA, Storey E (2011) Sensory neuropathy as part of the cerebellar ataxia neuropathy vestibular areflexia syndrome. Neurology 76(22):1903–1910. https://doi.org/10.1212/WNL.0b013e31821d746e
Migliaccio AA, Halmagyi GM, McGarvie LA, Cremer PD (2004) Cerebellar ataxia with bilateral vestibulopathy: description of a syndrome and its characteristic clinical sign. Brain 127(2):280–293. https://doi.org/10.1093/brain/awh030
Gebus O, Montaut S, Monga B, Wirth T, Cheraud C, Alves Do Rego C, Zinchenko I, Carré G et al (2017) Deciphering the causes of sporadic late-onset cerebellar ataxias: a prospective study with implications for diagnostic work. J Neurol 264(6), 1118–1126. https://doi.org/10.1007/s00415-017-8500-5
Szmulewicz DJ, McLean CA, MacDougall HG, Roberts L, Storey E, Halmagyi GM (2014) CANVAS an update: clinical presentation, investigation and management. J Vestib Res: Equil Orient 24(5–6):465–474. https://doi.org/10.3233/VES-140536
Krismer F, Wenning GK (2014) Autonomic failure in CANVAS syndrome. Brain 137(10):2625–2626. https://doi.org/10.1093/brain/awu220
Szmulewicz DJ, Roberts L, McLean CA, MacDougall HG, Halmagyi GM, Storey E (2016) Proposed diagnostic criteria for cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Neur Clin Pract 6(1):61–68. https://doi.org/10.1212/CPJ.0000000000000215
Coutinho P, Cruz VT, Tuna A, Silva SE, Guimarães J (2006) Cerebellar ataxia with spasmodic cough: a new form of dominant ataxia. Arch Neurol 63(4):553–555. https://doi.org/10.1001/archneur.63.4.553
Tatineni S, Jarrouj G, Gomez CM, Baird BJ (2023) Characterization of a chronic cough in cerebellar ataxia, neuropathy, vestibular areflexia syndrome. Laryngoscope Investig Oto 8(3):730–738. https://doi.org/10.1002/lio2.1067
Ghorbani F, de Boer-Bergsma J, Verschuuren-Bemelmans CC, Pennings M, de Boer EN, Kremer B, Vanhoutte EK, Vries JJ et al (2022) Prevalence of intronic repeat expansions in RFC1 in Dutch patients with CANVAS and adult-onset ataxia. J Neurol 269(11):6086–6093. https://doi.org/10.1007/s00415-022-11275-9
Gisatulin M, Dobricic V, Zühlke C, Hellenbroich Y, Tadic V, Münchau A, Isenhardt K, Bürk K et al (2020) Clinical spectrum of the pentanucleotide repeat expansion in the RFC1 gene in ataxia syndromes. Neurology 95(21):e2912–e2923. https://doi.org/10.1212/WNL.0000000000010744
Hadjivassiliou M, Currò R, Beauchamp N, Dominik N, Grunewald RA, Shanmugarajah P, Zis P, Hoggard N, Cortese A (2024) Can CANVAS due to RFC1 biallelic expansions present with pure ataxia? J Neurol Neurosurg Psychiatry 95(2):171–174. https://doi.org/10.1136/jnnp-2023-331381
Infante J, García A, Serrano-Cárdenas KM, González-Aguado R, Gazulla J, de Lucas EM, Berciano J (2018) Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS) with chronic cough and preserved muscle stretch reflexes: evidence for selective sparing of afferent Ia fibres. J Neurol 265(6):1454–1462. https://doi.org/10.1007/s00415-018-8872-1
Huin V, Coarelli G, Guemy CCD. sign©men, Boluda S, Debs R, Mochel F, Stojkovic T, Grabli D et al (2022) Motor neuron pathology in CANVAS due to RFC1 expansions. Brain, 145(6), 2121–2132. https://doi.org/10.1093/brain/awab449
Umeh CC, Polydefkis M, Chaudhry V, Zee DS (2017) Sweat gland denervation in cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Mov Disord Clin Pract 4(1):46–48. https://doi.org/10.1002/mdc3.12355
Jo M (2023) Pseudo-eye-of-the-tiger sign in cerebellar ataxia with neuropathy and vestibular areflexia syndrome ( CANVAS ). September, 1–4. https://doi.org/10.1002/ajmg.a.63419
Turner RD, Hirons B, Cortese A, Birring SS (2023) Chronic cough as a genetic neurological disorder? Insights from cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Lung 201(6):511–519. https://doi.org/10.1007/s00408-023-00660-4
Azevedo MPC, Lobo CC, Schmitt GS, Matos PCAAP, Barsottini OGP, Pedroso JL, Marques W Jr, Souza JP et al (2023) Nigrostriatal dysfunction in RFC1-related disorder/CANVAS. Parkinsonism Relat Disord 115:105854. https://doi.org/10.1016/j.parkreldis.2023.105854
da Silva Schmitt G, Martinez ARM, da Graça FF, de Lima FD, Bonadia LC, Amorim BJ, Nucci A, França MC Jr (2020) Dopa-responsive Parkinsonism in a patient with homozygous RFC1 expansions. Mov Disord 35(10):1889–1890. https://doi.org/10.1002/mds.28286. (PMID: 33068476)
Hovaguimian A (2023) Dysautonomia: diagnosis and management. Neurol Clin 41(1):193–213. https://doi.org/10.1016/j.ncl.2022.08.002
El Houjeiry E, Coudray S, Thouvenot E, Ion IM (2023) Spinal cord lesion mimicking a dysimmune myelitis revealing CANVAS syndrome. J Spinal Cord Med 46(2):332–336. https://doi.org/10.1080/10790268.2022.2033936
Szmulewicz DJ, McLean CA, Rodriguez ML, Chancellor AM, Mossman S, Lamont D, Roberts L, Storey E, et al (2014) Dorsal root ganglionopathy is responsible for the sensory impairment in CANVAS. Neurology 82(16), 1410 LP – 1415. https://doi.org/10.1212/WNL.0000000000000352
Dupré M, Hermann R, Froment Tilikete C (2021) Update on cerebellar ataxia with neuropathy and bilateral vestibular areflexia syndrome (CANVAS). Cerebellum 20(5):687–700. https://doi.org/10.1007/s12311-020-01192-w
Ishai R, Seyyedi M, Chancellor AM, McLean CA, Rodriguez ML, Halmagyi GM, Nadol JB, Szmulewicz DJ et al (2021) The pathology of the vestibular system in CANVAS. Otol Neurotol 42(3):E332–E340. https://doi.org/10.1097/MAO.0000000000002985
Pelosi L, Mulroy E, Leadbetter R, Kilfoyle D, Chancellor AM, Mossman S, Wing L, Wu TY et al (2018) Peripheral nerves are pathologically small in cerebellar ataxia neuropathy vestibular areflexia syndrome: a controlled ultrasound study. Eur J Neurol 25(4):659–665. https://doi.org/10.1111/ene.13563
Reyes-Leiva D, Aldecoa I, Gelpi E, Rojas-García R (2022) Motor neuron involvement expands the neuropathological phenotype of late-onset ataxia in RFC1 mutation (CANVAS). Brain Pathol (Zurich, Switzerland) 32(4):e13051. https://doi.org/10.1111/bpa.13051
Manto M, Bower JM, Conforto AB, Delgado-García JM, Da Guarda SNF, Gerwig M, Habas C, Hagura N et al (2012) Consensus paper: roles of the cerebellum in motor control-the diversity of ideas on cerebellar involvement in movement. Cerebellum 11(2):457–487. https://doi.org/10.1007/s12311-011-0331-9
Helmchen C, Kirchhoff JB, Göttlich M, Sprenger A (2017) Postural ataxia in cerebellar downbeat nystagmus: its relation to visual, proprioceptive and vestibular signals and cerebellar atrophy. PLoS ONE 12(1):1–16. https://doi.org/10.1371/journal.pone.0168808
Rinne T, Bronstein AM, Rudge P, Gresty MA, Luxon LM (1998) Bilateral loss of vestibular function: clinical findings in 53 patients. J Neurol 245(6–7):314–321. https://doi.org/10.1007/s004150050225
Vinther-Jensen T, Dunø M, Ingolfsdottir HM, Krarup C, Nielsen JE, Jakobsen JK (2023) Cerebellar ataxia-neuropathy-vestibular areflexia-syndrome. Ugeskr Laeger 185(19):V06220383
Barghigiani M, De Michele G, Tessa A, Fico T, Natale G, Saccà F, Pane C, Cuomo N et al (2022) Screening for RFC-1 pathological expansion in late-onset ataxias: a contribution to the differential diagnosis. J Neurol 269(10):5431–5435. https://doi.org/10.1007/s00415-022-11192-x
Tyagi N, Uppili B, Sharma P et al (2023) Investigation of RFC1 tandem nucleotide repeat locus in diverse neurodegenerative outcomes in an Indian cohort. Neurogenetics. https://doi.org/10.1007/s10048-023-00736-6
Beecroft SJ, Cortese A, Sullivan R, Yau WY, Dyer Z, Wu TY, Mulroy E, Pelosi L et al (2020) A Maori - Specific RFC1 pathogenic repeat configuration in CANVAS, likely due to a founder allele. Brain 143(9):2673–2680. https://doi.org/10.1093/brain/awaa203
Rafehi H, Szmulewicz DJ, Bennett MF, Sobreira NLM, Pope K, Smith KR, Gillies G, Diakumis P et al (2019) Bioinformatics-based identification of expanded repeats: a non-reference intronic pentamer expansion in RFC1 causes CANVAS. Am J Hum Genet 105(1):151–165. https://doi.org/10.1016/j.ajhg.2019.05.016
Arteche-López A, Avila-Fernandez A, Damian A, Soengas-Gonda E, de la Fuente RP, Gómez PR et al (2023) New cerebellar ataxia, neuropathy, vestibular areflexia syndrome cases are caused by the presence of a nonsense variant in compound heterozygosity with the pathogenic repeat expansion in the RFC1 gene. Clin Genet 103(2):236–241. https://doi.org/10.1111/cge.14249
Benkirane M, Da Cunha D, Marelli C, Larrieu L, Renaud M, Varilh J, Pointaux M, Baux D et al (2022) RFC1 nonsense and frameshift variants cause CANVAS: Clues for an unsolved pathophysiology. Brain 145(11):3770–3775. https://doi.org/10.1093/brain/awac280
Schrecker M, Castaneda JC, Devbhandari S, Kumar C, Remus D, Hite RK (2022) Multistep loading of a DNA sliding clamp onto DNA by replication factor C. eLife 11, e78253. https://doi.org/10.7554/eLife.78253
Pradhan S, Gao R, Bush K, Zhang N, Wairkar YP, Sarkar PS (2022) Polyglutamine expansion in huntingtin and mechanism of DNA damage repair defects in Huntington’s disease. Front Cell Neurosci 16(April):1–17. https://doi.org/10.3389/fncel.2022.837576
Shiomi Y, Nishitani H (2017) Control of genome integrity by RFC complexes; conductors of PCNA loading onto and unloading from chromatin during DNA replication. Genes 8(2). https://doi.org/10.3390/genes8020052
Kumari D, Gazy I, Usdin K (2019) Pharmacological reactivation of the silenced FMR1 gene as a targeted therapeutic approach for fragile X syndrome. Brain Sci 9(2):1–18. https://doi.org/10.3390/brainsci9020039
Yang Q, Jiao B, Shen L (2020) The development of C9orf72-related amyotrophic lateral sclerosis and frontotemporal dementia disorders. Front Genet 11(September):1–11. https://doi.org/10.3389/fgene.2020.562758
Kurosaki T, Ashizawa T (2022) The genetic and molecular features of the intronic pentanucleotide repeat expansion in spinocerebellar ataxia type 10. Front Gene 13(September). https://doi.org/10.3389/fgene.2022.936869
Wada T, Doi H, Okubo M, Tada M, Ueda N, Suzuki H, Tominaga W, Koike H et al (2023) RNA foci in two bi-allelic RFC1 expansion carriers. Annals Neurol n/a(n/a). https://doi.org/10.1002/ana.26848
Traschütz A, Cortese A, Reich S, Dominik N, Faber J, Jacobi H, Hartmann AM, Rujescu D (2021) Natural history, phenotypic spectrum, and discriminative features of multisystemic RFC1 disease. Neurology 96(9), e1369–e1382. https://doi.org/10.1212/WNL.0000000000011528
Wang Y, Wang J, Yan Z, Hou J, Wan L, Yi J, Guo P (2023) RFC1 AAGGG pentanucleotide repeats form parallel G-quadruplex : structural implications for aberrant molecular cascades in CANVAS. 1–27. https://doi.org/10.1101/2023.08.06.552146
Kudo K et al (2024) Structural polymorphism of the nucleic acids in pentanucleotide repeats associated with the neurological disorder CANVAS. J Biol Chem 300(4):107138. https://doi.org/10.1016/j.jbc.2024.107138
Akçimen F, Ross JP, Bourassa CV, Liao C, Rochefort D, Gama MTD, Dicaire MJ, Barsottini OG et al (2019) Investigation of the RFC1 Repeat Expansion in a Canadian and a Brazilian ataxia cohort: identification of novel conformations. Front Genet 10(November):1–5. https://doi.org/10.3389/fgene.2019.01219
Borsche M, Tadic V, König IR, Lohmann K, Helmchen C, Brüggemann N (2022) Head impulse testing in bilateral vestibulopathy in patients with genetically defined CANVAS. Brain Behavior 12(6), 3–7. https://doi.org/10.1002/brb3.2546
Verma AK, Khan E, Mishra SK, Kumar A (2022) Small molecule screening discovers compounds that reduce FMRpolyG protein aggregates and splicing defect toxicity in fragile X-associated tremor/ataxia syndrome. Mol Neurobiol 59(3):1992–2007. https://doi.org/10.1007/s12035-021-02697-z
Verma AK, Khan E, Mishra SK, Jain N, Kumar A (2019) Piperine modulates protein mediated toxicity in fragile X-associated tremor/ataxia syndrome through interacting expanded CGG repeat (r(CGG)exp) RNA. ACS Chem Neurosci 10(8):3778–3788. https://doi.org/10.1021/acschemneuro.9b00282
Verma AK, Khan E, Mishra SK, Mishra A, Charlet-Berguerand N, Kumar A (2020) Curcumin regulates the r(CGG)exp RNA hairpin structure and ameliorate defects in fragile X-associated tremor ataxia syndrome. Front Neurosci 14, 295. https://doi.org/10.3389/fnins.2020.00295
Jiang J, Ravits J (2019) Pathogenic mechanisms and therapy development for C9orf72 amyotrophic lateral sclerosis/frontotemporal dementia. Neurotherapeutics 16(4):1115–1132. https://doi.org/10.1007/s13311-019-00797-2
Quartesan I, Vegezzi E, Currò R, Heslegrave A, Pisciotta C, Iruzubieta P, Salvalaggio A, Fernande-Eulate G et al (2023) Serum neurofilament light chain in replication factor complex subunit CANVAS and disease spectrum. movement disorders, August, 1–7. https://doi.org/10.1002/mds.29680
Acknowledgements
SS would like to acknowledge the Ministry of Human Resource Development, Govt. of India, KG to Department of Biotechnology and KS to CSIR-UGC, Govt. of India, New Delhi for their individual fellowships. Figures employed the BioRender program (BioRender.com).
Funding
AK would like to acknowledge SERB, Department of Science & Technology, New Delhi for the grant CRG/2023/002978.
Author information
Authors and Affiliations
Contributions
A.K. conceived the idea. A.K., S.S. and K.G. wrote the draft. K.S. revised the manuscript and figures. A.M. provided the technical inputs. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shukla, S., Gupta, K., Singh, K. et al. An Updated Canvas of the RFC1-mediated CANVAS (Cerebellar Ataxia, Neuropathy and Vestibular Areflexia Syndrome). Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04307-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04307-0