
Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disorder with progressive memory and cognitive loss. Neuroinflammation is a central mechanism involved in the progression of AD. With the disruption of the blood-brain barrier (BBB), peripheral immune cells and inflammatory molecules enter into AD brain. However, the exact relationship between peripheral immune cells and AD remains unknown due to various challenges. This study aimed to investigate the potential causal association between peripheral immune cells and AD by using a two-sample Mendelian randomization (TSMR) analysis. We conducted a TSMR to decipher the causal relationship between AD and 731 types of peripheral immune cell parameters from the TBNK, regulatory T cell (Treg), myeloid cell, monocyte, maturation stages of T cell, dendritic cell (DC), and B cell panels. Various analytical methods were employed, including inverse variance weighting (IVW), MR Egger, and weighted median methods. The Cochran’s Q statistic, MR-Egger intercept, and MR-PRESSO tests were used to verify the heterogeneity and horizontal pleiotropy of the results. To further verify our results, we also conducted a replication analysis. The analysis identified CD33 on CD14 + monocyte (OR = 1.03; 95% CI, 1.01–1.04; p = 1.14E−04; adjust-p = 0.042) had an increased risk association for AD, which was verified by the replication study. CD33 on CD33dim HLA DR + CD11b- cell (OR = 1.02; 95% CI, 1.01–1.04; p = 2.87E−04; adjust-p = 0.035) had an increased risk association for AD, while secreting CD4 regulatory T cell %CD4 regulatory T cell (OR = 0.97; 95% CI, 0.96–0.99; p = 1.90E−04; adjust-p = 0.046) and CD25 on switched memory B cell (OR = 0.95; 95% CI, 0.92–0.98; p = 2.87E−04; adjust-p = 0.042) were discovered to be related to a lower risk of AD. However, the causal effect of these three immune cells on AD was insufficiently validated in the replication analysis. The MR analysis suggests a potential causal relationship between peripheral immune cells and the risk of AD. Further extensive research is needed on the specific role of peripheral immune cells in AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD), the most common type of dementia, is characterized by memory impairment and cognitive decline. AD is a growing global health concern with huge implications for individuals and society; it was estimated to increase to 131.5 million people living with dementia worldwide by 2050 [1]. The features of AD pathology are senile plaques of extracellular amyloid-beta (Aβ) protein, neurofibrillary tangles of intracellular tau protein, and neuron loss. In addition, chronic inflammation also plays a non-negligible role in the pathogenesis of AD [2]. The activation and accumulation of microglial cells around Aβ plaques have long been described and are believed to result in chronic inflammation [2]. The blood-brain barrier (BBB) restricts the entry of immunocompetent cells and peptides into the brain, making the brain an immune-privileged organ usually isolated from autoimmune reactions. The BBB is not an unvarying barrier between the brain and the immune system. Several factors such as multiple microtraumata, microvascular pathologies, and inflammation can change the integrity and permeability of the BBB. Thus, BBB dysfunction represents a possible connection between the immune system and AD [3, 4]. In recent studies, mounting evidence suggested that AD is a systemic disease and BBB is compromised at the early stage [5, 6]. In AD, the activation of brain-resident immune-competent cells and infiltration of activated peripheral immune cells may comprise the brain neuroinflammation [7]. Thus, AD-related inflammation is not restricted to the brain but also exists in the periphery. Since the immune system may not be activated solely in the brain, the peripheral immune system might contribute to the development and progression of AD. In AD, dysfunction in the immune system may be a primary factor, and infiltration of peripheral immune cells has been observed in AD brains. Neutrophil depletion or inhibition of neutrophil transport improved memory impairment in 3×Tg-AD transgenic mice [8]. Anti-NK1.1 antibody consumption of NK cells enhances neurogenesis, reduces neuroinflammation, and improves cognitive impairment in 3×Tg-AD mice [9]. These all suggested that peripheral immune cells had an interaction with the central nervous system in AD. However, studies to detect the role of peripheral immune cells in the AD brain are still at a relatively nascent stage. There are few studies about communication between peripheral immune cells and the pathogenesis of AD.
Mendelian randomization (MR) research performed genetic variants as instrumental variables (IVs) to conclude the genetic causality of exposure and outcome. We performed an MR study using summary statistics from the immunity-wide genome-wide association studies (GWAS) of immune cells and AD of European descent, which can mimic randomized controlled trials (RCTs) and is a robust statistical method [10, 11]. There are few studies about peripheral immune cells on the risk of AD by MR studies. Therefore, in here, an MR study was carried out to assess the causal association between immune cells and AD using the GWAS database which may provide new ideas for the treatment of AD.
Materials and Methods
Study Design
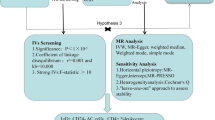
To examine the causal relationships between an extensive range of attributes (a total of 731) about peripheral immune cell parameters and AD, the present investigation employed a two-sample MR approach (TSMR) for our primary and replication analyses [12]. Immune cell parameters that were statistically significant in the primary MR study will be further validated by replication analysis with the same procedure. Within the MR framework, genetic variations were utilized as surrogates for risk factors. For IVs derived from these genetic markers to establish a robust causal inference, three essential criteria needed to be satisfied: (1) a direct correlation between the genetic variations and the exposure variable; (2) the absence of any associations between these genetic variants and potential confounding factors that could potentially influence both the exposure and the outcome; (3) the impact of genetic variations on the outcome being solely mediated through the exposure, thereby ruling out any alternative pathways [13].
GWAS Data Source
Immunity-Wide GWAS Data Sources
The accession numbers of GWAS summary statistics for each immune trait range from GCST0001391 to GCST0002121 [14]. In total, there are 731 immune cell parameters included in the analysis. The immune cells consist of seven distinct types, including the TBNK, regulatory T cell (Treg), myeloid cell, monocyte, maturation stages of T cell, dendritic cell (DC), and B cell panels. The parameters encompass various aspects such as absolute cell (AC) counts (n = 118), median fluorescence intensities (MFI) which reflect surface antigen levels (n = 389), morphological parameters (MP) (n = 32), and relative cell (RC) counts (n = 192). The original GWAS analysis on immune traits involved a database from 3757 individuals of European descent. It is important to note that there was no overlap between the cohorts. Approximately 22 million SNPs genotyped with high-density arrays were imputed with a Sardinian sequence-based reference panel and associations were tested after adjusting for covariates [15].
AD GWAS Data Sources
The primary GWAS summary statistics for AD were acquired from extensive research conducted by Bellenguez et al. based on the GWAS Catalog database [16]. This particular study thoroughly examined a database consisting of 487,511 individuals of European descent, from which 39,106 were cases and 46,828 were controls. To ensure accuracy, quality control measures were applied and missing data were imputed. As a result, the analysis involved a massive number of roughly 20.9 million genetic variants. For the replication analysis, we extracted summary statistics of the AD from the IEU Open GWAS database, including 63,926 European individuals, 21,982 cases, and 41,944 controls, to check the consistency of the findings across different databases [17].
Instrumental Variable Selection
The SNPs associated with immune cell traits at the genome-wide significance level (P < 1 × 10−5) were selected and assigned to distinct immune traits [14, 18]. If an SNP had multiple signals within one trait, the strongest signal was chosen. Then, the SNPs within each trait were clumped to retain only independent SNPs. The threshold for clumping based on linkage disequilibrium (LD) was set at R2 < 0.001, and the clumping window size was set to 10,000 kb [19]. SNPs exhibiting linkage disequilibrium or an F-statistic < 10 were excluded as the selection criteria of instrumental variables in the forward analysis [20].
Statistical Analysis
To evaluate the causal relationship between immune traits and AD, we utilized several methods including inverse variance weighting (IVW) [21, 22], MR-Egger [20, 23], and weighted median methods [24]. We assessed heterogeneity by employing Cochran’s Q method, considering heterogeneity to be significant at p < 0.05 [25]. We utilized the MR-Egger intercept test and the MR-PRESSO global test to identify and remove any potential outliers with horizontal pleiotropic effects that might have significantly influenced the estimation results [23, 26].
To reduce the risk of type 1 errors caused by testing multiple hypotheses, we employed the false discovery rate (FDR) correction to adjust the significance threshold, and the corrected p-value was named adjust-p [27]. Adjust-p < 0.05 was considered the significant association between immune traits and AD.
The software packages “TwoSampleMR” and “MRPRESSO” with R version 4.3.1 were performed to conduct our analyses.
Data Availability
All the data utilized in this study were obtained from the Medical Research Center-Integrative Epidemiology Unit (MRC-IEU) Open GWAS database (https://gwas.mrcieu.ac.uk/). Consequently, ethical approval or consent from patients was not required for the analysis.
Results
Selection for SNPs
For the primary analysis, a total of 728 types of immune cell parameters with 15,000 SNPs were extracted from the 731 traits based on the previously mentioned selection criteria, including 190 kinds of phenotypes from the B cell panel, 63 from the DC panel, 79 from the maturation stages of T cell panel, 43 from the monocyte panel, 63 from the myeloid cell panel, 123 from the TBNK panel, and 167 from the Treg panel. No weak IVs were selected (F-statistic for each SNP > 10). The data above can be viewed in Supplementary Tables Table S1. Detailed information on the selected SNPs for the replication analysis can be viewed in Supplementary Tables Table S7.
MR Analysis of Different Immune Phenotypes on AD
The TSMR analysis revealed that 69 kinds of immune cell parameters were associated with AD. Four types of cells passed the strict FDR test (Figs. 1 and 2), which were defined as significant cells, including CD33 on CD14 + monocyte from the myeloid cell panel, CD33 on CD33dim HLA DR + CD11b- cell from the myeloid cell panel, secreting CD4 regulatory T cell %CD4 regulatory T cell from the Treg panel, and CD25 on switched memory B cell from the B cell panel, whereas the other 65 kinds of cells as suggestively significant cells, which showed no significance after the FDR test (Supplementary Tables Table S4). Through the IVW method analysis (Fig. 1), both of the cells from the myeloid panel, CD33 on CD14 + monocyte (OR = 1.03; 95% CI, 1.01–1.04; p = 1.14E−04; adjust-p = 0.042) and CD33 on CD33dim HLA DR + CD11b- cell (OR = 1.02; 95% CI, 1.01–1.04; p = 2.87E−04; adjust-p = 0.035), showed detrimental effect on AD, while secreting CD4 regulatory T cell %CD4 regulatory T cell (OR = 0.97; 95% CI, 0.96–0.99; p = 1.90E−04; adjust-p = 0.046) and CD25 on switched memory B cell (OR = 0.95; 95% CI, 0.92–0.98; p = 2.87E−04; adjust-p = 0.042) had protective effect on AD. No association with AD was discovered in the four kinds of immune cells by MR Egger, weighted median methods after the FDR test (adjust-p > 0.05).

Forest plot showing the causal relationship between the genetically identified four types of immune cells and AD using the MR analysis by three methods. The p-value is revealed using scientific notation. Adjust-p, p-value that after the FDR test; blue square, OR value by MR Egger or weighted median method; CI, confidence interval; green square, OR value by IVW method; OR, odds ratio. *p < 0.05, **p < 0.001

Scatter plots for the causal association between the four immune cells and AD This image should be in color
In the replication analysis, CD33 on CD14 + monocyte (OR = 1.05; 95% CI, 1.03–1.08; p = 1.41E−04; adjust-p = 0.001) and CD33 on CD33dim HLA DR + CD11b- cell (OR = 1.05; 95% CI, 1.02–1.08; p = 0.003; adjust-p = 0.006) showed a similar significant causal association with AD through the IVW method (Supplementary Tables Table S8; Supplementary Figures Figure S1). However, the causal effects of secreting CD4 regulatory T cell %CD4 regulatory T cell and CD25 on switched memory B cell on AD were not significant in the replication study, whereas their effects on AD were in the same direction as in the primary study in the IVW method (Supplementary Tables Table S8; Supplementary Figures Figure S1). The detailed information on the MR analysis results for the replication study is listed in Supplementary Tables Table S8.
Sensitivity Analyses
The Cochran’s Q statistic, MR-Egger intercept, and MR-PRESSO tests for the four positive cells showed there was no heterogeneity and horizontal pleiotropy in both primary and replication analyses (Supplementary Tables Table S2-S3, Supplementary Tables Table S9-S10). The leave-one-out test demonstrated there was no causal effect caused by a particular SNP in the primary analysis (Fig. 3). For the replication analysis, the leave-one-out plot reveals that there is a potentially influential SNP (rs3865444) driving the causal link between CD33 on CD33dim HLA DR + CD11b- cell and AD, indicating the current finding needs to be interpreted carefully with caution (Supplementary Figure S2 (B)). The results of the other three cell parameters in the leave-one-out test for the replication study show no causal effect caused by a particular SNP (Supplementary Figure S2 (A, C-D)). Detailed information on sensitivity analyses for the suggestively positive cell parameters was listed in Supplementary Tables Table S5-S6.

The leave-one-out test for the four immune cells on AD
Discussion
We employed an MR study to explore the causal potential relationship between peripheral immune cell parameters and AD. We uncovered that the peripheral immune cells could affect AD. Especially, CD33 on CD14 + monocyte and CD33 on CD33 dim Lin-HLADR + CD11b- cell were found to be linked with a higher risk of AD, whereas secreting CD4 regulatory T cell %CD4 regulatory T cell and CD25 on switched memory B cell were discovered might be related with a lower risk of AD. Moreover, MR analyses of these genes were also implemented using these peripheral immune cell-associated SNPs paired on top of the genes, showing a causal connection between these genes and AD, which demonstrates that AD may affected by peripheral immune cells through these genes.
The transmembrane glycoprotein cluster of differentiation 33 (CD33) is expressed on myeloid progenitor cells, mature monocytes, and macrophages and is associated with AD susceptibility [28]. Griciuc et al. have reported that CD33 directly regulates Aβ uptake by microglial cells [29]. CD33 gene is a member of the sialic acid-binding Ig-like lectin family of receptors. Sialic acid activates CD33, stimulates the tyrosine phosphatases SHP1/SHP2, and finally results in the inhibition of phagocytosis, thus alleviating the clearance of Aβ protein deposits [30]. Microglia, the main resident immune cells in the central nervous system, plays a crucial role in the pathogenesis of AD. Monocytes are the counterparts of microglia in peripheral circulation, constituting an important part of the mononuclear phagocyte system [31]. Recent studies have found that blood monocytes in AD patients exhibit impaired Aβ phagocytosis [32, 33]. Enhancing blood monocytes Aβ phagocytosis by improving energy metabolism can alleviate brain Aβ deposition and improve cognitive function in AD mice [34]. In humans, according to the expression levels of CD14 and CD16, monocytes are divided into three subtypes, including the classical subset (CD14 + + CD16−), the intermediate subset (CD14 + + CD16+), and the nonclassical subset (CD14 + CD16++) [35]. The classical monocytes constitute up to 85% of circulating monocytes and differentiate into tissue macrophages, exerting phagocytic function [36]. In AD patients, there was an overall decline in Aβ uptake of all three monocyte subsets. Among the three subsets, the CD14 + CD16 + subset had the highest uptake of Aβ1−42 [32]. DCs share the same precursor cells with mononuclear phagocytes and they are the most potent professional antigen-presenting cells for stimulating immune response. The phenotypes of DCs defined as lineage-negative (Lin-) HLA-DR + cells have become widespread [37]. DCs regulate adaptive immune responses in two ways by modulating the expression of co-stimulatory and inhibitory molecules on DCs, which regulates the initiation of T and B cells by responding to cytokines produced by the pathogen that is largely responsible for determining the type of Th1/Th2/Th3 response [38]. Previous study found that monocyte-derived DCs (moDCs) obtained from AD patients showed more pronounced pro-inflammatory features, reduced antigen-presenting ability, and produced lower levels of brain-derived neurotrophic factor (BDNF) after stimulation with Aβ1−42. These suggest DCs may cause brain damage through mechanisms of over-activation of inflammatory response and Aβ-mediated reduction of neuronal nutritional support in AD conditions [39]. Collectively, these are consistent with our results; a higher expression of CD33 on CD14 + monocyte and CD33 dim Lin-HLADR + CD11b- cells had a higher risk of AD. Importantly, CD33 on CD14 + monocyte was also verified in the replication study. However, in our replication study, rs3865444 in the leave-one-out test shows a strong influence in the causal effect on CD33 on CD33 dim Lin-HLADR + CD11b- cell. Considering rs3865444 showed a significant association with CD33 protein and AD in European [40, 41], and it highly related to CD33 on CD33 dim Lin-HLADR + CD11b- cell with a high F-statistic (p = 2.40E−183, F = 1088.3, Supplementary Tables Table S7), we kept rs3865444 in the leave-one-out result of our replication analysis. We have also performed the MR analysis after eliminating rs3865444; the IVW method revealed no significant association between CD33 on CD33 dim Lin-HLADR + CD11b- cell and AD (OR = 0.99, 95% CI = 0.95–1.03, p = 0.51). Combined with the result in the primary study with a larger number of AD cases, we consider CD33 on CD33 dim Lin-HLADR + CD11b- cell had potential causal effects on AD, which needs to be verified by a database with a larger sample size.
Sustained inflammation secondary to accumulation of Aβ and tau protein is also a main feature of AD. Tregs, a subset of T cells that control inflammatory and immune response, play a critical role in the progression of animal models of AD. Previous studies found that early transient depletion of Tregs in animal models of AD can accelerate the onset of cognitive deficits [42]. In recent, increasing evidence suggests that Tregs have a protective effect on AD. Baek et al. reported that the depletion of Tregs, the deposition of Aβ plaque, and the number of microglia in the hippocampus of the animal models of AD were increased, and the deficiency of spatial learning was aggravated [43]. In addition, stimulation of Tregs proliferation by peripheral interleukin (IL)-2 therapy in animal models of AD can induce recruitment and activation of astrocytes in the hippocampus, increase microglia, improve synaptic plasticity, reduce spine density, and restore cognitive function [42, 44]. Studies have found that the levels of specific B cell subsets were reduced in some AD patients [45]. However, in moderate to severe AD patients, some pro-inflammatory receptors of B cells such as CCR6 and CCR7 have been reported to elevate [46]. Memory B cells (MBC) protect the body from repeated infections by rapidly differentiating into antibody-secreting cells. According to the expression of IgD, MBC can divide into switched and unswitched isotypes [47]. Switched MBCs have the potential for antigen memory and can differentiate into plasma cells upon reactivation [48]. CD25 is a marker of a regulatory B (Breg) cell that presents an immunosuppressive action [49]. The common feature of Bregs is inducing immunosuppression by secreting IL-10, transforming growth factor (TGF)-β, IL-35, and adenosine [50]. In mouse transplantation and tumor microenvironment, Bregs increased the number of Tregs by secreting TGF-β [51]. In a recent study, Feng et al. [52] found that B lymphocytes ameliorate AD-like neuropathology via IL-35. In our study, the primary TSMR analysis revealed that secreting CD4 regulatory T cell %CD4 regulatory T cell and CD25 on switched memory B cell had protective effects on AD, although replication analysis found that the protective effects of these two immune cell parameters on AD were not significant. Given the smaller sample size of AD in the replication analysis and the directions of OR in the IVW method were the same as the primary MR study, we consider secreting CD4 Treg %CD4 Treg and CD25 on switched memory B cells may have potential protective effects on AD.
To the best of our knowledge, our study is the first MR analysis on the association of extensive peripheral immune traits and AD. In this study, SNPs with genome-wide association, independent inheritance, and no LD were carefully selected as IVs to clarify the causal association between the peripheral immune system and AD. Additionally, the F-statistics in our analysis are substantially higher than 10, reflecting a minimal feasibility of weak IV bias. Lastly, the summary data for GWAS involving the peripheral immune system and AD were adjusted for many principal components, thus reducing potential bias.
However, this research does have some limitations. Firstly, the number of genetic loci identified in the peripheral immune system GWAS is limited, which may weaken the IVs and reduce the statistical power of our MR analysis. Secondly, the database mainly consists of European populations, so the universality of our findings to non-European groups is limited. Future studies should validate the applicability of these results across diverse ethnicities and populations. Therefore, it is imperative to conduct further research to build the pathogenesis between the relevant peripheral immune cells and AD conclusively.
Conclusion
The current MR analysis thoroughly evaluated the potential causal association between the peripheral immune system and AD. CD33 on CD14 + monocyte had an increased risk association with AD. CD33 on CD33 dim Lin-HLADR + CD11b- myeloid cells was found be potentially linked with a higher risk of AD, whereas secreting CD4 regulatory T cell %CD4 regulatory T cell and CD25 on switched memory B cell were discovered to have potentially protective effects on AD. Future studies are required to thoroughly investigate the exact mechanism of the peripheral immune cells in AD to explore potential therapeutic targets for AD.
Data Availability
The databases generated and/or analyzed during the current study are not publicly available due to privacy or ethical restrictions, but are available from the corresponding author on reasonable request.
References
Scheltens P, De Strooper B, Kivipelto M et al (2021) Alzheimer’s disease. Lancet 397:1577–1590
Prokop S, Miller KR, Heppner FL (2013) Microglia actions in Alzheimer’s disease. Acta Neuropathol 126:461–477
Sardi F, Fassina L, Venturini L et al (2011) Alzheimer’s disease, autoimmunity and inflammation. The good, the bad and the ugly. Autoimmun Rev 11:149–153
Tejera D, Heneka MT (2016) Microglia in Alzheimer’s disease: the good, the bad and the ugly. Curr Alzheimer Res 13:370–380
Cao W, Zheng H (2018) Peripheral immune system in aging and Alzheimer’s disease. Mol Neurodegener 13:51
Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12:723–738
Bettcher BM, Tansey MG, Dorothée G, Heneka MT (2021) Peripheral and central immune system crosstalk in Alzheimer disease - a research prospectus. Nat Rev Neurol 17:689–701
Zenaro E, Pietronigro E, Della Bianca V et al (2015) Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med 21:880–886
Zhang Y, Fung ITH, Sankar P et al (2020) Depletion of NK cells improves cognitive function in the Alzheimer disease mouse model. J Immunol 205:502–510
Davies NM, Holmes MV, Davey Smith G (2018) Reading mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 362:k601
Bennett DA, Holmes MV (2017) Mendelian randomisation in cardiovascular research: an introduction for clinicians. Heart 103:1400–1407
Pierce BL, Burgess S (2013) Efficient design for mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol 178:1177–1184
Glymour MM, Tchetgen Tchetgen EJ, Robins JM (2012) Credible mendelian randomization studies: approaches for evaluating the instrumental variable assumptions. Am J Epidemiol 175:332–339
Orrù V, Steri M, Sidore C et al (2020) Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat Genet 52:1036–1045
Sidore C, Busonero F, Maschio A et al (2015) Genome sequencing elucidates sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat Genet 47:1272–1281
Bellenguez C, Küçükali F, Jansen IE et al (2022) New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 54:412–436
Kunkle BW, Grenier-Boley B, Sims R et al (2019) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 51:414–430
Wang C, Zhu D, Zhang D et al (2023) Causal role of immune cells in schizophrenia: mendelian randomization (MR) study. BMC Psychiatry 23:590
Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G (2008) Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med 27:1133–1163
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR (2016) Assessing the suitability of summary data for two-sample mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol 45:1961–1974
Burgess S, Butterworth A, Thompson SG (2013) Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 37:658–665
Burgess S, Small DS, Thompson SG (2017) A review of instrumental variable estimators for mendelian randomization. Stat Methods Med Res 26:2333–2355
Bowden J, Davey Smith G, Burgess S (2015) Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 44:512–525
Bowden J, Davey Smith G, Haycock PC, Burgess S (2016) Consistent estimation in mendelian randomization with some Invalid instruments using a weighted median estimator. Genet Epidemiol 40:304–314
Greco MF, Minelli C, Sheehan NA, Thompson JR (2015) Detecting pleiotropy in mendelian randomisation studies with summary data and a continuous outcome. Stat Med 34:2926–2940
Stearns FW (2010) One hundred years of pleiotropy: a retrospective. Genetics 186:767–773
Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100:9440–9445
Malpass K (2013) Alzheimer disease: functional dissection of CD33 locus implicates innate immune response in Alzheimer disease pathology. Nat Rev Neurol 9:360
Griciuc A, Serrano-Pozo A, Parrado AR et al (2013) Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78:631–643
Bhattacherjee A, Jung J, Zia S et al (2021) The CD33 short isoform is a gain-of-function variant that enhances Aβ(1–42) phagocytosis in microglia. Mol Neurodegener 16:19
Nielsen MC, Andersen MN, Møller HJ (2020) Monocyte isolation techniques significantly impact the phenotype of both isolated monocytes and derived macrophages in vitro. Immunology 159:63–74
Chen SH, Tian DY, Shen YY et al (2020) Amyloid-beta uptake by blood monocytes is reduced with ageing and Alzheimer’s disease. Transl Psychiatry 10:423
Fiala M, Lin J, Ringman J et al (2005) Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J Alzheimers Dis 7:221–232 discussion 255–262
Liu ZH, Bai YD, Yu ZY et al (2023) Improving blood monocyte energy metabolism enhances its ability to phagocytose amyloid-β and prevents Alzheimer’s disease-type pathology and cognitive deficits. Neurosci Bull 39:1775–1788
Boyette LB, Macedo C, Hadi K et al (2017) Phenotype, function, and differentiation potential of human monocyte subsets. PLoS ONE 12:e0176460
Turcotte LM, Cao Q, Cooley SA et al (2019) Monocyte subpopulation recovery as predictors of hematopoietic cell transplantation outcomes. Biol Blood Marrow Transpl 25:883–890
MacDonald KP, Munster DJ, Clark GJ, Dzionek A, Schmitz J, Hart DN (2002) Characterization of human blood dendritic cell subsets. Blood 100:4512–4520
Kapsenberg ML (2003) Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol 3:984–993
Bossù P, Spalletta G, Caltagirone C, Ciaramella A (2015) Myeloid dendritic cells are potential players in human neurodegenerative diseases. Front Immunol 6:632
Javor J, Bucová M, Ďurmanová V et al (2022) Alzheimer's disease risk variant rs3865444 in the CD33 Gene: a possible role in susceptibility to multiple sclerosis. Life (Basel) 12(7):1094.
Li X, Shen N, Zhang S et al (2015) CD33 rs3865444 polymorphism contributes to Alzheimer’s disease susceptibility in Chinese, European, and North American populations. Mol Neurobiol 52:414–421
Dansokho C, Ait Ahmed D, Aid S et al (2016) Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain 139:1237–1251
Baek H, Ye M, Kang GH et al (2016) Neuroprotective effects of CD4 + CD25 + Foxp3 + regulatory T cells in a 3xTg-AD Alzheimer’s disease model. Oncotarget 7:69347–69357
Alsuliman A, Appel SH, Beers DR et al (2016) A robust, good manufacturing practice-compliant, clinical-scale procedure to generate regulatory T cells from patients with amyotrophic lateral sclerosis for adoptive cell therapy. Cytotherapy 18:1312–1324
Jiang Q, Jin S, Jiang Y et al (2017) Alzheimer’s disease variants with the genome-wide significance are significantly enriched in immune pathways and active in immune cells. Mol Neurobiol 54:594–600
Bulati M, Buffa S, Martorana A et al (2015) Double negative (IgG + IgD-CD27-) B cells are increased in a cohort of moderate-severe Alzheimer’s disease patients and show a pro-inflammatory trafficking receptor phenotype. J Alzheimers Dis 44:1241–1251
Edholm ES, Bengten E, Wilson M (2011) Insights into the function of IgD. Dev Comp Immunol 35:1309–1316
Kurosaki T, Kometani K, Ise W (2015) Memory B cells. Nat Rev Immunol 15:149–159
Ibrahim EH, Aly MG, Opelz G et al (2021) Higher CD19 + CD25(+) bregs are independently associated with better graft function in renal transplant recipients. BMC Nephrol 22:180
Rosser EC, Mauri C, Regulatory (2015) B cells: origin, phenotype, and function. Immunity 42:607–612
Lee KM, Stott RT, Zhao G et al (2014) TGF-β-producing regulatory B cells induce regulatory T cells and promote transplantation tolerance. Eur J Immunol 44:1728–1736
Feng W, Zhang Y, Ding S et al (2023) B lymphocytes ameliorate Alzheimer's disease-like neuropathology via interleukin-35. Brain Behav Immun 108:16–31
Acknowledgements
The authors thank the many participants and researchers for collecting, contributing to the GWAS dataset, and making their GWAS summary statistics publicly available.
Funding
This research was supported by Zhejiang Provincial Nature Science Foundation of China under Grant No. LQ23H090019.
Author information
Authors and Affiliations
Contributions
Houwen Zhang: present idea, perform MR analysis, and manuscript writing. Fangzheng Chao: manuscript writing and evaluate the quality of Mr. Yu Zhou: manuscript writing and evaluate the quality of Mr. Bin Wu: data collection, figure and table drawing. Chunrong Li: final approvement.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
This study received approval from the institutional review board of the Zhejiang Provincial People’s Hospital. For this type of study, formal consent was not required, and informed consent was waived. All methods were performed in accordance with the relevant guidelines and regulations.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, H., Cao, F., Zhou, Y. et al. Peripheral Immune Cells Contribute to the Pathogenesis of Alzheimer’s Disease. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04266-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04266-6