
Abstract
Hippocampal demyelination in multiple sclerosis (MS) has been linked with cognitive deficits, however, patients could benefit from treatment that induces oligodendroglial cell function and promotes remyelination. We investigated the role of A1 and A2A adenosine receptors (AR) in regulating oligodendrocyte precursor cells (OPCs) and myelinating oligodendrocyte (OL) in the demyelinated hippocampus using the cuprizone model of MS. Spatial learning and memory were assessed in wild type C57BL/6 mice (WT) or C57BL/6 mice with global deletion of A1 (A1AR-/-) or A2A AR (A2AAR-/-) fed standard or cuprizone diet (CD) for four weeks. Histology, immunofluorescence, Western blot and TUNEL assays were performed to evaluate the extent of demyelination and apoptosis in the hippocampus. Deletion of A1 and A2A AR alters spatial learning and memory. In A1AR-/- mice, cuprizone feeding led to severe hippocampal demyelination, A2AAR-/- mice had a significant increase in myelin whereas WT mice had intermediate demyelination. The A1AR-/- CD-fed mice displayed significant astrocytosis and decreased expression of NeuN and MBP, whereas these proteins were increased in the A2AAR-/- CD mice. Furthermore, Olig2 was upregulated in A1AR-/- CD-fed mice compared to WT mice fed the standard diet. TUNEL staining of brain sections revealed a fivefold increase in the hippocampus of A1AR-/- CD-fed mice. Also, WT mice fed CD showed a significant decrease expression of A1 AR. A1 and A2A AR are involved in OPC/OL functions with opposing roles in myelin regulation in the hippocampus. Thus, the neuropathological findings seen in MS may be connected to the depletion of A1 AR.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Extracellular adenosine is a purine nucleoside that is produced through the catabolism of adenosine triphosphate (ATP) via the CD39/CD73 ecto-nucleotidase axis by various cells including glial, neurons, and immune cells [1, 2]. Adenosine is important for modulation of neurotransmitter release [3], synaptic plasticity [4] and neuroprotection in ischemic, hypoxic and oxidative stress events [5]. It also controls T cell proliferation and cytokine production [6, 7]. The nucleoside also stimulates bronchoconstriction and inhibits lipolysis [8]. These effects are mediated through its 4 G-protein-coupled receptors: A1, A2A, A2B, and A3 [7]. These receptors vary in pharmacological profile, effector coupling, and distribution in tissue [2]. Apart from A3 adenosine receptor (AR) which exhibits significant variation between species, other ARs are highly conserved during evolution (i.e., 80–95% sequence homology) [9, 10]. All these AR subtypes are expressed by neurons and glial cells apart from microglia which do not express A2B [11, 12].
Adenosine receptor subtype A1 is the most abundant AR and it is widely expressed throughout the body with the highest expression in the brain [10]. It has been shown that activation of the A1 receptor appears to have the most profound neuroprotective role in the CNS. For example, A1 stimulation protects against neuroinflammation and demyelination in patients with multiple sclerosis and allergic encephalitis [13]. The neuronal A1 receptors also inhibit release of excitatory neurotransmitters and attenuate the propagation of their signaling thus contributing to neuroprotection in MS [14]. Additionally, A1 receptors stimulate neuronal growth factor release from astrocytes thus contributing to tissue repair [15].
High levels of A2 ARs are found in the striatum of the brain, pre- and post-synaptic nerve terminals, immune cells, leukocytes, thymus, and spleen [10, 16]. In mice, A2AR prevents tissue injury caused by inflammation, which represents the anti-inflammatory effect of these receptors [17]. The A2 ARs are subdivided into two subgroups A2A and A2B with high and low affinity for adenosine, respectively [10]. A2A ARs play a critical role in the modulation of inflammatory events in peripheral tissues [18].
The activation of A2A and A2B receptors leads to an increase in intracellular cAMP that has a general inhibitory effect on immune cells. While prevalent evidence suggests that the activation of A2A initiates potent anti-inflammatory responses, the role of this receptor in the CNS remains contentious as its activation is detrimental in some CNS diseases (e.g. cerebral ischemia) but beneficial in others such as lipopolysaccharide-induced meningitis [11]. There is therefore a need to investigate its role in regulating OPC and matured oligodendrocytes in the most common demyelinating disorder of the CNS, multiple sclerosis.
Multiple sclerosis (MS) is a debilitating, neuroinflammatory, and demyelinating disease that affects about 2.5 million people globally [19]. The symptoms caused by demyelination in MS result in long-term sensory changes, visual impairment, and motor dysfunctions leading to irreversible disability [20, 21]. More than 50% of patients with multiple sclerosis present with signs of cognitive dysfunction [22]. This cognitive decline and memory impairment in MS patients have been linked to hippocampal demyelination and atrophy. At present, there is no cure for MS, and available therapies do not improve associated cognitive decline [21, 23]. Previous studies from our lab showed that adenosine receptor (AR) signaling regulates encephalitogenic T-cell migration across the CNS barriers in experimental autoimmune encephalomyelitis (EAE), an animal model for MS [24, 25]. These revealed that ARs expressed on blood–brain barrier (BBB) endothelial cells regulate the migration of inflammatory immune cells into the CNS [24, 26]. It was also shown that antagonism of the A2A AR protected mice from EAE by blocking immune cell entry into the brain [26]. While our findings in EAE were consistent with a direct role for ARs action outside the hematopoietic lineage, we did not know which cells were involved. In the present study, we are interested in investigating how adenosine receptors modulate oligodendrocytes proliferation and maturation following hippocampal demyelination without the confounding superimposed inflammatory responses associated with T cell-mediated immune response; hence we used the cuprizone model.
Cuprizone (bis–cyclohexanone-oxaldihydrazone) is a copper-chelating agent that when fed to mice in chow causes rapid demyelination and gliosis [27-29]. We investigated how the inactivation of adenosine receptor subtypes A1 and A2A modulates behavior, cognition, oligodendrocyte precursor cell (OPC) proliferation, and/or oligodendrocyte (OL) following demyelination of the hippocampus by cuprizone. Our understanding of this may provide insights into a possible pharmacological target for the treatment of multiple sclerosis.
Material and Methods
Experimental Mice and Cuprizone Treatment
All the mice were housed in a sterile facility and maintained following National Institute of Health guidelines for the care and use of laboratory animals. The study was approved by our university's Institutional Animal Care and Use Committee (Protocol # 2008–0092). To examine the contribution of adenosine receptor (subtypes A1 and A2A) to the regulation of demyelination we used transgenic mice that are homozygous for the knockout of the Adora1 gene and Adora2a genes purchased from the Jackson Laboratories. In the A1R KO strain, exon 2 of the endogenous Adora1 gene was replaced by a neomycin resistance cassette eliminating its function. C57/BL6 from our breeding colony was used as wild-type control. Breeding pairs of the transgenic mice were on the C57BL/6 background and purchased from the Jackson Laboratories. To validate the strain of the litter used in our study, we analyzed toe genomic DNA with PCR using the corresponding primers.
Male wildtype C57BL/6 mice (WT), A1 or A2A adenosine receptor knockout mouse lines on C57BL/6 genetic backgrounds aged 8–10 weeks were fed standard or cuprizone diet (0.2% w/w, #TD.150233, Envigo®, WI) in pellets for 4 weeks. Animal weights were monitored throughout the experiment. Control, untreated C57BL/6 mice were maintained on a standard diet for the duration of the experiment.
By day 25 most of the A1 KO cuprizone-treated mice developed severe sickness behavior and emaciation that we had to abort the experiment after 4 weeks of cuprizone feeding.
Behavioral Assessments
Cognitive difficulties are a common feature accompanying neurological manifestations in multiple sclerosis (MS); hence, we explored a range of behavior in WT and transgenic mice after feeding with cuprizone (0.2%) pellets.
Elevated Plus Maze Test
The elevated plus maze (EPM) has (about 100 cm high) four interconnecting arms (two open and two closed arms). Mice were placed at the intersection of the maze with the head facing one of the closed arms. A camera linked to computer software was placed above the EPM to track mouse positioning and movement during the 5-min test as previously described [30]. The time spent and the number of entries in both the open and closed zones of the maze were thereafter analyzed by the Topscan Clever Systems Video Tracking Software.
Barnes Maze Test
The Barnes maze was used to assess spatial memory as documented [31] between days 23–28 after the start of the cuprizone diet. Mice were placed at the center of a plastic circular platform (91 cm in diameter, 75 cm high) containing twelve peripheral circular holes. For each mouse, a target hole was randomly selected, and an escape box (10 × 8.5 × 4 cm) was placed below a particular hole. During the trial, the target hole was randomized but stayed constant for each mouse throughout the duration of the test. The time taken by each mouse to find the escape box (latency to find) and the amount of time taken to enter the escape box (latency to enter) were recorded. The session ended when the mouse entered the escape box or after 5 min. If a mouse did not find the escape hole within 5 min, it was placed into the escape box and left there for at least 1 min. Two trials separated by a 15-min inter-trial interval were performed daily for five consecutive days. The average latencies to find and enter the escape box were used for analysis.
Spatial memory was evaluated on day 28 by removing the escape box and observing the search behavior of the mice for 5 min. The time spent in the target quadrant was recorded. For this analysis, the maze was divided into 4 quadrants; the quadrant with the escape box was designated as the target. The other quadrants going clockwise from the target were labeled: positive, opposite, and negative.
Open Field Test
To assess locomotion and explorative activity we used the open field boxes as described [32]. At the commencement of the test, each mouse was positioned at the center of the box (18″ × 18″ × 18″). The movement of each mouse was captured for 5 min with a USB webcam (Logitech HD-1820p) placed above the arena and a PC-based video capture software. The average speed (ms−1), total distance traveled (m), distance traveled in the periphery and corners of the box were analyzed using the Topscan, Clever Systems Video Tracking Software.
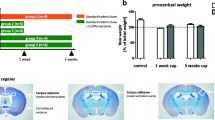
The plan of the behavioral assessments is depicted in Fig.1.

Design of behavioral assessments
Tissue Preparation
The mice were deeply anaesthetized intraperitoneally with ketamine/xylazine (ketamine: 50 mg/kg; xylazine: 10 mg/kg body weight) and perfused by intracardiac perfusion with 20 ml 0.01 M PBS (pH = 7.4) followed by 150 ml 4% paraformaldehyde in 0.1 M PB (pH = 7.4). Thereafter, brains were removed and cryoprotected in 20% sucrose in PBS at 4 °C overnight, then frozen on dry ice. 8 μm thick sagittal sections were cut by cryostat and serially mounted on Superfrost Plus glass slides and stored at − 80 °C until used.
Luxol Fast Blue (LFB) and Luxol Fast Blue–Periodic Acid Schiff (LFB–PAS) Staining
To investigate the extent of myelin staining, brain sections were stained with Luxol Fast Blue (LFB) using 0.1% LFB Solution (Electron Microscopy Sciences #26,681–01) according to the manufacturer’s instructions followed by differentiation in 0.05% aqueous solution of lithium carbonate. We thereafter performed LFB-PAS staining which provides a contrast between myelinated and demyelinated areas. This involved staining of LFB sections with 0.5% solution of periodic acid before transferring into Schiff ‘s reagent (Electron Microscopy Sciences #26,920–04). Sections were dehydrated with graded ethanol and mounted with Permount (Fisher Scientific, Waltham, MA). Whole brain images were scanned using Leica Biosystems Scanscope and regions of the hippocampus and corpus callosum were analyzed.
Nissl Staining
Brain sections (20 µm) were mounted onto glass slides and placed directly into alcohol/chloroform (1:1) overnight and then rehydrated through 100% and 95% alcohol to distilled water. The slides were later stained with 0.1% cresyl violet solution (26,681–02, Electron Microscopy Sciences) for 10 min. Sections were then rinsed with distilled H2O, dehydrated in a series of ascending concentrations of ethanol, and cleared in xylene before cover slipping in Permount.
Terminal Deoxynucleotidyl Transferase DUTP Nick End Labeling (TUNEL) Assay
TUNEL assays were performed as previously described [33] using a commercially available in situ apoptosis detection kit (ApopTag®, S7100; Millipore) to detect apoptotic cells by labeling and detecting DNA strand breaks. Brain sections were incubated with TdTenzyme; color development was done with 0.05% diaminobenzidine and counterstained with 0.5% methyl green. The number of TUNEL-positive cells (TPC) was manually counted in 10 separate fields of view per section at the lesion center at × 100 magnification (n = 5 per group). The average TPC per mm2 brain tissue was thereafter calculated.
Immunofluorescence
Immunofluorescence was performed by fixing (100% cold acetone, 10 min at room temperature) and blocking frozen sections in blocking buffer (2X casein/10% goat serum/0.1% Triton X-100; pH 7.4) at 4 ºC overnight. The sections were incubated with the following primary antibodies: anti-GFAP mouse polyclonal antibody (1:500; #556,330, BD Pharmingen®), anti-MBP rabbit polyclonal antibody (1:500; #PA5-78,397, Invitrogen®), Adenosine A1 receptor mouse monoclonal (1;500, #SC-514337, Santa Cruz®), olig2 (1:500, #MABN50, Millipore®), NeuN mouse monoclonal antibody (1:250, #GTX30773), overnight at 4 ºC; sections with Adenosine A1 receptor antibodies were incubated for 72 h. The sections were rinsed 3 times (5 min each) in PBS and then incubated with secondary antibodies conjugated with Alexa dyes (Invitrogen ™; AF-488, A11001; AF-647, A21245) at room temperature for 1 h. Sections were then washed 3 times (5 min each) followed by the addition of 2 drops of mounting media with DAPI (Invitrogen #P36935) before sections were coverslipped.
Gliosis, Axonal, Microglial/Macrophage, and Apoptosis Quantification
For calculating the area fluorescence of GFAP, NeuN, MBP, and A1AR antibody staining, the slide with representative slices of the entire mouse brain was scanned on a Zeiss fluorescence microscope. Area fluorescence in specific brain regions was calculated after thresholding to eliminate the background and nonspecific staining using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Area fluorescence of GFAP, NeuN, MBP, and A1AR staining was analyzed thereafter, and the parameters (brightness, pinhole, and contrast) were kept constant for all the images. The counting of cells was evaluated by two experimenters blinded to the study group and was expressed as “number of cells/mm2.
Western Blot Assay
Mice brains were dissected and homogenized in ice-cold lysis buffer (25 mM Tris⋅HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, pH 8.0, 5 mM EGTA, pH 8.0, 0.5% Triton X-100, 1% phosphatase inhibitor, 1% protease inhibitor). Protein concentrations were measured using a protein assay kit (RC DC®, Life Science, #5,000,122). Twenty micrograms of total protein extracts were resolved on 12% Bis–Tris acrylamide gels and transferred to a nitrocellulose membrane. MOG primary antibody (1:1,000; Millipore, #MAB5680) and horseradish-conjugated secondary antibody (1:250; Southern Biotech #1070–05) were used. Chemiluminescent signals were detected by using Supersignal® West Pico Luminol/Enhancer Solution (#1,856,136). To normalize the samples for equal loading and protein transfer, we re-probed the nitrocellulose membrane with an antibody against GAPDH.
Statistical Analysis
Data are presented as mean ± Standard Error of Mean. Data were analyzed by one-way analysis of variance (ANOVA) for repeated exposures and subsequently, Tukey post-hoc using Graph pad Prism version 8 (Windows® GraphPad software) for mean comparisons. p-values ≤ 0.05 were considered statistically significant.
Results
Mice Lacking A1 Adenosine Receptors Displayed Sickness Behavior With Emaciation
Although the cuprizone-fed mice lost weight during the experiment, by day 17, the body weight loss in the A1 AR-/- mice became significant (F1,54 = 8.194; p < 0.0001). In addition to the loss of weight, these mice displayed signs of lethargy, showed a lack of interest in their surroundings, and shivered vigorously starting from day 24. We had to abort the experiment at 4 weeks instead of the proposed 6 weeks as the A1AR-/- mice had lost about 16% (16.12 ± 0.63) of their initial body weight by day 28 as recommended by the Institutional Animal Care and Use Committee. Nonetheless, significant weight loss in the WT mice was not evident until day 27 (F 1, 54 = 3.695 p = 0.0005) (Fig. 2).

Daily body weight in comparison to initial body weight in experimental mice. At day 17 after the start of cuprizone diet (CD), A1 AR-/- mice began to significantly lose body weight relative to WT mice on standard and CD chow. Depicted are Mean ± SEM. (n = 8) ***P < 0.05 vs WT SD. Significant weight loss was only observed in the WT CD mice 25 days after the start of CD diet. WT SD: wild type mice fed standard diet: WT CD: wild type mice fed cuprizone diet, A1 AR-/- CD: A1 adenosine receptor knock out mice fed cuprizone diet, A2a AR-/- CD: A2A adenosine receptor knock out mice fed cuprizone diet
A1A Mice Show Anxiogenic Responses and Impairment in Spatial Memory After Cuprizone diet
In the elevated plus maze, similarities were observed in the time spent in the closed arms, however, the significant decrease in number of entries and duration in the open arm (p = 0.01185, 0.0005) without alterations (p = 0.8234 and 0.8754) in the time spent and number of entries in the closed arms in A1AR-/- CD compared to the control mice fed standard pellets (WT SD) is indicative of anxiogenic responses (Fig. 3a-d).

Effect of cuprizone feeding on exploration, anxiety-like behavior, spatial memory and learning. Elevated plus-maze test: (a) Time spent in open arms (b) number of entries into the open arms (c) Time spent in closed arms (d) number of entries into the closed arms. Open-field test: (e) Total distance travelled (f) Distance travelled in the periphery (g) Distance travelled in the center (h) Average velocity during 5-min test periods. (i). Latency to enter escape platform during the probe trial of the Barnes maze test (j) Duration in the zone where the escape platform was previously located. Data are presented as the mean ± standard error. WT SD: wild type mice fed standard diet: WT CD: wild type mice fed cuprizone diet, A1 AR-/- CD: A1 adenosine receptor knock out mice fed cuprizone diet, A2A AR-/- CD: A2A adenosine receptor knock out mice fed cuprizone diet p-values were calculated using the one-way ANOVA, @significant difference compared to the wild type mice on standard diet (WT SD) (p < 0.05); # significant difference compared to the A2A AR-/- mice on cuprizone diet A2A AR-/- CD (p < 0.05)
Additionally, we used the open field test (OFT) as a control to evaluate the general activity levels in the Barnes maze and elevated plus maze. Our data revealed no significant difference in the overall locomotor activity of the mice (as observed by the average travel speed, total distance traveled, and distance traveled in the periphery and center of the open field box) across the experimental groups (Fig. 3e-h).
During the acquisition phase of the Barnes maze test, we observed a decrease in escape latency as the number of trials increases across all groups. However, by the 5th day of the trial, A1AR-/- CD and A2AAR-/- CD showed a significant increase in time to enter the escape box compared to WT SD. Additionally, during the probe trial of the test, all the groups on cuprizone diet (A1AR-/- CD, A2AAR-/- CD, WT CD) spent significantly less time in the zone where the escape box was previously located when compared to the control (WT SD). Statistical analysis by two-way ANOVA revealed that this decrease was significant in WT CD and A1AR-/- CD groups when compared to the WT SD group (Fig. 3i-j).
Variability in the Extent of Demyelination in A1AR-/-, A2AAR-/- Mice and WT Mice on Cuprizone Diet
Assessment of hippocampal demyelination by quantification of Luxol fast blue (LFB) staining showed that the intensity was decreased from 192.45 ± 8.75 AU in the WT-SD group to 43.98 ± 1.03AU in the WT-CD group (p = 0.032). This reduction in LFB intensity was further reduced in the A1AR-/-CD group (23.17 ± 0.78 AU; p = 0.016), whereas the A2AR-/-CD group (247.04 ± 5.19 AU; p = 0.047) was resistant to demyelination compared to WT-CD group (Fig. 4a). We thereafter performed LFB-PAS staining which provided contrast by staining myelinated areas blue and demyelinated areas pink (Fig. 4b). Analogous to results obtained in the LFB staining widespread demyelination was observed in the A1AR-/- CD mice while moderate demyelination was seen in WT CD mice. Quantification of the LFB-stained sections further revealed a significant decrease (p = 0.0136) in the staining intensity in the A1AR-/- fed cuprizone. These data showed a remarkable correlation with results obtained from the Nissl stain (Fig. 4c).

A1 AR-/- mice developed severe demyelination while A2A AR-/- mice exhibit intense myelination at 4 weeks on cuprizone diet. a Luxol Fast Blue staining for detection of myelin content; b Representative images of the LFB-PAS stained hippocampus. Myelin displays as blue stain, and the demyelinated area displays as pink (black arrows). LFB-PAS staining reveals relatively mild demyelination in WT mice in comparison to A1 AR-/- on cuprizone diet while no evidence of demyelination was observed in the A2A AR-/- on cuprizone diet. Note A1 AR-/- cuprizone-fed mice sections show more pink and less blue coloration compared to other groups c Representative overview showing Cresyl violet staining in the hippocampus in the standard diet (SD) or cuprizone diet (CD) fed mice d Quantification of myelin in the hippocampus of mice by pixel count using ImageJ®software (five sections/mice were analyzed). WT SD: wild type mice fed standard diet: WT CD: wild type mice fed cuprizone diet, A1 AR-/- CD: A1 adenosine receptor knock out mice fed cuprizone diet, A2A AR-/- CD: A2A adenosine receptor knock out mice fed cuprizone diet @ signifies p ≤ 0.05 compared to WT SD # signifies p ≤ 0.05 compared to A2a AR-/- CD by one-way ANOVA followed by Tukey post hoc test for comparison of multiple means
We next examined the expression of myelin basic protein (MBP) across all the treatment groups. Our results revealed abundant MBP-positive oligodendrocytes in A2AAR-/- CD mice, but a reduction was observed in the WT and A1AR-/- CD mice when compared to WT SD (Fig. 5). This finding is consistent with the severe demyelination seen in the LFB and LFB-PAS stains in WT CD and A1AR-/- CD mice. Furthermore, we examined the expression of myelin oligodendrocyte glycoprotein (MOG), a protein exclusively expressed at the external surface of oligodendrocyte membranes and myelin. We normalized the band with GAPDH and observed significant decreases in WT CD (0.56, p < 0.0001) and A1AR-/- CD (0.28, p < 0.0001) mice while A2AAR-/- CD mice presented significant increases (1.28, p < 0.0001) in the intensity of the band when compared to WT SD (Fig. 5c).

a Immunofluorescence staining with MBP and Olig2 antibodies: order of demyelination: A2AAR-/- < WT < A1 AR -/- (b) Quantification of expression of MBP and (c) Olig2 immunofluorescence intensity @ denotes statistical significance compared with controls at p < 0.05; # denotes statistical significance compared with A2AAR -/-CD 5 mice/ group. Scale bar: 50 µm (d) Western blot analysis of MOG revealed major bands at approximately 28 kDa (e) the ratio of densities of MOG-specific to GAPDH bands. WT CD: wild type mice fed cuprizone diet, A1 AR-/- CD: A1 adenosine receptor knock out mice fed cuprizone diet, A2AAR-/- CD: A2A adenosine receptor knock out mice fed cuprizone diet @ signifies p ≤ 0.05 compared to WT SD # signifies p ≤ 0.05 compared to A2AAR-/- CD by one-way ANOVA followed by Tukey post hoc test for comparison of multiple means
To verify if oligodendrocytes progenitor cells (OPCs) were still present in the hippocampus, we performed immunofluorescence staining for Olig2 and further quantified its levels of expression. Our results showed a significant (p < 0.01) increase in immature oligodendrocytes in WT CD and A1AR-/ CD compared to WT SD mice (Fig. 5b). This finding suggests that the loss of mature oligodendrocytes triggered a compensatory increased activity of OPCs in a bid to replenish lost myelin sheaths or induce remyelination.
A1 Adenosine Receptor Expression is Linked to NeuN Protein Expression and Could Determine the Extent of Demyelination Following Cuprizone Diet
To examine the role of adenosine receptors in modulating neuronal population following demyelination we performed double immunofluorescence staining using anti-NeuN and anti-A1 AR antibodies. As expected, the A1AR-/- mice did not express A1AR receptor (Fig. 6a), in addition, NeuN-positive neurons were only intermittently seen in the hippocampus and corpus callosum of these mice as against WT SD mice (Fig. 4). Also, we observed a significant downregulation of A1AR with concurrent loss of NeuN protein expression in the hippocampus of WT CD compared to WT SD mice. On the contrary, the expression of A1AR and NeuN in A2AAR-/- CD mice was similar to WT SD (Fig. 5). These outcomes could suggest a direct relationship between A1AR and NeuN expression suggesting that the A1AR receptor is implicated in neuronal nuclei protein expression and that disruption in its signaling could exacerbate demyelination.

a Immunofluorescence double labeling of NeuN and A1 adenosine receptor in the hippocampus showing neuronal loss in the hippocampus of A1 AR -/-and WT cuprizone-fed mice (b) Quantification of expression of NeuN and A1 AR -/- immunofluorescence in the hippocampus, Images were captured from stained frozen sections using a fluorescence microscope equipped with 20 × objectives. Scale bar, 20 µm. WT CD: wild type mice fed cuprizone diet, A1 AR-/- CD: A1 adenosine receptor knock out mice fed cuprizone diet, A2a AR-/- CD: A2A adenosine receptor knock out mice fed cuprizone diet @ signifies p ≤ 0.05 compared to WT SD # signifies p ≤ 0.05 compared to A2a AR-/- CD by one-way ANOVA followed by Tukey post hoc test for comparison of multiple means
Uprizone-Induced Demyelination was Accompanied by astrocytosis and Apoptosis in WT and A1AR -/- Mice
We thereafter investigated if the observed changes in neuronal expression and myelinating oligodendrocytes extend to other glial cells by performing immunofluorescence to quantify GFAP-positive astrocytes. In WT SD mice, GFAP-positive cells were observed with fine processes compared to high-density, hypertrophic GFAP immunolabeled cells with enlarged cell bodies and disorganized process in all the groups fed CD (WT, A1AR-/-and A2AAR-/-mice) (Fig. 7a). The quantitative count also revealed the expression of GFAP was significantly increased in these groups (Fig. 7b).

a Hippocampal astrocytosis in WT, A1 AR-/- and A2A AR-/- mice fed cuprizone diet. b Quantification of GFAP-positive astrocyte cell number in the hippocampus of experimental mice. Few reactive astrocytes were seen in control mice compared to swollen and ramified, (white arrow heads) in WT, A1 AR-/- and A2A AR-/- mice fed cuprizone. Data presented as mean ± SEM n = 5; WT CD: wild type mice fed cuprizone diet, A1 AR-/- CD: A1 adenosine receptor knock out mice fed cuprizone diet, A2a AR-/- CD: A2A adenosine receptor knock out mice fed cuprizone diet @ signifies p ≤ 0.05 compared to WT SD by one-way ANOVA followed by Tukey post hoc test for comparison of multiple means
Using Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay we verified apoptosis in the experimental mice. Quantitative analysis revealed 1.80 ± 0.02, 2.80 ± 0.01, 8.80 ± 0.01, and 3.36 ± 0.15 apoptotic cells per mm2 in WTSD, WTCD, A1AR-/-CD and A2AAR-/-CD groups respectively (Fig. 8). This indicates about a fivefold and twofold increase in apoptotic cells in A1AR -/-CD and A2AAR-/- CD respectively when compared to WT SD group.

(A) TUNEL-positive cells in the hippocampus of control and cuprizone fed diet in WT, A1AR-/- and A2AAR-/- mice. (B) TUNEL-positive cells quantification. Scale Bars = 50 μm. WT CD: wild type mice fed cuprizone diet, A1 AR-/- CD: A1 adenosine receptor knock out mice fed cuprizone diet, A2a AR-/- CD: A2A adenosine receptor knock out mice fed cuprizone diet Data presented as mean ± SEM n = 5; @ significantly different from WT at p < 0.05; # significantly different from WT (CD) A2AAR-/- CD group at p < 0.05
Discussion
Adenosine and adenosine receptors (AR) are involved in the modulation of neuroinflammation in multiple sclerosis and other neurodegenerative diseases. Our lab has previously shown that antagonism of the A2A AR protected mice from experimental autoimmune encephalomyelitis (EAE) by blocking immune cell entry into the brain [24]. Using the EAE model we initially demonstrated a direct role for AR action outside the hematopoietic lineage, however, there remains a need to identify the cells that are involved. In the present study, we set out to study these receptors’ roles in oligodendrocyte precursor cell (OPC) and/or oligodendrocyte (OL) function, in a bid to understand how they contribute to myelin regulation [27, 29]. It has been established that some of the clinical signs in MS are associated with focal hippocampal demyelination as well as disconnection of the hippocampus from several brain networks. The hippocampus likewise contributes to neurogenesis and plasticity, both of which possibly play a role in functional preservation and restoration. These underline the importance of assessing the hippocampus to improve our understanding of the clinical manifestations of multiple sclerosis, as well as a potential target for treatment.
We had intended to feed the mice for 6 weeks at the commencement of our study, but we had to terminate the experiment at 4 weeks when we observed that the A1AR-/- mice had lost a significant percentage of their initial body weight by day 28 with signs of sickness behavior. This unanticipated shorter duration of cuprizone feeding nevertheless has been documented to result in demyelination and has been used extensively to model multiple sclerosis [34-36]. Even shorter duration of 3 weeks cuprizone diet has also been described in the literature to cause acute demyelination [37]. The sickness behavior observed in A1AR-/- mice three weeks after the start of cuprizone diet in our study is similar to what is observed in Alzheimer’s patients [28, 38]. This has been described as a cluster of behavioral and neurological signs that occur when the levels of pro-inflammatory cytokines increase in the brain [39, 40]. Besides, A1AR-/- CD mice exhibited similar severe demyelination in the corpus callosum and hippocampus. Although, this is outside the scope of the present study, we hypothesized that the rapid and extensive demyelination in A1AR-/- mice may have triggered damage/danger-associated signaling molecules that elicited a cytokine response in the CNS that resulted in the altered behavior in the mice.
We investigated spatial learning using the less stressful Barnes maze that showed that cuprizone-fed WT and A1AR-/-mice exhibited longer escape latency and were less accurate in locating the escape platform compared to control and A2AAR-/- groups. The lack of significant differences in motor and locomotor performance in the open field test illustrates clearly that the cognitive deficits displayed in the Barnes maze test were not due to impairment in exploration but rather revealing memory disruption in the cuprizone-fed WT and A1AR-/-.
We monitored demyelination by MBP immunofluorescence, LFB, and LFB-PAS which allowed the assessment of myelin integrity [41]. We focused on the hippocampus because of its prior implication and cognitive decline in MS pathologies [42]. LFB quantification revealed approximately 82% reduction in myelin in A1AR-/- cuprizone-fed mice, a 50% reduction in WT cuprizone-fed mice contrary to the 30% increase seen in A2AAR-/- cuprizone-fed mice. This might be an indication that demyelination induced by the cuprizone diet is amplified in A1AR-/- mice leading to severe demyelination in contrast to the intense myelination observed in A2AAR-/- cuprizone-fed mice while WT cuprizone-fed mice present with an intermediate demyelination phenotype. The hippocampus is crucial for spatial working memory acquisition and studies have shown that it is susceptible to demyelination by cuprizone as seen in the A1AR-/- and WT mice. Conversely, the A2AAR-/- cuprizone-fed mice showed no evidence of myelin depletion in the hippocampus/can withstand cuprizone-induced demyelination demonstrating a role for the A1 adenosine receptor in maintaining myelin integrity despite cuprizone intoxication. Furthermore, the cognitive impairment observed in the A1AR-/- and WT cuprizone-fed mice correlate with demyelination of the hippocampus while the A2AAR-/- showed no such impairment. The correlation between hippocampal myelin status and spatial memory impairment observed in the present study is similar to previous reports [43].
The A1AR receptors immunofluorescence staining confirms the lack of this receptor in the A1 AR -/- mice, we observed that cuprizone feeding in WT mice led to a significant depletion of A1 adenosine receptors while A2AAR-/- CD mice had profuse A1 adenosine receptors. These findings showed that the higher the expression of A1 AR in cuprizone-fed mice, the more resistant they are to demyelination, raising the possibility that A1 AR receptors positively regulate myelin formation in the hippocampus. It also becomes obvious that disruption of A1 AR could be connected to demyelination and failure of remyelination reported in multiple sclerosis (MS) patients. This finding is in line with those of Johnston et al. [44] who observed decreased expression of A1 AR in the peripheral blood of MS patients. In general, these observations demonstrated that cuprizone-induced neuro histopathological changes were significantly more severe in A1AR -/- compared to WT mice.
Olig2 is an important determinant for the specification of neural precursor cells into oligodendrocyte progenitor cells (OPCs) while MBP is absolutely expressed on the surface of myelin sheaths and matured oligodendrocyte processes [45]. It is well understood that OPCs are responsible for remyelinating lesions in the CNS [46] and thus failure of myelin repair has been attributed to deficiencies in the generation of mature oligodendrocytes. However, in the present study, despite the over-expression of OPCs in cuprizone-fed mice there was a deficiency in the generation of mature oligodendrocytes. This finding support claims that despite the presence of endogenous OPCs in multiple sclerosis [20, 47], they often fail to remyelinate axons, suggesting a failure in differentiation [48]. This failure in remyelination is suggestive of the protagonist role of A1 AR in various steps (such as migration, differentiation, and maturation) in the transition of OPCs to mature oligodendrocytes. In agreement, A1 receptor knockout mice develop severe demyelination and oligodendrocyte cytotoxicity as evidenced by increased cuprizone-induced apoptosis. This apoptosis precedes massive demyelination caused by cuprizone administration [49]; no wonder oligodendrocyte degeneration and demyelination were directly proportional to the number of apoptotic cells. Oligodendrocyte increases the speed of nerve conduction; myelin plays a significant role in maintaining the integrity and long-term survival of axons [50]. The myelin proteins play a role in providing this trophic support to neurons and this function appears independent of any role in myelin sheath formation [51]. For example, myelin basic protein (MBP) is an important adhesion molecule on the myelin sheath of the CNS providing structural integrity for neuronal axons [52]. We observed that decreased expression of MBP was accompanied by neuronal loss in WT and A1AR-/- CD-fed mice in line with the findings of Frid et al. [53], who established that prion-like aggregation of MBP following demyelination may contribute to neuronal damage that occurs in MS. Thus, the disruption of myelin as evidenced by a significant reduction in MBP may explain the neuronal loss in the different brain regions in mice in this study.
The observed astrogliosis in our results correlates with the severity of demyelination, this is in congruence with reports of increasing astrocytic expression during acute demyelination and other CNS diseases [54]. Astrocytes become reactive and hypertrophied leading to the upregulation of filament proteins such as vimentin and GFAP [55]. These reactive astrocytes secrete several factors proposed to induce a drastic change in the environment of the demyelinated lesion, which is sensed by OPCs, to induce proliferation and differentiation [56]. Thus, the increased astrocytosis observed in this study could be in a bid to clear myelin debris. Other authors [27], however, argued that in addition to the direct effect of cuprizone on oligodendrocytes, the involvement of the brain’s resident neuroinflammatory cells such as astrocytes is also believed to be responsible for the massive demyelination occurring following cuprizone exposure.
In conclusion, it is evident that deletion of A1 AR in mice exacerbates lesions of MS while A2A knockout mice resisted neurotoxicity/demyelination by CD. This corroborates our earlier reports that the absence of the A2A receptor protected mice from multiple sclerosis-like diseases [24]. This study also demonstrated that the lack of A1 or A2A adenosine receptors in mice altered OPC and/or OL function suggesting their critical role in myelin regulation and OPC/OL function. Concurrently, this trend of events depicts that A1 and A2A adenosine receptors play opposing roles in the maintenance of myelin sheath integrity. We posit that A1 ÁR is a positive regulator of myelin synthesis and future research could be targeted at investigating the possibility of agonists as potential therapies in hippocampal demyelination in multiple sclerosis.
Data Availability
The supporting data of this study are available from the corresponding author upon reasonable request.
References
Stone TW, Ceruti S, Abbracchio MP (2009) Adenosine receptors and neurological disease: neuroprotection and neurodegeneration. Handb Exp Pharmacol 193:535–587. https://doi.org/10.1007/978-3-540-89615-9_17
Lanser AJ, Rezende RM, Rubino S, Lorello PJ, Donnelly DJ, Xu H, Lau LA, Dulla CG, Caldarone BJ, Robson SC, Weiner HL (2017) Disruption of the ATP/adenosine balance in CD39-/- mice is associated with handling-induced seizures. Immunol 152(4):589–601
Sebastião AM, Ribeiro JA (2009) Adenosine receptors and the central nervous system. Handb Exp Pharmacol 193:471–534. https://doi.org/10.1007/978-3-540-89615-9_16
Kerkhofs A, Canas PM, Timmerman AJ, Heistek TS, Real JI, Xavier C, Cunha RA, Mansvelder HD, Ferreira SG (2018) Adenosine A2A Receptors Control Glutamatergic Synaptic Plasticity in Fast Spiking Interneurons of the Prefrontal Cortex. Front Pharmacol 9:133. https://doi.org/10.3389/fphar.2018.00133
Cunha RA (2005) Neuroprotection by adenosine in the brain: From A(1) receptor activation to A (2A) receptor blockade. Purinergic Signal 1(2):111–134. https://doi.org/10.1007/s11302-005-0649-1
Linden J, Cekic C (2012) Regulation of lymphocyte functions by adenosine. Arterioscler Thromb Vasc Biol 32(9):2097–2103. https://doi.org/10.1161/ATVBAHA.111.226837
Kashfi S, Ghaedi K, Baharvand H, Nasr-Esfahani MH, Javan M (2017) A1 Adenosine Receptor Activation Modulates Central Nervous System Development and Repair. Mol Neurobiol 54(10):8128–8139. https://doi.org/10.1007/s12035-016-0292-6
Jacobson KA, Gao ZG (2006) Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 5(3):247–264. https://doi.org/10.1038/nrd1983
Haskó G, Pacher P, Deitch EA, Vizi ES (2007) Shaping of monocyte and macrophage function by adenosine receptors. Pharmacol Ther 113(2):264–275. https://doi.org/10.1016/j.pharmthera.2006.08.003
Sachdeva S, Gupta M (2013) Adenosine and its receptors as therapeutic targets: An overview. Saudi Pharm J 21(3):245–253. https://doi.org/10.1016/j.jsps.2012.05.011
Haskó G, Pacher P, Vizi ES, Illes P (2005) Adenosine receptor signaling in the brain immune system. Trends Pharmacol Sci 26(10):511–516. https://doi.org/10.1016/j.tips.2005.08.004
Abbracchio MP, Ceruti S (2007) P1 receptors and cytokine secretion. Purinergic Signal 3(1–2):13–25. https://doi.org/10.1007/s11302-006-9033-z
Lintermans LL, Stegeman CA, Heeringa P, Abdulahad WH (2014) T cells in vascular inflammatory diseases. Front Immunol 5:504. https://doi.org/10.3389/fimmu.2014.00504
Deuchars SA, Brooke RE, Deuchars J (2001) Adenosine A1 receptors reduce release from excitatory but not inhibitory synaptic inputs onto lateral horn neurons. J Neurosci 21(16):6308–6320. https://doi.org/10.1523/JNEUROSCI.21-16-06308.2001
Ciccarelli R, Di Iorio P, Bruno V, Battaglia G, D’Alimonte I, D’Onofrio M, Nicoletti F, Caciagli F (1999) Activation of A1 adenosine or mGlu3 metabotropic glutamate receptors enhances the release of nerve growth factor and S-100beta protein from cultured astrocytes. Glia 27(3):275–281
Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W (2000) Structure and function of adenosine receptors and their genes. Naunyn-Schmiedeberg’s Arch Pharmacol 362(4–5):364–374. https://doi.org/10.1007/s002100000313
Conigrave AD, Fernando KC, Gu B, Tasevski V, Zhang W, Luttrell BM, Wiley JS (2001) P2Y(11) receptor expression by human lymphocytes: evidence for two cAMP-linked purinoceptors. Eur J Pharmacol 426(3):157–163. https://doi.org/10.1016/s0014-2999(01)01222-5
Varani K, Vincenzi F, Ravani A, Pasquini S, Merighi S, Gessi S, Setti S, Cadossi M, Borea PA, Cadossi R (2017) Adenosine Receptors as a Biological Pathway for the Anti-Inflammatory and Beneficial Effects of Low Frequency Low Energy Pulsed Electromagnetic Fields. Mediators Inflamm 2017:2740963. https://doi.org/10.1155/2017/2740963
DeLuca J, Chiaravalloti ND, Sandroff BM (2020) Treatment and management of cognitive dysfunction in patients with multiple sclerosis. Nat Rev Neurol 16(6):319–332. https://doi.org/10.1038/s41582-020-0355-1
Boyd A, Zhang H, Williams A (2013) Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol 125(6):841–859. https://doi.org/10.1007/s00401-013-1112-y
Grzegorski T, Losy J (2019) Multiple sclerosis-the remarkable story of a baffling disease. Rev Neurosci 30(5):511–526. https://doi.org/10.1515/revneuro-2018-0074
Baltan S, Jawaid SS, Chomyk AM et al (2021) Neuronal hibernation following hippocampal demyelination. Acta Neuropathol Commun 9:34. https://doi.org/10.1186/s40478-021-01130-9
Dubey D, Sguigna P, Stüve O (2016) Managing Disability in Progressive Multiple Sclerosis. Curr Treat Options Neurol 18(6):27. https://doi.org/10.1007/s11940-016-0412-7
Mills JH, Alabanza LM, Mahamed DA, Bynoe MS (2012) Extracellular adenosine signaling induces CX3CL1 expression in the brain to promote experimental autoimmune encephalomyelitis. J Neuroinflammation 9:193. https://doi.org/10.1186/1742-2094-9-193
Mills JH, Thompson LF, Mueller C, Waickman AT, Jalkanen S, Niemela J, Airas L, Bynoe MS (2008) CD73 is required for efficient entry of lymphocytes into the central nervous system during experimental autoimmune encephalomyelitis. PNAS 105(27):9325–9330. https://doi.org/10.1073/pnas.0711175105
Mills JH, Kim DG, Krenz A, Chen JF, Bynoe MS (2012) A2A adenosine receptor signaling in lymphocytes and the central nervous system regulates inflammation during experimental autoimmune encephalomyelitis. J Immunol 188(11):5713–5722. https://doi.org/10.4049/jimmunol.1200545
Praet J, Guglielmetti C, Berneman Z, Van der Linden A, Ponsaerts P (2014) Cellular and molecular neuropathology of the cuprizone mouse model: clinical relevance for multiple sclerosis. Neurosci Biobehav Rev 47:485–505. https://doi.org/10.1016/j.neubiorev.2014.10.004
Bihler K, Kress E, Esser S, Nyamoya S, Tauber SC, Clarner T, Stope MB, Pufe T, Brandenburg LO (2017) Formyl Peptide Receptor 1-Mediated glial cell activation in a mouse model of cuprizone-induced demyelination. J Mol Neurosci 62:232–243. https://doi.org/10.1007/s12031-017-0924-y
Vega-Riquer JM, Mendez-Victoriano G, Morales-Luckie RA, Gonzalez-Perez O (2019) Five Decades of Cuprizone, an Updated Model to Replicate Demyelinating Diseases. Curr Neuropharmacol 17(2):129–141. https://doi.org/10.2174/1570159X15666170717120343
Komada M, Takao K, Miyakawa T (2008) Elevated Plus Maze for Mice. J Vis Exp (22):1088
Ingersoll J, Moody M, Holland R, Kuegler W, Murrah C Jr, Winslow J, Reynolds N, Lloyd N, Dugan B, Hammock M, Houlé K, Clabough E (2018) Analyzing Spatial Learning and Prosocial Behavior in Mice Using the Barnes Maze and Damsel-in-Distress Paradigms. J Vis Exp 17(141). https://doi.org/10.3791/58008
Seibenhener ML, Wooten MC (2015) Use of the open field maze to measure locomotor and anxiety-like behavior in mice. J Vis Exp 96(e52434):1–6. https://doi.org/10.3791/52434
Jang M, Lee MJ, Kim CS, Cho IH (2013) Korean Red Ginseng Extract Attenuates 3-Nitropropionic Acid-Induced Huntington’s-Like Symptoms. Evid Based Complement Alternat Med 2013:237207. https://doi.org/10.1155/2013/237207
Hiremath MM, Saito Y, Knapp GW, Ting JP, Suzuki K, Matsushima GK (1998) Microglial/macrophage accumulation during cuprizone-induced demyelination in C57BL/6 mice. J Neuroimmunol 92(1–2):38–49
Tanaka T, Murakami K, Bando Y, Yoshida S (2013) Minocycline reduces remyelination by suppressing ciliary neurotrophic factor expression after cuprizone-induced demyelination. J Neurochem 127(2):259–270. https://doi.org/10.1111/jnc.12289
Yamate-Morgan H, Lauderdale K, Horeczko J, Merchant U, Tiwari-Woodruff SK (2019) Functional Effects of Cuprizone-Induced Demyelination in the Presence of the mTOR-Inhibitor Rapamycin. Neurosci 406:667–683. https://doi.org/10.1016/j.neuroscience.2019.01.038
Toomey LM, Papini M, Lins B, Wright AJ, Warnock A, McGonigle T, Hellewell S, Bartlett CA, Anyaegbu C, Fitzgerald M (2021) Cuprizone feed formulation influences the extent of demyelinating disease pathology. Sci Rep 11:22594. https://doi.org/10.1038/s41598-021-01963-3
Kelley KW, Bluthé RM, Dantzer R, Zhou JH, Shen WH, Johnson RW, Broussard SR (2003) Cytokine-induced sickness behavior. Brain Behav Immun 17(Suppl 1):S112–S118. https://doi.org/10.1016/s0889-1591(02)00077-6
Perry VH, Cunningham C, Holmes C (2007) Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 7(2):161–167. https://doi.org/10.1038/nri2015
Holmes C, Cunningham C, Zotova E, Culliford D, Perry VH (2011) Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurol 77(3):212–218. https://doi.org/10.1212/WNL.0b013e318225ae07
Moharregh-Khiabani D, Blank A, Skripuletz T, Miller E, Kotsiari A, Gudi V, Stangel M (2010) Effects of fumaric acids on cuprizone induced central nervous system de- and remyelination in the mouse. PloS One 5(7):e11769. https://doi.org/10.1371/journal.pone.0011769
Sicotte NL, Kern KC, Giesser BS, Arshanapalli A, Schultz A, Montag M, Wang H, Bookheimer SY (2008) Regional hippocampal atrophy in multiple sclerosis. Brain 131(4):1134–1141. https://doi.org/10.1093/brain/awn030
Nooraei A, Khazaeel K, Darvishi M, Ghotbeddin Z, Basir Z (2022) Dimorphic evaluation of hippocampal changes in rat model of demyelination: A comparative functional, morphometric, and histological study. Brain Behav 12:e2723. https://doi.org/10.1002/brb3.2723
Johnston JB, Silva C, Gonzalez G, Holden J, Warren KG, Metz LM, Power C (2001) Diminished adenosine A1 receptor expression on macrophages in brain and blood of patients with multiple sclerosis. Ann Neurol 49(5):650–658
Delarasse C, Gaspera B, Lu CW, Lachapelle F, Gelot A, Rodriguez D, Dautigny A, Genain C, Pham-Dinh D (2006) Complex alternative splicing of the myelin oligodendrocyte glycoprotein gene is unique to human and non-human primates. J Neurochem 98(6):1707–1717. https://doi.org/10.1111/j.1471-4159.2006.04053.x
Fernandez-Castaneda A, Gaultier A (2016) Adult oligodendrocyte progenitor cells - Multifaceted regulators of the CNS in health and disease. Brain Behav Immun 57:1–7. https://doi.org/10.1016/j.bbi.2016.01.005
Chang A, Tourtellotte WW, Rudick R, Trapp BD (2002) Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med 346(3):165–173. https://doi.org/10.1056/NEJMoa010994
Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Brück W (2008) Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131(7):1749–1758. https://doi.org/10.1093/brain/awn096
Torkildsen O, Brunborg LA, Myhr KM, Bø L (2008) The cuprizone model for demyelination. Acta Neurol Scand Suppl 188:72–76. https://doi.org/10.1111/j.1600-0404.2008.01036.x
Nave KA, Trapp BD (2008) Axon-glial signaling and the glial support of axon function. Ann Rev Neurosci 31:535–561. https://doi.org/10.1146/annurev.neuro.30.051606.094309
Nave KA (2010) Myelination and the trophic support of long axons. Nat Rev Neurosci 11(4):275–283. https://doi.org/10.1038/nrn2797
Boggs JM (2006) Myelin basic protein: a multifunctional protein. Cell Mol Life Sci 63(17):1945–1961. https://doi.org/10.1007/s00018-006-6094-7
Frid K, Einstein O, Friedman-Levi Y, Binyamin O, Ben-Hur T, Gabizon R (2015) Aggregation of MBP in chronic demyelination. Ann Clin Transl Neurol 2(7):711–721. https://doi.org/10.1002/acn3.207
Gudi V, Moharregh-Khiabani D, Skripuletz T, Koutsoudaki PN, Kotsiari A, Skuljec J, Trebst C, Stangel M (2009) Regional differences between grey and white matter in cuprizone induced demyelination. Brain Res 1283:127–138. https://doi.org/10.1016/j.brainres.2009.06.005
Traiffort E, Kassoussi A, Zahaf A, Laouarem Y (2020) Astrocytes and Microglia as Major Players of Myelin Production in Normal and Pathological Conditions Front. Cell Neurosci 14:79. https://doi.org/10.3389/fncel.2020.00079
Franklin RJM, Ffrench-Constant C (2017) Regenerating CNS myelin-from mechanisms to experimental medicines. Nat Rev Neurosci 18:753–769. https://doi.org/10.1038/nrn.2017.136
Funding
This study received support (for subsistence only) from the US Department of States, Bureau of Educational and Cultural Affairs via the Fulbright Foreign Program awarded to OEA for a Research Visit to Cornell University.
Author information
Authors and Affiliations
Contributions
OEA carried out the experiment, analyzed the data, drafted the manuscript, and designed the figures. MSB conceived the original idea and supervised the experiments. All authors discussed the results and contributed to the final manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
We have no conflict of interest to declare.
Ethics Approval
All the mice were housed in a sterile facility and maintained following National Institute of Health guidelines for care and use of laboratory animals. The study was approved by the Institutional Animal Care and Use Committee of our university (Protocol # 2008–0092).
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Adebiyi, O.E., Bynoe, M.S. Roles of Adenosine Receptor (subtypes A1 and A2A) in Cuprizone-Induced Hippocampal Demyelination. Mol Neurobiol 60, 5878–5890 (2023). https://doi.org/10.1007/s12035-023-03440-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03440-6