
Abstract
The adult neocortex is a six-layered structure, consisting of nearly continuous layers of neurons that are generated in a temporally strictly coordinated order. During development, cortical neurons originating from the ventricular zone migrate toward the Reelin-containing marginal zone in an inside-out arrangement. Focal adhesion kinase (FAK), one tyrosine kinase localizing to focal adhesions, has been shown to be phosphorylated at tyrosine 925 (Y925) by Src, an important downstream molecule of Reelin signaling. Up to date, the precise molecular mechanisms of FAK and its phosphorylation at Y925 during neuronal migration are still unclear. Combining in utero electroporation with immunohistochemistry and live imaging, we examined the function of FAK in regulating neuronal migration. We show that phosphorylated FAK is colocalized with Reelin positive Cajal-Retzius cells in the developing neocortex and hippocampus. Phosphorylation of FAK at Y925 is significantly reduced in reeler mice. Overexpression and dephosphorylation of FAK impair locomotion and translocation, resulting in migration inhibition and dislocation of both late-born and early-born neurons. These migration defects are highly correlated to the function of FAK in regulating cofilin phosphorylation and N-Cadherin expression, both are involved in Reelin signaling pathway. Thus, fine-tuned phosphorylation of focal adhesion kinase at Y925 is crucial for both glia-dependent and independent neuronal migration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The adult mammalian cerebral cortex consists of six layers that are formed by the precisely coordinated proliferation, migration and lamination of cortical neurons during development, thereby providing a basis for the establishment of functional neuronal connectivity [1]. These neurons originate from radial glia cells in the ventricular zone (VZ) via asymmetric proliferation, migrate toward the pial surface and finally localize their cell bodies in their final destination and form the six aligned layers of the cortical plate (CP) [2,3,4,5].
The neurons of layer VI are established first and layers II–V are subsequently formed in an inside-out manner. Two distinct forms of cell movement: locomotion and translocation, are responsible for the migration of cortical neurons [6]. During the early neocortical development, early-born neurons (neurogenesis peaked at embryonic day (E)12.5) destined to form deep layers migrate in radial glia-independent manner [7], which means they extend leading processes to the superficial marginal zone (MZ) and then shorten their leading processes to lift their cell bodies. This migration model is called somal translocation. Migration of late-born neurons (neurogenesis peaked at E14.5) is more complex, consisting of multipolar migration, morphology transformation, glia-dependent locomotion and terminal translocation [7,8,9]. When the leading process of locomoting neurons reaches MZ, they switch to terminal translocation mode, in which the somata move rapidly in a radial glia-independent manner [10]. Even though early-born and late-born neurons apply different migrating strategies, precisely regulated adhesion and cytoskeleton dynamics are involved in controlling lamination [11,12,13]. Impairment in any of these processes has been related to cortical malformation and neurological disorders, which shows life-long effects on daily life and cognitive function of human beings, as well as of mice [14,15,16,17]. Therefore, investigation on the molecular mechanisms underlying cortical lamination, although fairly well established, is still the subject of ongoing refinements.
Among all the known signaling cascades, the Reelin signaling pathway is one of the most studied. Reelin, an extracellular matrix protein, is crucial for controlling layer formation of brain structures, such as the neocortex, hippocampus and cerebellum [18,19,20,21]. Reeler mice lacking Reelin expression, mutants deficient in Reelin signaling molecules as well, show severe defects of neuronal migration and layer lamination, as a consequence of which, dampened learning and memory and cognitive and behavioral abnormalities [22,23,24,25,26,27]. It has been shown that α3β1 integrins bind Reelin and control neuronal migration [28]. Moreover, treatment of cortical tissue with Reelin induces phosphorylation of actin depolymerizing protein Cofilin and proto-oncogene tyrosine-protein kinase (Src), resulting in stabilization of the actin cytoskeleton [18]. The involvement of Cofilin and Src in Reelin signaling and in neuronal migration was experimentally confirmed by our subsequent studies [29, 30]. Focal adhesion kinase (FAK), one tyrosine kinase localizing to focal adhesions, has been shown to be a downstream of Src. FAK expression is related to cell motility, thus elevated FAK activity in human cancers promotes cell invasion through matrix and tissue barriers [31]. By contrast, FAK-deficient cells have more focal adhesions, spread slower than control cells and respond poorly to multiple cues [32]. It was shown that phosphorylation of FAK by Cdk5 at serine 732 is critical for microtubule organization, nuclear movement and neuronal migration [33]. Consistently, conditional FAK knockdown and ablation in migrating cells display abnormal morphology and defective turnover of Connexin 26, suggesting that FAK is required for radial glia-dependent neuronal migration [34]. To be noted, only a minor change in FAK tyrosine 397 phosphorylation was seen in response to Src overexpression, but strong Src-dependent phosphorylation of FAK at tyrosine 925 (Y925) [30]. Recently, impairment of neuronal migration by overexpressing FAK or a mutated form of FAK was reported during corticogenesis [35].
Despite this wealth of studies, it has remained unclear how FAK and its phosphorylation at Y925 are involved in neuronal migration and whether the function of FAK is regulated by Reelin. Here, we address these knowledge gaps and investigate the role of FAK in neuronal migration. The present results show that FAK phosphorylation at Y925 is highly related to Reelin in both the neocortex and hippocampus. The migration of both late-born and early-born cortical neurons is inhibited by overexpressing FAK and its dominant-negative mutant. We found that FAK controls the speed of neuronal locomotion within the CP and the final migration step: terminal translocation and somal translocation of late-born and early-born neurons, respectively. Furthermore, our findings suggest that regulation of these processes by FAK is mediated at the level of actin and cell-cell adhesion, possibly through regulation of activity of cofilin and expression of N-Cadherin through phosphorylation of FAK by Reelin.
Materials and Methods
Animals
All experiments were performed in compliance with the German laws and the guidelines of the European Community for the use of animals in research and were approved by the local ethical committee (48/13). Reeler (B6C3Fe-a/a-Relnrl) were obtained from Dr. Joachim Herz (University of Texas Southwestern, Dallas, USA) and bred in the University Medical Center Hamburg-Eppendorf animal facility. Timed-pregnant wild-type C57BL/6 J mice from the animal facility and reeler mice were housed individually at a 12 h light/12 h dark cycle and were given access to water and food ad libitum. The day of vaginal plug detection was considered E0.5, the day of birth was considered postnatal day (P) 0. All experiments were performed on pups of both sexes.
Plasmid Construction
Plasmid pCAG-GFP, a mammalian expression green fluorescent protein (GFP) vector driven by the chicken actin promoter (Addgene, plasmid 11,150), was used as a control (CON). pCAG-GFP-FAK (FAK-WT) encoding mouse FAK (GenBank: M95408.1) was constructed by reverse transcription PCR (RT-PCR) and In-Fusion (Takara, Japan). The used primers were designed as follows (S, sense; A, anti-sense; underlines are the sites of restriction enzyme): (S)5′-GCAAAGAATTGAATTCGCCACCATGGCAGCTGCTTATCTTGACCC-3′; (A)5′-TGCTCGAGGCAAGCTTGCGTGTGGCCGTGTCTGCCCTAGC-3′. A dominant-negative mutant pCAG-FAK (Y925F)-GFP (FAK-DN) mimics the dephosphorylated form by replacing tyrosine 925 into phenylalanine was generated by overlap PCR. The mutant primers were designed as follows: (S) 5′-GACAAGGTATTTGAGAATGTGAC-3′; (A) 5′-GTCACATTCTCAAATACCTTGTC-3′. Underlines are the sites of the gene mutant. The constructs were further tested by sequence analysis and enzyme restriction. The resulting plasmids were then purified using an Endo-free maxi prep kit from Qiagen.
Cell Culture and Transfection
The neuro-2a mouse neuroblastoma cell line (ATCC, CCL-131) were cultured as monolayer cultures in Dulbecco’s modified minimal essential medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS, Life Technologies), penicillin (100 units/ml) and streptomycin (100 mg/ml; Life Technologies) in a humidified atmosphere containing 95% air and 5% CO2 at 37 °C. When used for western blotting, cells were seeded in 60-mm cell/tissue culture dishes 36 h before transfection. Cells were transfected with liposome (Merck) according to the manufacturer’s instructions transfection. For CON and the expression of FAK and its mutant, cells were transfected at a similar dose and transfection conditions.
Western Blot Analysis
Total lysates were obtained from 48-h transfected neuro-2a cells or the cortex tissue of the brains of P0. Transfected cells were washed with 2 ml of ice-cold phosphate buffered saline (PBS) and harvested with a cell scraper. For the preparation of brain tissues, hemispheres of newborn pups were isolated in ice-cold PBS and the tissues were chopped and resuspended in ice-cold PBS. The harvested cells or tissues were centrifuged at 2300 rpm for 5 min and lysed in RIPA lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 2 mM NaF, 5 mM EDTA and 1 mM sodium orthovanadate. The protease inhibitor phenylmethylsulfonyl fluoride (PMSF, 1 mM) and phosphatase inhibitor Cocktail-1 (FIVEphoton Biochemicals) were added to the RIPA buffer in advance. After 1-h incubation on ice, the lysates were centrifuged at 10,000 rpm for 10 min at 4 °C to remove cell debris. The supernatants were mixed with SDS sample buffer (50 mM Tris–HCl (pH 6.8), 2% SDS, 10% glycerol, 100 mM dithiothreitol, or 5% 2-mercaptoethanol and bromophenol blue). Protein concentrations were measured and equal amounts were loaded in each condition. The extracted protein was electrophoresed in 12% SDS-PAGE gels and transferred to 0.45-μm polyvinylidene fluoride membranes (PVDF, Millipore). The PVDF membranes were blocked in TBST buffer (20-mM Tris–HCl (pH 8.0), 150-mM NaCl and 0.05% Tween 20) containing 5% w/v skimmed milk proteins for 3 h at room temperature (RT) and then incubated with primary antibodies (rabbit polyclonal phospho-FAK (Y925) antibody (1:1000, Cell Signaling Technology), rabbit anti-phospho-cofilin Ser3 (1:1000, Santa Cruz), rabbit anti-Actin (1:5000, Millipore), followed by treatment with horseradish peroxidase (HRP)-conjugated secondary antibodies for 2 h at RT. After washing three times with TBST, targeted proteins were detected using Enhanced Chemi-luminescence (ECL) Plus Western blotting detection reagents (Pierce) according to the manufacturer’s instruction. At least three Western blots were analyzed for each experiment. The stained bands were scanned and the relative optical densities were measured for quantization of the relative expression levels of each protein using Fiji analysis software (National Institutes of Health).
In Utero Electroporation (IUE)
Timed-pregnant mice (E12.5 for target early-born neurons or E14.5 for targeting late-born neurons) were injected subcutaneously with buprenorphine (0.05 mg/kg body weight) 30 min before surgery. Surgery was performed on a heating blanket, toe pinch and breathing were monitored throughout. Under isoflurane anesthesia (induction: 5%, maintenance: 3.5%), the eyes of the dam were covered with eye ointment to prevent damage before dehairing and disinfecting the abdomen with iodine tincture. A, ~ 3-cm midline laparotomy was performed and the uterine horns were exposed and moistened with warm sterile phosphate buffered saline (PBS, 37 °C, Ph 7.4). Solution containing 1.0 µg/µl pCAG-GFP (CON), pCAG-FAK-GFP (FAK-WT) or dominant-negative pCAG-FAK (Y925F)-GFP (FAK-DN) and 0.1% fast green dye at a volume of 0.75–1.25 µl was injected into the right lateral ventricle of individual embryos using pulled borosilicate glass capillaries (GB100TF-10; Science Products, Hofheim, Germany), with a sharp and long tip. Plasmid DNA was purified with NucleoBond (Macherey-Nagel, Germany). IUE was performed as described previously [29, 30, 36]. Each embryo within the uterus was placed between the electroporation tweezer-type paddles (5-mm diameter, Protech, TX, USA). Electrode pulses (30 V, 50 ms) were applied six times at intervals of 950 ms controlled by an electroporator (ECM 830 BTX; Harvard Apparatus). Uterine horns were placed back into the abdominal cavity after electroporation. The abdominal cavity was filled with warm sterile PBS and abdominal muscles and skin were sutured individually with absorbable and non-absorbable suture thread, respectively. After recovery, pregnant mice were returned to their home cages, which were half placed on a heating blanket for 2 days after surgery and received on a daily basis additional wet food supplemented with 2–4 drops Metacam (0.5 mg/ml, Boehringer-Ingelheim, Germany). Dams were sacrificed and the brains of the embryos were used for the preparation of slice cultures (see below). Alternatively, 3, 5, or 7 days after IUE, brains were fixed in 4% paraformaldehyde (PFA) and sectioned coronally into 50-μm-thick sections.
Preparation of Embryonic Cortical Slice Cultures
Three days after electroporation, pregnant mice at E17.5 were decapitated under hypothermic anesthesia. The embryos at E17.5 (CON, n = 6; FAK-WT, n = 6; FAK-DN, n = 8) were collected and rapidly placed in ice-cold Hank’s balanced salt solution (HBSS; Invitrogen). The brains were dissected from the skull and checked under a fluorescence microscope. GFP-positive cerebral cortices were then dissected and sectioned (300 μm) perpendicular to the longitudinal axis of the cerebral cortex using a McIlwain tissue chopper. The slices were placed onto culture inserts (Millipore) and transferred to 6-well plates with 1 ml/well nutrition medium containing 25% heat-inactivated horse serum, 25% Hank’s balanced salt solution, 50% minimal essential medium and 2-mM glutamine (pH 7.2; Invitrogen) and incubated in 5% CO2 at 37 °C for at least 3 h. After recovery, the culture inserts were placed in Petri dishes (30-mm diameter) with a glass-bottom containing fresh medium and then transferred for live imaging.
Live Imaging of Slice Cultures
For live imaging of slice cultures, an Improvision confocal spinning-disc microscope (Zeiss) and 20 × air immersion objective was used to acquire z-series of cortical slices at 5-μm intervals through a tissue depth of about 20 μm. The z-series were then visualized as single optical scans with concurrent orthogonal views using Volocity6 software (Perkin Elmer). The time interval was 10 min. Duration of imaging was up to 15 h. This microscope was equipped with a chamber for the control of temperature, humidity and CO2. Results of time-lapse imaging of the slice cultures were analyzed using Volocity6 software and the average speed of neuronal cell migration was measured by using Imaris software. Measurements from 220 GFP-expressing cells were collected from three videos of each different plasmid transfection and stage (3 days after IUE). Only neurons whose migration could be tracked for a substantial distance (> 20 μm) were included. In these cells, the average speed of movement and the direction of migration (toward the marginal zone or ventricular zone) were measured in the x- and y-axes.
Histology and Immunohistochemistry and Imaging
Brains were prepared for histological analysis on 3, 5, and 7 days after IUE or from non-treated pregnant dams or newborn pups at the desired date. Pregnant wild-type mice were anesthetized with isoflurane and were decapitated under hypothermic anesthesia and the embryos were dissected in ice-cold HBSS. Five days after IUE, corresponding to the day of birth or 7 days after IUE, newborn mice were sacrificed by cervical dislocation under hypothermia and the brains were dissected and rapidly placed in HBSS. Brains after IUE were viewed under a fluorescence microscope to verify transfection. Brains were fixed in 4% PFA overnight at 4 °C.
After washing in 0.1-M PBS at RT for several hours, brains were embedded in 5% agar and cut transversally into 50-μm-thick slices on a Leica VT 1000S vibratome (Leica Microsystems) and the sections were placed in a 24-well plate containing 0.1 M PBS. For immunohistochemistry, free floating slices were permeabilized and blocked with PBS containing 0.3% Triton X-100 (Sigma Aldrich), 5% normal bovine serum albumin (BSA, Jackson ImmunoResearch). Subsequently, slices were incubated overnight with mouse monoclonal anti-Reelin G10 (1:1000, Millipore), rabbit polyclonal anti-phospho-FAK (Y925) (1:1000, Cell Signaling Technology), mouse monoclonal anti-NeuN (1:1000, Millipore) or rabbit monoclonal anti-Brn2 (1:1000, Cell Signaling Technology) diluted in PBS containing 0.03% Triton X-100, 0.5% BSA and 1% normal goat serum (NGS, Sigma), followed by 2-h incubation with secondary antibody Alexa Fluor-568 goat anti-mouse (1:500, Invitrogen), Alexa Fluor-488 goat anti-rabbit (1:500, Invitrogen), Alexa Fluor-568 donkey anti-mouse (1:500, Invitrogen), Alexa Fluor-647 donkey anti-rabbit (1:500, Invitrogen). DAPI (1:5000) was added to the second antibody for the nuclear labeling. Finally, slices were transferred to glass slides and covered with Vecta-Shield (Vector Laboratories). Sections were photographed using a confocal laser-scanning microscope (Leica TCS SP5) and 20 × air or 63 × oil immersion objectives. z-series of brain sections at 0.5-μm intervals through a tissue depth of 9 μm were acquired and visualized as single optical scans with concurrent orthogonal views using Leica LAS software (Leica).
Data Analysis
Of the embryos transfected with the different constructs, at least three brains per different construct and stage were used. Statistical analyses were performed in Graphpad Prism environment. Student’s t-tests were used to determine the statistical significance of differences between the two groups. Comparisons among multiple groups were evaluated by one-way analysis of variance (ANOVA) followed by Bonferroni-corrected post hoc analysis. Data are presented as mean ± SEM. Significance levels of p < 0.05 (*,#), p < 0.01 (**,##) or p < 0.001 (***,###) were used.
Results
Phosphorylation of FAK at Y925 is Related to Reelin Expression
FAK is involved in cell motility through regulating different molecules and plays a critical role in corticogenesis [33,34,35]. However, the function of FAK, especially its phosphorylation at Y925 (p-FAK925) during neuronal migration in vivo and the underlying molecular mechanism is still poorly understood.
To close this knowledge gap, we first performed immunostaining in the developing brain (Fig. 1A, B). FAK925 was highly phosphorylated in both neocortex and hippocampal formation at E17.5 (Fig. 1A). p-FAK925 did not show layer preference but was ubiquitously distributed throughout the cortical plate (CP) and subareas of the hippocampus. Exceptions were marginal zone (MZ) of the neocortex, the MZ of the dentate gyrus (DG), the prospective outer molecular layer (oml) of the DG, where populate Reelin-positive Cajal-Retzius cells (CRs). Intense immunostaining against p-FAK highly overlapped with Reelin positive CRs. A similar expression profile was noticed in neocortical and hippocampal slices from P0 mice (Fig. 1B). Reelin immunostaining was found in the cortical plate, however, phosphorylation of FAK was not as strong as in CRs (Fig. 1B, circles). This might be related to the much weaker expression level of Reelin in these cells. In addition, we performed Western blots of p-FAK925 in cortical lysates prepared from P0 wild-type mice (rl+/+) or their reeler (rl−/−) littermates (Fig. 1C). Densitometric analysis from three independent experiments showed that the protein level of p-FAK925 was significantly downregulated in reeler mutant mice (Fig. 1D, rl+/+: 1.00 ± 0.0, rl−/−: 0.66 ± 0.038, p < 0.0001). Our observations suggest that FAK phosphorylation at Y925 might be highly related to the Reelin signaling pathway, which is one of the most important signaling pathways regulating neuronal migration.

FAK925 was highly phosphorylated in Reelin-positive Cajal-Retzius cells in the marginal zone of developing neocortex and hippocampus. A, Representative fluorescent images of phosphorylated focal adhesion kinase (p-FAK925, green) in neocortex (up row) and hippocampus (bottom row) slices from E17.5 mice when co-stained with Reelin (red) and DAPI (blue). Insets, colocalization of p-FAK and Reelin (arrows) shown at higher magnification. Dotted lines mark area borders. Scale bar, 100 μm. MZ: marginal zone, CP: cortical plate, IZ: intermediate zone, VZ: ventricular zone, CA1: Cornu Ammonis 1, DG: dentate gyrus, H: hilus, ML: molecular layer. B, Same as A, for P0 mice. Circles: Reelin positive cells in the cortical plate. Scale bar, 50 μm. C, Western blotting for p-FAK925 expression in cortical lysates from P0 wild-type mice (rl+/+) or their reeler (rl−/−) littermates. Actin was used as a loading control. D, Densitometric analysis of relative p-FAK925 expression level (normalized to Actin) from three independent experiments. Data are presented as mean ± SEM. Student’s t-test, ***p < 0.001
FAK Disrupts the Radial Neuronal Migration and Reduces the Migratory Speed of Late-Born Neurons
To further gain insight into the function of FAK during neuronal migration, plasmids overexpressing wild-type FAK (FAK-WT) and a dominant-negative mutant (FAK-DN) by changing tyrosine 925 into phenylalanine were constructed (Fig. 2A). These plasmids were transfected into cultured Neuro-2a cells, exogenous p-FAK was detected by immunostaining after 24 h. The elevated phosphorylation of FAK was only seen in the FAK-WT expressing cells but not in GFP (CON) and FAK-DN expressing cells (Fig. 2B). In line with the nature distribution of FAK, GFP-labeling was only detected in the cytoplasm and neruite after transfection of FAK-WT and FAK-DN. Neuro-2a cells were harvested 48 h after transfection for Western blot. Strong endogenous p-FAK was found in FAK-WT lysate (Fig. 2C). These results indicate FAK and the mutated form of FAK were expressed and functioned.

Construction and verification of wild-type and dominant-negative FAK plasmids. A, Schematic diagrams of FAK function motifs and the mutated amino acid (Phe925). All constructs express a fusion protein of FAK or its mutants and GFP. B, Representative images of neuro-2a cells after 24 h transfection with different constructs. GFP (green) expression was detected in the whole cells transfected with control plasmid expressing GFP (up row), while only in the cytoplasm of cells transfected with plasmids expressing FAK-WT (middle row) and FAK-DN (bottom row). Cells were co-stained with p-FAK (red) and DAPI (blue). Scale bar, 20 μm. C, Western blotting p-FAK925 expression from neuro-2a cells after 48-h transfection with different constructs. Actin (upper band) was used as a loading control
To study the effects of FAK on neuronal migration, in utero electroporation [9] was employed to overexpress different plasmids in mouse brains at E14.5. Plasmids were transfected into newly generated late-born neurons in the ventricular zone (VZ). After 3 days at E17.5, transfected brains were sectioned. In CON (n = 10), numerous GFP-positive cells migrated into both the upper region of the CP (UCP) and the deeper region of the CP (DCP) (Fig. 3A). However, neurons in brains transfected with the plasmid expressing FAK-WT (n = 8) and FAK-DN (n = 10) had fewer neurons invaded the CP, but more accumulated in the intermediate zone (IZ) and ventricular zone/subventricular zone (VZ/SVZ) (Fig. 3B, Table 1). In CON group, the transfected cells that were migrating in the CP showed a typical bipolarity morphology in vivo (Fig. 3C), which is a long and thick leading process directed toward the pial surface and a short, thin trailing process (arrowhead) pointing to the VZ. Similar to Neuro-2a cells in vitro, overexpression of FAK-WT and FAK-DN was mainly located in processes and the perinuclear region of migrating neurons. Moreover, compared to the even distribution of GFP signal in CON neurons, aggregates of FAK-WT and FAK-DN were found in the unsmooth-looking membrane of neurons (Fig. 3C, arrows), which might be the sites of focal adhesions between neurons or neurons with glial cells (Fig. 3C). Given the polarity of migrating neurons was barely affected, we checked the migratory behavior of neurons transfected with the different constructs using time-lapse imaging. CON cells revealed their characteristic bipolar shape and moved smoothly by nuclear translocation (n = 227 cells, 18.93 ± 0.646 μm/h, Fig. 3D–E, Table 1, Movie 1, 2). By contrast, neurons transfected with FAK-WT showed slower forward movement (n = 256 cells, 16.14 ± 0.515 μm/h, p = 0.0025, Fig. 3D–E, Table 1, Movie 3, 4), as well as in FAK-DN neurons (n = 338 cells, 15.92 ± 0.510 μm/h, p = 0.0004, Fig. 3D–E, Table 1, Movie 5, 6). Thus, overexpression and mutation of FAK disrupt the radial neuronal migration of late-born neurons, at least partially by reducing their migratory speed. Of note, since the plasmids we used were fused FAK with GFP, so GFP represented the distribution changing of FAK during locomotion circles. In FAK-WT transfected neurons, the dynamic GFP signal was observed inside the migrating neurons. FAK-WT either spread relatively more ‘even’ in the cytoplasm and the leading process (Fig. 3D, FAK-WT, 0–40 min) or aggregated in the perinuclear area of the leading process and tail process (Fig. 3D, FAK-WT, 60–100, 180–200 min) or localized on one side of the cell body (possible the interaction side between neuron and radial glial cell, Fig. 3D, FAK-WT, 120–160 min). In contrast, the FAK-DN transfected cells showed a more stable GFP signal (Fig. 3D, FAK-DN 40–200 min), especially in the FAK-DN high-expressing migrating neuron (Movie 7).

Overexpression of FAK-WT and FAK-DN disrupted the migration of late-born neurons in the neocortex. A, Representative images showing the distribution of late-born cortical neurons (green) in DAPI (blue) stained coronal slices from E17.5 mice electroporated with CON (left), FAK-WT (middle), FAK-DN (right) at E14.5. Scale bar, 100 μm. B, Quantitative analysis of the distribution pattern of electroporated cortical neurons across four different cortical zones at E17.5. C, Higher magnifications of migrating neurons in CP of E17.5 cortical sections electroporated at E14.5. Sections were counter stained with DAPI (red). White arrows indicate aggregates of FAK-WT and FAK-DN expression. White arrowhead indicates the trailing process. Scale bar, 10 μm. D, Migratory behavior of neurons transfected with the different constructs 3 days after IUE. Individual neurons were monitored over a period of 200 min (selected neurons from Movies 1–7). White arrowheads indicate the somata. Scale bar, 10 μm. E, Quantitative assessment (left) and cumulative distribution (the probability distribution of speed evaluated at the values between 20 and 60 μm/h, right) of locomotion speed of migrating neurons 3 days after IUE. Data are presented as mean ± SEM. One-way ANOVA test followed by Bonferroni-corrected post hoc test, *p < 0.05, **p < 0.01, ***p < 0.001
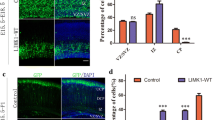
The migration defects observed above led to the question of whether the final location of late-born neurons is affected as well. Next, we examined the transfected brains after birth at P0. At this time point, most neurons have terminated their migration and reached their destinations in the CP. More than 90% of all labeled CON cells (n = 6, 91.02 ± 1.945%) formed a compact layer subjacent to the MZ, which is layer II/III of the cortex (Fig. 4A, B, Table 1). Layer IV, V/VI and IZ contained a few neurons (Fig. 4A, B, Table 1). Notably, the distribution pattern of FAK-WT transfected cells was quite different from that in the CON group (Fig. 4A). Significantly fewer cells (n = 8, 59.39 ± 4.156%, p < 0.0001) were found in the layer II/III of the CP, while significantly more cells in layer IV, V/VI and IZ (Fig. 4B, Table 1). The majority of FAK-DN neurons (n = 5, 73.73 ± 1.881%, p = 0.0118) occupied layer II/III, whose number was significantly lower than in the CON group. Consequently, there were much more neurons stacked in layer IV (Fig. 4B, Table 1). Furthermore, the neurons that reached layer II/III were significantly less (p = 0.0273) in the FAK-WT group when compared to the FAK-DN group as well. These migration impairments in the FAK-WT and FAK-DN groups lasted till P2, when there were less transfected neurons reaching their final destination in layer II/III compared to CON (Fig. 4C, D, CON: n = 4, 91.71 ± 1.71%; FAK-WT: n = 7, 80.08 ± 2.352%, p = 0.0386; FAK-DN: n = 12, 78.92 ± 2.662%, p = 0.0292, Table 1), but more neurons stayed in layer IV and V/VI (Fig. 4C, D, Table 1). These mislocated neurons gave rise to several branches (Fig. 4C, insets), suggesting their migration stopped and differentiation started.

Late-born neurons transfected with FAK-WT and FAK-DN were unable to reach their final destination. Retarded and inhibited neuronal migration of late-born neurons after transfection of FAK-WT and FAK-DN. A, Representative images showing the distribution of the transfected late-born cortical neurons (green) from P0 mice electroporated with CON (left), FAK-WT (middle), FAK-DN (right) at E14.5. The sections were counterstained with DAPI (blue). Scale bar, 100 μm. B, Quantitative analysis of the distribution pattern of electroporated cortical neurons across four different cortical zones at P0. C–D, Same as A–B, for P2 mice. Scale bar, 100 μm. White arrowheads in C indicate neurons with multiple branches. Insets: branching neurons shown at higher magnification. Scale bar, 20 μm. Data are presented as mean ± SEM. One-way ANOVA test followed by Bonferroni-corrected post hoc test, *, #p < 0.05, **p < 0.01, ***p < 0.001
These data indicate that overexpression of FAK and its mutant induces migration defects of late-born neurons, not only delaying the locomotion but the final destination.
FAK is Required for Glia-Independent Terminal Translocation in Late-Born Neurons and Somal Translocation in Early-Born Neurons
The abnormal location of FAK-WT and FAK-DN neurons observed above suggests that their migration disturbance was not solely related to the reduction of locomotion speed, but was contributed by other processes regulated by FAK. One possible process might be the glia-independent terminal translocation. Locomoting neurons directionally migrate toward the pia surface and when they arrive beneath MZ, they apply their final migration mode, in which they extend processes to the MZ and shorten their leading processes to move their cell bodies to their final positions [10].
To directly test this hypothesis, we then first monitored the effect of FAK-WT and FAK-DN on terminal translocation in late-born neurons. These neurons transiently pause and accumulate, forming into the primitive cortical zone (PCZ). At P0, PCZ could be identified by dual staining of NeuN and Brn2 (Fig. 5A). Quantification showed that, in the CON group, 47.72 ± 1.695% (n = 4) transfected neurons located their somata in the NeuN−Brn2+ PCZ region (Fig. 5A, B), whereas there was a much lower number of neurons in FAK-WT (n = 4, 39.34 ± 2.391%, p = 0.0403, Table 1) and FAK-DN group (n = 4, 30.39 ± 1.920%, p = 0.0006, Table 1). Of note, the difference of this number in FAK-WT and FAK-DN was significant as well (p = 0.0365). These results suggest that the terminal translocation of late-born neurons was affected by FAK manipulation.

Overexpression of FAK-WT and FAK-DN disrupted the translocation of late-born and early-born neurons in the neocortex. A, Representative images showing the soma location of transfected late-born cortical neurons (green) in upper layers of coronal slices from P0 mice electroporated with CON (left), FAK-WT (middle), FAK-DN (right) at E14.5. Brn2 (cyan) and NeuN (magenta) were used to identify the primitive cortical zone (PCZ) (Brn2+NeuN−). Scale bar, 100 μm. B, Quantitative analysis of the percentage of electroporated cortical neurons in PCZ at P0. C, Representative images showing the distribution of the transfected early-born cortical neurons (green) in neocortex from E17.5 mice electroporated with CON (left), FAK-WT (middle), FAK-DN (right) at E12.5. The sections were counterstained with DAPI (blue). Scale bar, 100 μm. D, Bin analysis of the distribution pattern of electroporated cortical neurons across cortex at E17.5. The area from the outer margin of the CP to the inner margin of the VZ was divided into 10 equally spaced bins and the transfected neurons in each bin were counted. Data are presented as mean ± SEM. One-way ANOVA test followed by Bonferroni-corrected post hoc test, *, #p < 0.05, **p < 0.01, ***p < 0.001
Second, we studied the migration of early-born neurons in the early developmental stages since they undergo somal translocation mode, which is morphologically similar to the final terminal translocation mode of late-born neurons [7]. To target early-born neurons, IUE experiments were performed at E12.5. Transfected brains were sectioned and examined 5 days later at E17.5. Most neurons in all groups migrated into the CP by E17.5 (Fig. 5C, D), while significantly less occupation of FAK-WT (n = 4) and FAK-DN (n = 4) neurons in the upper region of CP (Bin 2–3) when compared to CON (n = 4). More neuronal accumulation was observed around the border between the CP and the IZ (Fig. 5D, Bin 10). These observations suggest that FAK deregulation affected the somal translocation of early-born neurons, similar to the phenotype of FAK-WT and FAK-DN at a later stage (electroporation at E14.5).
Collectively, these series of data suggest that FAK is required for the neuronal migration of both early-born and late-born neurons. However, different mechanisms might be involved in these processes.
FAK Regulates the Phosphorylation of Cofilin and the Expression of N-Cadherin
To investigate the molecular mechanisms of how FAK affects neuronal migration, molecules control cytoskeleton dynamics and cellular adhesion, both processes that are highly involved in regulating neuronal migration, were first examined. Previous results showed that Cofilin and N-Cadherin are critical genes in regulating neuronal migration, whose function was closely related to Reelin signaling cascade [18, 29, 37, 38]. In the present study, FAK phosphorylation at Y925 was found to be highly related to Reelin expression. Therefore, Neuro-2a cells were transfected with FAK-WT and its mutant FAK-DN and then subjected to immunoblot analysis with antibodies against p-cofilin and N-Cadherin (Fig. 6A).

Reelin and FAK regulate cofilin phosphorylation and N-Cadherin expression. A, Western blotting for p-cofilin and N-Cadherin expression from neuro-2a cells after 48-h transfection with different constructs. Actin was used as a loading control. B, Densitometric analysis of relative p-cofilin and N-Cadherin expression level from three independent experiments (t-tests, ***p < 0.001). C, Western blotting for p-cofilin and N-Cadherin expression in cortical lysates from P0 wild-type mice (rl+/+) or their reeler (rl−/−) littermates. Actin was used as a loading control. D, Densitometric analysis of relative (normalized to Actin) p-cofilin and N-Cadherin expression level from three independent experiments. Data are presented as mean ± SEM. Student’s t-test, ***p < 0.001
The results showed that the overexpression of FAK-WT did not affect the phosphorylation of cofilin. However, phosphorylation of cofilin was significantly inhibited in cells transfected with FAK-DN (Fig. 6A, B, CON: 1.00 ± 0.0, FAK-WT: 0.95 ± 0.081, FAK-DN: 0.72 ± 0.060, F(2,12) = 6.451, P = 0.012, *p = 0.017, #p = 0.049). In addition, the expression of N-Cadherin was significantly downregulated in the FAK-DN group, while no change was found in cells transfected with FAK-WT (Fig. 6A, B, CON: 1.00 ± 0.0, FAK-WT: 1.05 ± 0.054, FAK-DN: 0.63 ± 0.097, F(2,6) = 12.26, P = 0.008, *p = 0.022, #p = 0.012). Given that p-FAK925 was significantly lower in reeler mice (Fig. 1D), we next examined whether cofilin and N-Cadherin expression was affected by reelin knockout. Western blots of N-Cadherin in cortical lysates prepared from P0 wild-type mice (rl+/+) or their reeler (rl−/−) littermates showed that the protein level of N-Cadherin was downregulated in reeler mutant mice (Fig. 6C, D, rl+/+: 1.00 ± 0.0, rl−/−: 0.52 ± 0.019, p < 0.0001). Phosphorylation of cofilin was lower in reeler as well (Fig. 6C, D, rl+/+: 1.00 ± 0.0, rl−/−: 0.66 ± 0.036, p < 0.0001), which confirm previous investigations that Reelin enhances cofilin activity by regulating its phosphorylation [18].
Thus, our observations suggest that the phosphorylation of FAK at Y925 is crucial for different migrating processes via coordinate cytoskeleton dynamics and adhesion, which are highly related to cofilin and N-Cadherin through the Reelin signaling pathway (Fig. 7).

Locomotion and terminal translocation is regulated by Reelin-FAK mediated actin arrangement and cell-cell adhesion via cofilin and N-Cadherin. Radial glia cell (RGC, gray) extends long process from VZ to MZ. Migrating neuron (green) experiences glial-dependent locomotion in bipolar morphology. Of note, the N-Cadherin (purple) mediated cell-cell adhesion is zoomed in between migrating neuron and its parental RGC. The neuron detaches from RGC and applies terminal translocation to lift its cell body to the destination. Note that the red background schema represents the density of Reelin in ECM, shading from MZ toward VZ. Binding of Reelin to the transmembrane receptor ApoER2 and VLDLR induces reciprocal activation of Dab1 and Src. This leads to the phosphatidylation of FAK at Y925; which is required for the phosphorylation of cofilin. The subsequent cofilin phosphorylation at serine 3 disables its function in depolymerizing F-actin, thereby stabilize the F-actin in the leading process of migrating neuron. FAK phosphorylation at Y925 increases the expression of N-Cadhenrin, which maintains a proper cell-cell adhesion during locomotion
Discussion
Coordinated corticogenesis during early development has been proposed to underlie precise connectivity and higher brain functions such as learning, memory and cognition [1]. Neuronal migration, as a core aspect of corticogenesis, is especially being the interest of research in the last decades. However, until recently, the molecular signaling cascades for controlling neuronal migration during development were not fully uncovered. In the present study, we reported that overexpression of FAK (FAK-WT) and its dominant-negative mutant (FAK-DN) in cortical neurons in vivo disrupts the normal radial migration. This phenotype is likely to be due to inhibited glia-dependent locomotion speed revealed by time-lapse imaging and glia-independent translocation defects, in both aFAK-WT and FAK-DN conditions. We show that FAK controls actin polymerization and cell adhesion by regulating cofilin phosphorylation and expression of N-Cadherin, respectively. These processes involve phosphorylation of FAK at Y925 and are correlated with Reelin. Our findings reveal a new Reelin regulated mechanism, through which FAK controls actin dynamics and cell adhesion, thus neuronal migration during development.
Detailed expression and localization investigation revealed ubiquitously presence of FAK in all regions of developing and adult brains in rat, with enrichment in specific neuronal populations of neocortex, hippocampus and cerebellum during development [39]. In contrast to restricted to focal adhesions in astrocytes, FAK was found in cell bodies, axons, dendrites of neurons and showed no enrichment in any compartment [39, 40], which was confirmed by the results of FAK-WT and FAK-DN transfection in cell lines in vitro and in migrating neurons in vivo. A wealth of studies documented FAK plays a pivotal role in the regulation of cell proliferation, survival and migration in various cell types [41,42,43,44,45,46]. FAK-null mice showed an embryonic lethal phenotype due to defects in the axial mesodermal tissues and cardiovascular system [32], which limited its use for studying the function of FAK in embryonic development. Conditional deletion of FAK in migrating pyramidal neurons did not show any obvious migratory alteration [47]. While Xie et al. showed Cdk5 dependent FAK phosphorylation at S732 is critical for the organization of microtubules based nuclear translocation and, in turn, neuronal migration [33]. In addition, FAK-deficiency induced abundant branching and swelling of the leading process of migrating neurons by a defective assembly of Connexin 26 contact points in the adhesion surface between neurons and radial glial cells [34]. Overexpression of FAK in vivo impaired neuronal migration and layer formation of the neocortex possibly is related to its relationship with vinculin and F-actin cytoskeleton [35]. The involvement of FAK in brain development was also confirmed in another study of Inositol hexakisphosphate kinase 1 (IP6K1) [48]. IP6K1 physiologically interacts with α-actinin and localizes to focal adhesions, whose deletion abolishes FAK autophosphorylation and α-actinin and leads to brain malformation and abnormalities of neuronal migration. Collectively, increasing evidence showed FAK participate in neuronal migration in a cell-autonomous manner.
Our observations of colocalization between Reelin and p-FAK in CRs and downregulated FAK phosphorylation in reeler mutant experimentally provided evidence for the involvement of FAK in the Reelin signaling pathway. CRs tangentially migrate from the hem to the marginal zone of the cortex around embryonic date (E) 10.5–E12.5 [49]. Of note, the positioning and density of CRs in marginal zone is comparable in normal and reeler mice [50], suggesting that the migration and positioning of CRs might not be regulated by Reelin signaling pathway. Canonical Reelin signaling involves the very low density lipoprotein receptor (VLDLR) and apolipoprotein E receptor 2 (ApoER2) [27, 51]. The cellular adaptor protein Disabled 1 (Dab1) is phosphorylated by SFK upon binding of Reelin to its receptors, which further activates Src family kinases (SFK) and the subsequent downstream molecules [23, 52]. Tyrosine phosphorylation is a critical regulator of FAK and is mainly regulated by SFKs [53]. Although FAK hypophosphorylation was only found in fyn mutants [40], other members of SFK: src and yes were proved to activate FAK in various studies [30, 54, 55]. Conditional ablation and knockdown of FAK showed that FAK is required for radial migration of pyramidal cells, but not for tangential migration of cortical interneurons [34]. Since Reelin is only expressed by interneurons in the postnatal cortex [56, 57], when most neurons have stopped migrating, we believed that these Reelin-positive interneurons have little effect on neuronal migration. Phosphorylation of FAK at Y925 was not fully abolished in reeler P0 cortex, indicating that Reelin is not unique in regulator of FAK Y925 activity. Other candidates might be Netrin, Semaphorin, which have been shown to be involved in neuronal migration [58, 59].
In contrast to that FAK promotes cancer cell migration thus enhancing invasion [31, 60], we showed overexpressing FAK strongly inhibited neuronal migration in vivo. Although striking similarities in neuronal and cancer cells, there are more contrasts between cancer metastasis and neuronal motility, e.g., migrating substrates, microenvironment, intracellular forces and cytoskeleton arrangement [61]. Neuronal migration is a more intricate process including synergistic cooperation between neurons, glial cells and the extracellular cues. In the present study, overexpression of FAK-WT and FAK-DN induced similar phenotype at E17.5, the extend of neuronal inhibition reviewed at P0 was much severer in FAK-DN. Overexpression of FAK-WT resulted in partial exogenous FAK phosphorylation, whereas overexpression of FAK-DN led to complete unphosphorylatable FAK that was capable of binding to Src but without any further phosphorylation of endogenous FAK and other downstream molecules. The results of western blot after FAK overexpression provide first insights into the mechanisms of how FAK Y925 phosphorylation is involved in different stages of radial migration. Our previous study has shown that Reelin induces cofilin phosphorylation at Serine3 [18, 29], thus stabilizing actin dynamics and controlling the orientation and speed of migrating neurons. In reeler mutants, cofilin phosphorylation was also significantly reduced in our observation. In the present study, we found that FAK-DN strongly inhibited cofilin phosphorylation, suggesting that FAK regulates neuronal migration through influencing actin cytoskeleton dynamics. This mechanism not only underlies the reduced migrating speed, as well as the final translocation—both processes were disturbed by inhibiting FAK. Compared to other phosphorylation sites of FAK, e.g., Y397, Y576/577; S732; Y861 [62], Y925 is of particular especial. Phosphorylated FAK at Y925 creates a specific SH2-binding site for the GRB2 adaptor protein [63]. It has been shown that only FAK Y925 is required for Strain-stimulated AKT phosphorylation for controlling cell migration [64]. Src-dependent phosphorylation of FAK at Y925 specifically promotes detachment of focal adhesions at the trailing edge of migrating neurons [54]. FAK Y925 phosphorylation is critical for focal adhesions disassembly rate at the cell rear and cell protrusion [44].
In addition, N-Cadherin expression was significantly reduced in reeler mice, as well as after FAK dephosphorylation. N-Cadherin is a single-pass transmembrane adhesion receptor required for cell-cell adhesion [65, 66]. N-Cadherin controls neuronal migration and lamination in the neocortex under the control of Reelin transduction [12, 37, 38, 67]. Moreover, knockdown of Myosin X abolishes neuronal N-Cadherin expression and disturbs the adherence of migrating neurons to radial glial cells, resulting in neuronal migration defects [68]. N-Cadherin regulates the migration of late-born neurons by regulating their radial orientation of multipolar stage and forms a cis and trans asymmetric bridge complex with astrotactin (ASTN1) that is required for glial-guided migration [12, 67]. In our results, dynamics of FAK distribution in different neuronal subregions were observed by time-lapse imaging, suggesting adhesion sites between neurons and other cells are not stabilized during the one locomotion circle, which is consistent with the function of FAK in regulating focal adhesion turnover [44]. Apparently, N-Cadherin stabilizes the leading process in the MZ and facilitates the somal translocation of early-born cortical neurons [37]. In line with these results, glia-independent terminal translocation of late-born neurons was inhibited after FAK overexpression and dephosphorylation in the present results. FAK dephosphorylation at Y925 seems strongly inhibit the dynamic transporting of FAK. Reelin transiently enhances the adhesiveness of neurons to N-Cadherin and promotes neuronal aggregation, which is important for correct stopping and positioning of neurons during migration [38]. Unlike this observation, there was no neuronal aggregation observed after manipulating FAK function in the present study, suggesting Reelin-FAK-mediated N-Cadherin expression might not regulate neuron-neuron adhesion but glia-neuron interaction. The above-mentioned function of N-Cadherin in controlling both radial orientation and translocation involves Rap1 GTPases [37, 67], which provide a key nexus for activating other small GTPases, which regulate cytoskeleton and cell motility [69,70,71]. Hence, function of FAK Y925 phosphorylation on N-Cadherin regulates not only cell-cell adhesion, but also cytoskeleton dynamics.
Noncanonical Reelin signaling can refer to the involvement of transmembrane proteins other than VLDLR and ApoER2, e.g., integrins. Reelin binds directly to the extracellular domain of integrins and interacts with Dab1 through its NPXY motif in the cytoplasmic tail [13, 28, 72], thus facilitating cell-cell and cell-extracellular matrix (ECM) adhesion [73]. Reelin-α3β1 integrin interactions inhibit cortical neuronal migration [28] and are required for the formation of the radial glial scaffold necessary for neuronal migration in the hippocampus [20]. As a non-receptor protein tyrosine kinase, FAK is activated by interactions with integrins and functions as a ‘scaffold’ for the recruitment signaling proteins needed for proliferation [74], extracellular stimulated cell migration [41], neurites outgrowth and axon pathfinding [75, 76]. Moreover, the SH2 binding site for GRB2 at FAK Y925 partially overlaps with binding sites for paxillin [77], which is an important integrin-binding protein that recruits FAK and vinculin to focal contacts [78,79,80]. Overexpression of a Y925F mutant of FAK resulted in strong focal contact distribution and blocks the turnover of focal contacts [54, 81]. A point mutation in the Y925 of FAK selectively disrupts GRB2 but promotes paxillin binding, then the subsequent association of FAK with β1-integrin and prevents the dissociation of FAK from focal contacts and leads to inefficient focal turnover [79], which is consistent with our observation of the relatively stable distribution of FAK-DN signal in the migrating neurons. Therefore, although the direct relation between FAK and integrin was not examined in the present study, it strongly suggests that Reelin-integrin-FAK mediated cell-ECM adhesion might be one underlying mechanism for glial-independent translocation. Moreover, the function of FAK in regulating microtubule organization cannot be neglected during the glial-independent translocation process, during which microtubules generate mechanical forces and drive nuclear translocation [82].
In the present study, we show that FAK-mediated Reelin signaling regulates cortical lamination by controlling two key events during embryonic development: glia-dependent locomotion and glia-independent translocation. Both processes involve dynamic regulating of the actin cytoskeleton and focal adhesions, which might be related to the activity of cofilin and N-Cadherin. Overall, these data expand the known Reelin signal pathway and provide new insights in understanding neuronal migration mechanisms.
Data Availability
The data and material supporting the findings of this study are available with the article or is available from the corresponding author upon request.
Code Availability
Not applicable.
References
Guy J, Staiger JF (2017) The functioning of a cortex without layers. Front Neuroanat 11:54. https://doi.org/10.3389/fnana.2017.00054
Berry M, Rogers AW (1965) The migration of neuroblasts in the developing cerebral cortex. J Anat 99(Pt 4):691–709
Rakic P, Lombroso PJ (1998) Development of the cerebral cortex: I Forming the cortical structure. Journal of the American Academy of Child and Adolescent Psychiatry 37(1):116–117. https://doi.org/10.1097/00004583-199801000-00026
Ohtaka-Maruyama C, Okado H (2015) Molecular pathways underlying projection neuron production and migration during cerebral cortical development. Front Neurosci 9:447. https://doi.org/10.3389/fnins.2015.00447
Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR (2004) Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 7(2):136–144. https://doi.org/10.1038/nn1172
Hirota Y, Nakajima K (2017) Control of neuronal migration and aggregation by reelin signaling in the developing cerebral cortex. Front Cell Dev Biol 5:40. https://doi.org/10.3389/fcell.2017.00040
Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL (2001) Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci 4(2):143–150. https://doi.org/10.1038/83967
Nadarajah B, Parnavelas JG (2002) Modes of neuronal migration in the developing cerebral cortex. Nat Rev Neurosci 3(6):423–432. https://doi.org/10.1038/nrn845
Tabata H, Nakajima K (2003) Multipolar migration: the third mode of radial neuronal migration in the developing cerebral cortex. J Neurosci 23(31):9996–10001. https://doi.org/10.1523/jneurosci.23-31-09996.2003
Sekine K, Honda T, Kawauchi T, Kubo K, Nakajima K (2011) The outermost region of the developing cortical plate is crucial for both the switch of the radial migration mode and the Dab1-dependent “inside-out” lamination in the neocortex. J Neurosci 31(25):9426–9439. https://doi.org/10.1523/JNEUROSCI.0650-11.2011
Frotscher M, Zhao S, Wang S, Chai X (2017) Reelin signaling inactivates cofilin to stabilize the cytoskeleton of migrating cortical neurons. Front Cell Neurosci 11:148. https://doi.org/10.3389/fncel.2017.00148
Horn Z, Behesti H, Hatten ME (2018) N-cadherin provides a cis and trans ligand for astrotactin that functions in glial-guided neuronal migration. Proc Natl Acad Sci U S A 115(42):10556–10563. https://doi.org/10.1073/pnas.1811100115
Schmid RS, Maness PF (2008) L1 and NCAM adhesion molecules as signaling coreceptors in neuronal migration and process outgrowth. Curr Opin Neurobiol 18(3):245–250. https://doi.org/10.1016/j.conb.2008.07.015
Kubo KI, Deguchi K, Nagai T, Ito Y, Yoshida K, Endo T, Benner S, Shan W, Kitazawa A, Aramaki M, Ishii K, Shin M, Matsunaga Y, Hayashi K, Kakeyama M, Tohyama C, Tanaka KF, Tanaka K, Takashima S, Nakayama M, Itoh M, Hirata Y, Antalffy B, Armstrong DD, Yamada K, Inoue K, Nakajima K (2017) Association of impaired neuronal migration with cognitive deficits in extremely preterm infants. JCI Insight 2 (10). https://doi.org/10.1172/jci.insight.88609
Stouffer MA, Golden JA, Francis F (2016) Neuronal migration disorders: Focus on the cytoskeleton and epilepsy. Neurobiol Dis 92(Pt A):18–45. https://doi.org/10.1016/j.nbd.2015.08.003
Subramanian L, Calcagnotto ME, Paredes MF (2019) Cortical malformations: lessons in human brain development. Front Cell Neurosci 13:576. https://doi.org/10.3389/fncel.2019.00576
Gleeson JG, Walsh CA (2000) Neuronal migration disorders: from genetic diseases to developmental mechanisms. Trends Neurosci 23(8):352–359. https://doi.org/10.1016/s0166-2236(00)01607-6
Chai X, Forster E, Zhao S, Bock HH, Frotscher M (2009) Reelin stabilizes the actin cytoskeleton of neuronal processes by inducing n-cofilin phosphorylation at serine3. J Neurosci 29(1):288–299. https://doi.org/10.1523/JNEUROSCI.2934-08.2009
D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T (1995) A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374(6524):719–723. https://doi.org/10.1038/374719a0
Förster E, Tielsch A, Saum B, Weiss KH, Johanssen C, Graus-Porta D, Müller U, Frotscher M (2002) Reelin, Disabled 1, and beta 1 integrins are required for the formation of the radial glial scaffold in the hippocampus. Proc Natl Acad Sci U S A 99(20):13178–13183. https://doi.org/10.1073/pnas.202035899
Miyata T, Ono Y, Okamoto M, Masaoka M, Sakakibara A, Kawaguchi A, Hashimoto M, Ogawa M (2010) Migration, early axonogenesis, and Reelin-dependent layer-forming behavior of early/posterior-born Purkinje cells in the developing mouse lateral cerebellum. Neural Dev 5(1):23. https://doi.org/10.1186/1749-8104-5-23
Beffert U, Weeber EJ, Durudas A, Qiu S, Masiulis I, Sweatt JD, Li WP, Adelmann G, Frotscher M, Hammer RE, Herz J (2005) Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron 47(4):567–579. https://doi.org/10.1016/j.neuron.2005.07.007
Howell BW, Herrick TM, Cooper JA (1999) Reelin-induced tyrosine [corrected] phosphorylation of disabled 1 during neuronal positioning. Genes Dev 13(6):643–648. https://doi.org/10.1101/gad.13.6.643
Iafrati J, Orejarena MJ, Lassalle O, Bouamrane L, Gonzalez-Campo C, Chavis P (2014) Reelin, an extracellular matrix protein linked to early onset psychiatric diseases, drives postnatal development of the prefrontal cortex via GluN2B-NMDARs and the mTOR pathway. Mol Psychiatry 19(4):417–426. https://doi.org/10.1038/mp.2013.66
Ishii K, Kubo KI, Nakajima K (2016) Reelin and neuropsychiatric disorders. Front Cell Neurosci 10:229. https://doi.org/10.3389/fncel.2016.00229
Teixeira CM, Martin ED, Sahun I, Masachs N, Pujadas L, Corvelo A, Bosch C, Rossi D, Martinez A, Maldonado R, Dierssen M, Soriano E (2011) Overexpression of Reelin prevents the manifestation of behavioral phenotypes related to schizophrenia and bipolar disorder. Neuropsychopharmacology 36(12):2395–2405. https://doi.org/10.1038/npp.2011.153
Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J (1999) Reeler/disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97(6):689–701. https://doi.org/10.1016/s0092-8674(00)80782-5
Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, Anton ES (2000) Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron 27(1):33–44. https://doi.org/10.1016/s0896-6273(00)00007-6
Chai X, Zhao S, Fan L, Zhang W, Lu X, Shao H, Wang S, Song L, Failla AV, Zobiak B, Mannherz HG, Frotscher M (2016) Reelin and cofilin cooperate during the migration of cortical neurons: a quantitative morphological analysis. Development 143(6):1029–1040. https://doi.org/10.1242/dev.134163
Wang JT, Song LZ, Li LL, Zhang W, Chai XJ, An L, Chen SL, Frotscher M, Zhao ST (2015) Src controls neuronal migration by regulating the activity of FAK and cofilin. Neuroscience 292:90–100. https://doi.org/10.1016/j.neuroscience.2015.02.025
Lim ST, Mikolon D, Stupack DG, Schlaepfer DD (2008) FERM control of FAK function: implications for cancer therapy. Cell cycle (Georgetown, Tex) 7(15):2306–2314. https://doi.org/10.4161/cc.6367
Ilić D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T (1995) Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377(6549):539–544. https://doi.org/10.1038/377539a0
Xie Z, Sanada K, Samuels BA, Shih H, Tsai L-H (2003) Serine 732 Phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell 114(4):469–482. https://doi.org/10.1016/s0092-8674(03)00605-6
Valiente M, Ciceri G, Rico B, Marin O (2011) Focal adhesion kinase modulates radial glia-dependent neuronal migration through connexin-26. J Neurosci 31(32):11678–11691. https://doi.org/10.1523/JNEUROSCI.2678-11.2011
An L, Li W, Hu X, Zhang W, Zhao S (2018) Abundant focal adhesion kinase causes aberrant neuronal migration via its phosphorylation at Tyr925. J Mol Neurosci 64(1):102–110. https://doi.org/10.1007/s12031-017-1010-1
Saito T, Nakatsuji N (2001) Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol 240(1):237–246. https://doi.org/10.1006/dbio.2001.0439
Franco SJ, Martinez-Garay I, Gil-Sanz C, Harkins-Perry SR, Muller U (2011) Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron 69(3):482–497. https://doi.org/10.1016/j.neuron.2011.01.003
Matsunaga Y, Noda M, Murakawa H, Hayashi K, Nagasaka A, Inoue S, Miyata T, Miura T, Kubo KI, Nakajima K (2017) Reelin transiently promotes N-cadherin-dependent neuronal adhesion during mouse cortical development. Proc Natl Acad Sci U S A 114(8):2048–2053. https://doi.org/10.1073/pnas.1615215114
Burgaya F, Menegon A, Menegoz M, Valtorta F, Girault JA (1995) Focal adhesion kinase in rat central nervous system. Eur J Neurosci 7(8):1810–1821. https://doi.org/10.1111/j.1460-9568.1995.tb00700.x
Grant SG, Karl KA, Kiebler MA, Kandel ER (1995) Focal adhesion kinase in the brain: novel subcellular localization and specific regulation by Fyn tyrosine kinase in mutant mice. Genes Dev 9(15):1909–1921. https://doi.org/10.1101/gad.9.15.1909
Sieg DJ, Hauck CR, Schlaepfer DD (1999) Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J Cell Sci 112(Pt 16):2677–2691
Sonoda Y, Matsumoto Y, Funakoshi M, Yamamoto D, Hanks SK, Kasahara T (2000) Anti-apoptotic role of focal adhesion kinase (FAK) Induction of inhibitor-of-apoptosis proteins and apoptosis suppression by the overexpression of FAK in a human leukemic cell line, HL-60. J Biol Chem 275(21):16309–16315. https://doi.org/10.1074/jbc.275.21.16309
Zhao J, Guan JL (2009) Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev 28(1–2):35–49. https://doi.org/10.1007/s10555-008-9165-4
Deramaudt TB, Dujardin D, Hamadi A, Noulet F, Kolli K, De Mey J, Takeda K, Ronde P (2011) FAK phosphorylation at Tyr-925 regulates cross-talk between focal adhesion turnover and cell protrusion. Mol Biol Cell 22(7):964–975. https://doi.org/10.1091/mbc.E10-08-0725
Liu H, Yue J, Huang H, Gou X, Chen SY, Zhao Y, Wu X (2015) Regulation of focal adhesion dynamics and cell motility by the EB2 and Hax1 protein complex. J Biol Chem 290(52):30771–30782. https://doi.org/10.1074/jbc.M115.671743
Abbi S, Ueda H, Zheng C, Cooper LA, Zhao J, Christopher R, Guan JL (2002) Regulation of focal adhesion kinase by a novel protein inhibitor FIP200. Mol Biol Cell 13(9):3178–3191. https://doi.org/10.1091/mbc.e02-05-0295
Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF (2003) FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron 40(3):501–514. https://doi.org/10.1016/s0896-6273(03)00666-4
Fu C, Xu J, Cheng W, Rojas T, Chin AC, Snowman AM, Harraz MM, Snyder SH (2017) Neuronal migration is mediated by inositol hexakisphosphate kinase 1 via alpha-actinin and focal adhesion kinase. Proc Natl Acad Sci U S A 114(8):2036–2041. https://doi.org/10.1073/pnas.1700165114
Takiguchi-Hayashi K, Sekiguchi M, Ashigaki S, Takamatsu M, Hasegawa H, Suzuki-Migishima R, Yokoyama M, Nakanishi S, Tanabe Y (2004) Generation of reelin-positive marginal zone cells from the caudomedial wall of telencephalic vesicles. J Neurosci 24(9):2286–2295. https://doi.org/10.1523/jneurosci.4671-03.2004
Derer P (1985) Comparative localization of Cajal-Retzius cells in the neocortex of normal and reeler mutant mice fetuses. Neurosci Lett 54(1):1–6. https://doi.org/10.1016/s0304-3940(85)80109-9
D’Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T (1999) Reelin is a ligand for lipoprotein receptors. Neuron 24(2):471–479. https://doi.org/10.1016/s0896-6273(00)80860-0
Bock HH, Herz J (2003) Reelin activates SRC family tyrosine kinases in neurons. Curr Biol 13(1):18–26. https://doi.org/10.1016/s0960-9822(02)01403-3
Parsons SJ, Parsons JT (2004) Src family kinases, key regulators of signal transduction. Oncogene 23(48):7906–7909. https://doi.org/10.1038/sj.onc.1208160
Brunton VG, Avizienyte E, Fincham VJ, Serrels B, Metcalf CA 3rd, Sawyer TK, Frame MC (2005) Identification of Src-specific phosphorylation site on focal adhesion kinase: dissection of the role of Src SH2 and catalytic functions and their consequences for tumor cell behavior. Cancer Res 65(4):1335–1342. https://doi.org/10.1158/0008-5472.CAN-04-1949
Chatterji T, Varkaris AS, Parikh NU, Song JH, Cheng CJ, Schweppe RE, Alexander S, Davis JW, Troncoso P, Friedl P, Kuang J, Lin SH, Gallick GE (2015) Yes-mediated phosphorylation of focal adhesion kinase at tyrosine 861 increases metastatic potential of prostate cancer cells. Oncotarget 6(12):10175–10194. https://doi.org/10.18632/oncotarget.3391
Ma J, Yao XH, Fu Y, Yu YC (2014) Development of layer 1 neurons in the mouse neocortex. Cereb Cortex 24(10):2604–2618. https://doi.org/10.1093/cercor/bht114
Lopez-Bendito G, Sturgess K, Erdelyi F, Szabo G, Molnar Z, Paulsen O (2004) Preferential origin and layer destination of GAD65-GFP cortical interneurons. Cereb Cortex 14(10):1122–1133. https://doi.org/10.1093/cercor/bhh072
Gehler S, Compere FV, Miller AM (2017) Semaphorin 3A Increases FAK phosphorylation at focal adhesions to modulate MDA-MB-231 cell migration and spreading on different substratum concentrations. International journal of breast cancer 2017:9619734. https://doi.org/10.1155/2017/9619734
Moore SW, Zhang X, Lynch CD, Sheetz MP (2012) Netrin-1 attracts axons through FAK-dependent mechanotransduction. J Neurosci 32(34):11574–11585. https://doi.org/10.1523/jneurosci.0999-12.2012
Schlaepfer DD, Mitra SK, Ilic D (2004) Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim Biophys Acta 1692(2–3):77–102. https://doi.org/10.1016/j.bbamcr.2004.04.008
Heine P, Ehrlicher A, Kas J (2015) Neuronal and metastatic cancer cells: unlike brothers. Biochim Biophys Acta 1853 (11 Pt B):3126–3131. https://doi.org/10.1016/j.bbamcr.2015.06.011
Mitra SK, Hanson DA, Schlaepfer DD (2005) Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6(1):56–68. https://doi.org/10.1038/nrm1549
Schlaepfer DD, Hunter T (1996) Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol 16(10):5623–5633. https://doi.org/10.1128/mcb.16.10.5623
Gayer CP, Chaturvedi LS, Wang S, Alston B, Flanigan TL, Basson MD (2009) Delineating the signals by which repetitive deformation stimulates intestinal epithelial migration across fibronectin. Am J Physiol Gastrointest Liver Physiol 296(4):G876-885. https://doi.org/10.1152/ajpgi.90648.2008
Fujimori T, Takeichi M (1993) Disruption of epithelial cell-cell adhesion by exogenous expression of a mutated nonfunctional N-cadherin. Mol Biol Cell 4(1):37–47. https://doi.org/10.1091/mbc.4.1.37
Gumbiner BM (1996) Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell 84(3):345–357. https://doi.org/10.1016/s0092-8674(00)81279-9
Jossin Y, Cooper JA (2011) Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat Neurosci 14(6):697–703. https://doi.org/10.1038/nn.2816
Lai M, Guo Y, Ma J, Yu H, Zhao D, Fan W, Ju X, Sheikh MA, Malik YS, Xiong W, Guo W, Zhu X (2015) Myosin X regulates neuronal radial migration through interacting with N-cadherin. Front Cell Neurosci 9:326. https://doi.org/10.3389/fncel.2015.00326
Park HO, Bi E (2007) Central roles of small GTPases in the development of cell polarity in yeast and beyond. Microbiol Mol Biol Rev 71(1):48–96. https://doi.org/10.1128/MMBR.00028-06
Gartner A, Fornasiero EF, Dotti CG (2015) Cadherins as regulators of neuronal polarity. Cell Adh Migr 9(3):175–182. https://doi.org/10.4161/19336918.2014.983808
Lambert M, Choquet D, Mege RM (2002) Dynamics of ligand-induced, Rac1-dependent anchoring of cadherins to the actin cytoskeleton. J Cell Biol 157(3):469–479. https://doi.org/10.1083/jcb.200107104
Calderwood DA, Fujioka Y, de Pereda JM, García-Alvarez B, Nakamoto T, Margolis B, McGlade CJ, Liddington RC, Ginsberg MH (2003) Integrin beta cytoplasmic domain interactions with phosphotyrosine-binding domains: a structural prototype for diversity in integrin signaling. Proc Natl Acad Sci U S A 100(5):2272–2277. https://doi.org/10.1073/pnas.262791999
Sekine K, Kawauchi T, Kubo K, Honda T, Herz J, Hattori M, Kinashi T, Nakajima K (2012) Reelin controls neuronal positioning by promoting cell-matrix adhesion via inside-out activation of integrin alpha5beta1. Neuron 76(2):353–369. https://doi.org/10.1016/j.neuron.2012.07.020
Shibue T, Weinberg RA (2009) Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci U S A 106(25):10290–10295. https://doi.org/10.1073/pnas.0904227106
Ivankovic-Dikic I, Gronroos E, Blaukat A, Barth BU, Dikic I (2000) Pyk2 and FAK regulate neurite outgrowth induced by growth factors and integrins. Nat Cell Biol 2(9):574–581. https://doi.org/10.1038/35023515
Robles E, Gomez TM (2006) Focal adhesion kinase signaling at sites of integrin-mediated adhesion controls axon pathfinding. Nat Neurosci 9(10):1274–1283. https://doi.org/10.1038/nn1762
Liu G, Guibao CD, Zheng J (2002) Structural insight into the mechanisms of targeting and signaling of focal adhesion kinase. Mol Cell Biol 22(8):2751–2760. https://doi.org/10.1128/MCB.22.8.2751-2760.2002
Brown MC, Perrotta JA, Turner CE (1996) Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J Cell Biol 135(4):1109–1123. https://doi.org/10.1083/jcb.135.4.1109
Klingbeil CK, Hauck CR, Hsia DA, Jones KC, Reider SR, Schlaepfer DD (2001) Targeting Pyk2 to beta 1-integrin-containing focal contacts rescues fibronectin-stimulated signaling and haptotactic motility defects of focal adhesion kinase-null cells. J Cell Biol 152(1):97–110. https://doi.org/10.1083/jcb.152.1.97
Liu S, Thomas SM, Woodside DG, Rose DM, Kiosses WB, Pfaff M, Ginsberg MH (1999) Binding of paxillin to alpha4 integrins modifies integrin-dependent biological responses. Nature 402(6762):676–681. https://doi.org/10.1038/45264
Katz BZ, Romer L, Miyamoto S, Volberg T, Matsumoto K, Cukierman E, Geiger B, Yamada KM (2003) Targeting membrane-localized focal adhesion kinase to focal adhesions: roles of tyrosine phosphorylation and SRC family kinases. J Biol Chem 278(31):29115–29120. https://doi.org/10.1074/jbc.M212396200
Nakazawa N, Kengaku M (2020) Mechanical regulation of nuclear translocation in migratory neurons. Front Cell Dev Biol 8:150. https://doi.org/10.3389/fcell.2020.00150
Acknowledgements
We are most grateful to Dr. Lei An for helping with the construction of the plasmids. We thank Dr. Joachim Herz for providing reeler mice.
Funding
This work was supported by the Foundation for top talent recruitment of Xi’an Medical College (No. 2018RCYJ04 to X.C.), General project of basic research of Shaanxi Province (2020JM-603 to X.C.), National Key Research and Development Program of China (2018YFE0127000 to S.Z.) and German Research Foundation (FR 620/13–1 to M.F.) and the China Scholarship Council to L.S.
Author information
Authors and Affiliations
Contributions
L.S. M.F. and X.C. designed the experiments, L.S. performed experiments and analyzed the data, X.C. helped with IUE and living imaging, S.Z. discussed the data and contributed to unpublished analytic tools, L.S. interpreted the data and wrote the original draft of the paper. L.S. S.Z. and X.C. discussed and commented on the manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
The animal study was reviewed and approved by the local ethical committee of the University of Hamburg. Welfare of animals were considered during experiments.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
1. FAK and phosphorylation of FAK at Y925 are required for the migration of both early-born and late-born neurons.
2. Fine-tuned phosphorylation of FAK is crucial for regulating migratory speed and soma translocation.
3. The function of FAK in neuronal migration is mediated via cofilin and N-Cadherin, which are related to Reelin signaling pathway.
Michael Frotscher is deceased
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (AVI 89 KB)
Supplementary file2 (AVI 55 KB)
Supplementary file3 (AVI 151 KB)
Supplementary file4 (AVI 57 KB)
Supplementary file5 (AVI 84 KB)
Supplementary file6 (AVI 43 KB)
Supplementary file7 (AVI 61 KB)
Rights and permissions
About this article

Cite this article
Song, L., Zhao, S., Frotscher, M. et al. Phosphorylation of Focal Adhesion Kinase at Y925: Role in Glia-Dependent and Independent Migration through Regulating Cofilin and N-Cadherin. Mol Neurobiol 59, 3467–3484 (2022). https://doi.org/10.1007/s12035-022-02773-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-02773-y