
Abstract
LDL receptor-related protein (LRP) 10 was recently identified as a Parkinson’s disease gene through genome-wide linkage and sequencing analysis, but its role in Parkinson’s disease in various populations is still unclear. The aim of this study was to determine the frequency and spectrum of LRP10 mutations in a cohort of Parkinson’s disease patients from mainland China. All LRP10 exons and their flanking intron regions were screened by direct sequencing in 567 unrelated Parkinson’s disease patients and 600 unrelated controls. We detected 29 exonic or splicing variants in 79 patients with Parkinson’s disease. Five variants (c.A181C:p.I61L, c.C652T:p.Q218X, c.C833T:p.T278I, c.T1592G:p.I531S, c.T1697C:p.L566P) were predicted to be disease-causing or damaging by multiple in silico tools. Our study provides genetic evidence that LRP10 defects may correlate with sporadic Parkinson’s disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD), whose prevalence and incidence increase with age, is the second most frequently occurring neurodegenerative disorder after Alzheimer’s disease [1]. PD is predicted to cause increasing economic and social burden as the global population ages [1,2,3]. This condition involves progressively debilitating motor symptoms such as resting tremor, bradykinesia, rigidity, and postural instability [4]. The pathological hallmarks of Parkinson’s disease are loss of nigrostriatal dopaminergic neurons, with intracellular inclusions containing α-synuclein protein (i.e., Lewy bodies and Lewy neurites) in surviving neurons [5].
Genetic risk factors have been described for PD; pedigree and genome-wide association studies have established links with mutations in SNCA, DJ-1, GBA, LRRK2, CHCHD2, and other genes [6, 7]. However, many open questions remain about the etiology of PD.
Recently, a genome-wide linkage and sequencing study provided evidence for a role of the LRP10 gene in α-synucleinopathies [8]. Furthermore, that study reported that brains of Caucasian patients with different LRP10 variants contained abundant α-synuclein aggregation in the form of brainstem and cortical Lewy bodies and Lewy neurites. The role of LRP10 protein in α-synucleinopathies remains controversial, and its frequency and expression in non-White ethnicities are poorly understood [9,10,11,12,13].
Therefore in this work, we sequenced the entire LRP10 coding region, exon-intron boundaries, as well as untranslated and flanking regions in order to clarify the potential role of LRP10 in Han Chinese patients with sporadic PD.
Materials and Methods
Study Subjects
The study protocol was approved by the Ethics Committee of West China Hospital, Sichuan University (Chengdu, China). Written informed consent from the participants/their parents/legally authorized representative of participants was obtained. PD was diagnosed based on MDS Clinical Diagnostic Criteria [14]. A total of 567 Han Chinese patients (289 men and 278 women) diagnosed with PD were recruited from among inpatients at the Department of Neurology of West China Hospital, Sichuan University, in southwest China. Patients had a mean age of 62.9 (SD, 10.7; range, 24–95). As controls, 600 unrelated healthy Han Chinese subjects without a history of neurodegenerative disease were recruited.
DNA Sequencing and Statistical Analysis
Genomic DNA was collected from peripheral blood leukocytes using a standard phenol-chloroform procedure. All seven exons of LRP10 (NM_014045) were PCR-amplified as previously reported [15]. PCR products were directly sequenced using a targeted approach using an ABI3730 automated DNA sequencing system (BGI, Shenzhen, China). LRP10 was analyzed by direct nucleotide sequencing (Shanghai Genesky Bio-Tech, Shanghai, China), and variants were identified using Complete Genomic Analysis Tools (version 2.2.0.26; www.completegenomics.com/analysis-tools/cgatools). The detected mutations were validated as known or novel by comparison with direct sequencing of controls and with sequences in the ExAC (http://exac.broadinstitute.org/) and 1000 Genomes Project databases (http://www.1000genomes.org/). The potential function of genetic variants was predicted using the following in silico tools: MutationTaster (http://www.mutationtaster.org/), SIFT (http://sift.jcvi.org/), Polymorphism Phenotyping version 2 (http://genetics.bwh.harvard.edu/pph/pph_help_text.html), Cadd_Raw (http://cadd.gs.washington.edu.), and Dann (http://bioinformatics.oxfordjournals.org/content/31/5/761). The frequencies of allele and genotype were determined by direct counting of LRP10 alleles. Minor allele frequencies were compared using the chi-squared test in SPSS 21.0 (IBM, Chicago, USA).
Results
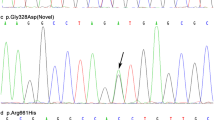
A total of 29 exonic or splicing single-nucleotide variants (SNVs) were detected in 79 patients with PD (Supplement 1), all of whom were heterozygous for the variants. Seven variants were identified in the previous reports [9, 13, 16], although in these studies the allele frequencies did not differ between PD patients and controls. Of the 29 variants identified in the present study, 10 were novel. Two of the novel variants (p.T278I, p.I531S) were predicted to be disease-causing or damaging by the five in silico tools mentioned above.
Minor allele frequencies of the other 12 variants were smaller than 0.05 in the ExAC and 1000 Genomes Project databases. Six of 12 variants were detected only in PD patients but not in healthy controls, and three (p.I61L, p.Q218X, p.L566P) were predicted to be disease-causing or damaging. Interestingly, variant c.T1697C(p.L566P), identified in our study, and variant c.C1696 > T(p.L566F), previously identified [13], were predicted to alter the same residue in the LRP10 protein sequence. The other six variants were detected in controls, and their frequencies were similar between cases and controls.
The clinical features of patients carrying the two novel variants and three known variant are listed in Table 1, and the flow chart for analyzing the 29 SNVs is shown in Fig. 1 and Supplement 1. All five mutated residues are highly conserved among species (Fig. 2).

Flow chart of the analysis of 29 LRP10 single-nucleotide variants (SNVs)

Alignment of partial LRP10 protein sequences from various species, showing conservation of the mutations p.I61L, p.Q218X, p.T278I, p.I531S, and p.L566P
Discussion
In the present study, we sequenced the LRP10 gene in 567 Han Chinese PD patients and identified five rare, potentially pathogenic exonic variants. LRP10 mutations were previously recognized as a potential cause of PD, Parkinson’s disease dementia, and dementia with Lewy bodies in different populations [8]. Two previous studies in Chinese populations similarly reported evidence for a role of LRP10 in patients with sporadic or familial PD, and identified three potentially pathogenic variants independently (Fig. 3) [13, 16]. However, other work based on the International Parkinson’s Disease Genomics Consortium database that includes 2835 individuals with PD and 5343 healthy individuals, mainly of northern European ancestry, did not identify LRP10 as a disease-causing gene in PD or dementia with Lewy bodies [10]. Similarly, studies on dementia with Lewy bodies and multiple system atrophy found no association between LRP10 and synucleinopathies [9, 11, 12]. These differences may be explained by ethnic variations among the study populations.

LRP10 protein structure showing the different domains and repeats. Blue arrows indicate the positions of the coding variants identified in the present study. (The splicing variant c.C652T: p.Q218X, 496 is not shown). Brown arrows indicate the positions of the coding variants identified in Chen et al [16]. Light green arrows indicate the positions of the coding variants identified in Shi et al [13]
Interestingly, we found five rare, potentially pathogenic exonic variants in five PD patients by in silico analysis. Clinical manifestations of all five patients carrying these variants included all typical Parkinson’s symptoms, similar to a previous study in another Chinese population [13, 16]. In the present study, the age at PD onset was over 50 years, and all patients showed good response to treatment with levodopa (> 30%). All five PD patients lacked a family history of PD, which may indicate that the variants are de novo mutations. However, incomplete penetrance or non-pathogenic variants cannot be completely ruled out. Since parental samples are not available, it is difficult to clarify the origins of the mutations.
LRP10 is a member of the low-density lipoprotein receptor family, although little is known about its function. Previous studies have shown that LRP10 is localized in vesicular structures closely associated with proteins implicated in trafficking between the trans-Golgi network and endosomes, including vacuolar protein sorting-associated protein 35 (VPS35) [8]. VPS35 has been associated with autosomal dominant PD through its ability to enhance mitochondrial fragmentation [17, 18]. Some studies suggested that LRP10 may play a role in the canonical Wnt/β-catenin signaling pathway, which regulates mitochondria morphology and function [19, 20]. Mitochondrial dysfunction appears to play an important role in the pathogenesis of PD [21, 22]. Collectively, these results may support a role for LRP10 in the pathogenesis of PD.
Our research has some limitations. Firstly, although we applied the MDS criteria, the diagnosis of these patients was not confirmed by pathology. Secondly, the number of patients recruited in our study was not large, and we could not include an adequate number of patients with familial PD. Thirdly, the potential function of genetic variants was only predicted by in silico tools. Finally, we were unable to obtain blood samples from parents of the patients with the five mutations. Analysis of such samples might help clarify the origins of the mutations, such as incomplete penetrance. Further studies, such as in vitro experiments, are needed in order to clarify whether these variants are functionally important.
Despite these limitations, our results suggest that LRP10 may play a role in sporadic Parkinson’s disease in Han Chinese patients. These findings should be verified and extended in larger samples, preferably from different ethnic groups, and analyzed in conjunction with familial pedigree data. Such work may help to elucidate how LRP10 contributes to the pathogenesis of neurodegenerative diseases.
Data Availability
The datasets used and/or analyzed in this study are available from the corresponding author on reasonable request.
References
Janca A (2002) Parkinson's disease from WHO perspective and a public health point of view. Parkinsonism Relat Disord 9(1):3–6
Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM et al (2007) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 68(5):384–386
Findley LJ (2007) The economic impact of Parkinson's disease. Parkinsonism Relat Disord 13(Suppl):S8–S12
Hoehn MM, Yahr MD (1998) Parkinsonism: onset, progression, and mortality. 1967. Neurology 50(2):318–316
Obeso JA, Stamelou M, Goetz CG, Poewe W, Lang AE, Weintraub D, Burn D, Halliday GM et al (2017) Past, present, and future of Parkinson's disease: a special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord 32(9):1264–1310
Unger EL, Eve DJ, Perez XA, Reichenbach DK, Xu Y, Lee MK, Andrews AM (2006) Locomotor hyperactivity and alterations in dopamine neurotransmission are associated with overexpression of A53T mutant human alpha-synuclein in mice. Neurobiol Dis 21(2):431–443
Jansen IE, Bras JM, Lesage S, Schulte C, Gibbs JR, Nalls MA, Brice A, Wood NW et al (2015) CHCHD2 and Parkinson’s disease. Lancet Neurol 14(7):678–679
Quadri M, Mandemakers W, Grochowska MM, Masius R, Geut H, Fabrizio E, Breedveld GJ, Kuipers D et al (2018) LRP10 genetic variants in familial Parkinson’s disease and dementia with Lewy bodies: a genome-wide linkage and sequencing study. Lancet Neurol 17(7):597–608
Guerreiro R, Orme T, Neto JL, Bras J, International DLBGC (2018) LRP10 in alpha-synucleinopathies. Lancet Neurol 17(12):1032–1033
Kia DA, Sabir MS, Ahmed S, Trinh J, Bandres-Ciga S (2018) International Parkinson's Disease Genomics C. LRP10 in alpha-synucleinopathies. Lancet Neurol 17(12):1032
Pihlstrom L, Schottlaender L, Chelban V, Houlden H, Consortium MSAE (2018) LRP10 in alpha-synucleinopathies. Lancet Neurol 17(12):1033–1034
Tesson C, Brefel-Courbon C, Corvol JC, Lesage S, Brice A (2018) French Parkinson’s disease genetics study G. LRP10 in alpha-synucleinopathies. Lancet Neurol 17(12):1034
Shi CH, Luo HY, Fan Y, Li YS, Xu YM (2018) LRP10 in alpha-synucleinopathies. Lancet Neurol 17(12):1034–1035
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, Obeso J, Marek K et al (2015) MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 30(12):1591–1601
Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG et al (2015) Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 7(270):270ra6
Chen Y, Cen Z, Zheng X, Pan Q, Chen X, Zhu L, Chen S, Wu H et al (2019) LRP10 in autosomal-dominant Parkinson’s disease. Mov Disord 34(6):912–916
Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ et al (2011) VPS35 mutations in Parkinson disease. Am J Hum Genet 89(1):162–167
Zhou L, Wang W, Hoppel C, Liu J, Zhu X (2017) Parkinson's disease-associated pathogenic VPS35 mutation causes complex I deficits. Biochim Biophys Acta Mol basis Dis 1863(11):2791–2795
Wang J, Li Y, Gao L, Yan F, Gao G, Li L (2018) GSK-3beta inhibitor alsterpaullone attenuates MPP(+)-induced cell damage in a c-Myc-dependent manner in SH-SY5Y cells. Front Cell Neurosci 12:283
Jeong YH, Sekiya M, Hirata M, Ye M, Yamagishi A, Lee SM, Kang MJ, Hosoda A et al (2010) The low-density lipoprotein receptor-related protein 10 is a negative regulator of the canonical Wnt/beta-catenin signaling pathway. Biochem Biophys Res Commun 392(4):495–499
Wang B, Cai Z, Tao K, Zeng W, Lu F, Yang R, Feng D, Gao G et al (2016) Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy. 12(8):1215–1228
Perez Carrion M, Pischedda F, Biosa A, Russo I, Straniero L, Civiero L, Guida M, Gloeckner CJ et al (2018) The LRRK2 variant E193K prevents mitochondrial fission upon MPP+ treatment by altering LRRK2 binding to DRP1. Front Mol Neurosci 11:64
Acknowledgments
We are very grateful to You Chen, Wei Luo, and colleagues for providing the date for joint analysis. We thank Shanghai Genesky Bio-Tech for technical support. We also thank the patients and controls in this study.
Funding
This work was funded by the National Natural Science Foundation of China (grant numbers 81471300, 81960242) and the Basic Conditions Platform Construction Project of Sichuan Science and Technology Department (grant number 2019JDPT0015) and the Key Research and Development Project of Sichuan Province (grant number 2020YFS0259).
Author information
Authors and Affiliations
Contributions
YMX(Yanming Xu), QZZ, PPN, and XLY participated in conceiving and designing the study, analyzing and interpreting the data, as well as drafting and reviewing the manuscript. CHS and YMX(Yuming Xu) collected, analyzed, provided relevant information of the patients and controls in another Chinese population, and participated in reviewing the manuscript. QYS participated in collecting the data and reviewing the manuscript. HYH, DX and YLC participated in collecting clinical data and helped review the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Ethical Approval
The protocol of this study was approved by the Ethics Committee of West China Hospital of Sichuan University (Chengdu, China), and the study was performed in accordance with the ethical standards in the 1964 Declaration of Helsinki and its amendments.
Consent to Participate
On admission, each participant signed written informed consent for their data to be analyzed and published anonymously for research purposes.
Consent to publication
On admission, each participant signed written informed consent for their data to be analyzed and published anonymously for research purposes.
Code Availability
The software applications used and/or analyzed in this study are available from the corresponding author on reasonable request.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOC 114 kb).
Rights and permissions
About this article

Cite this article
Zhao, Q., Ning, P., Yang, X. et al. LRP10 Mutations May Correlate with Sporadic Parkinson’s Disease in China. Mol Neurobiol 58, 1212–1216 (2021). https://doi.org/10.1007/s12035-020-02186-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02186-9