
Abstract
In the past two decades, genetic studies of familial forms of Parkinson’s disease (PD) have shown evidence that PD has a significant genetic component. Indeed, 12 genes are strongly involved in PD causality, three of them having dominant inheritance and 9 causing early-onset autosomal recessive forms, including 3 with a typical PD and 6 with an atypical parkinsonism. The aim of this study was to determine the genetic basis of familial PD in Moroccan patients. We selected 18 Moroccan index case with familial forms of PD. Patients were first screened for exon-rearrangements by MLPA kit. They were then analyzed by gene panel next-generation sequencing (NGS). Functional variants with minor allele frequencies < 0.5% in public databases were considered potential candidate variants to PD. In the 18 PD patients with a positive family history that were analyzed, MLPA assays identified PRKN deletions in two patients: a homozygous exon 3–5 deletion and a heterozygous exon 4 deletion. Sixteen rare SNV were identified by NGS, four of them were novel. Seven mutations were categorized as pathogenic, five as likely pathogenic, two to be of uncertain significance, and 3 were predicted to be likely benign but may give a weaker pathogenic effect and could contribute to PD since they were found in late-onset PD patients. Rare or novel mutations that could be related to the disease were identified in 72% of these patients (13/18), including nine with bi-allelic pathogenic/likely pathogenic variants in genes causing recessive PD, particularly PRKN and PINK1. Mutations in genes with dominant inheritance were found in 4/18 patients (22%).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer disease. PD prevalence increases with age affecting 2% of individuals over 60 years of age and 3% of the population older than age 80 (Tysnes and Storstein 2017). The most typical PD motor features are bradykinesia, rest tremor, rigidity, and postural instability, whereas the most common non-motor features comprise cognitive impairment, olfactory dysfunction, autonomic dysfunction, and psychiatric symptoms (De Virgilio et al. 2016).
In the past two decades, genetic studies of familial forms of PD gave evidence that PD has a significant genetic component. Indeed, 12 genes are strongly involved in PD causality (Lunati et al. 2018); three of them are associated with autosomal dominant inheritance (ADPD) (LRRK2, SNCA and VPS35) and nine cause early-onset autosomal recessive forms (ARPD), including three with a typical PD (PRKN, PINK1, DJ-1) and six with an atypical parkinsonism (ATP13A2, PLA2G6, FBXO7, DNAJC6, SYNJ1, VPS13C). Generally, ADPD presents with a clinical phenotype identical to that found in idiopathic PD (Trinh et al. 2018) and the LRRK2 p.G2019S mutation was reported to be the most common mutation in PD worldwide (Monfrini and Di Fonzo 2017). Recessive forms of PD are mainly due to mutations in the PRKN gene that are essentially exon deletions, followed by point mutations in PINK1and DJ-1, all of them causing early-onset PD with phenotypes resembling typical PD with good Levodopa response. Other rare pathogenic mutations in ATP13A2, PLA2G6, FBXO7, DNAJC6, SYNJ1, and VPS13C cause a more severe disease with an enriched clinical picture and a poor response to levodopa. Furthermore, many other genes are suspected to be responsible for both ADPD and ARPD but these results need replications in order to be confirmed (Lunati et al. 2018).
All these genes have been discovered by studying familial forms of PD, exhibiting a classical Mendelian type of inheritance, which account for approximately 5–10% of PD patients (Deng et al. 2018; Lunati et al. 2018). In Maghreb countries, this percentage seems to be relatively larger. The LRRK2 p.G2019S mutation alone accounts for about 40% in the region of North Africa (Lesage et al. 2006). In an earlier study in Morocco, we have shown a higher frequency of p.G2019S of 41% overall, with 30% for sporadic cases and 76% for patients with an autosomal dominant mode of inheritance. These LRRK2 p.G2019S carriers showed a typical PD phenotype with dystonia and sleep disorders (Bouhouche et al. 2017b). In another series of 18 Moroccan patients with positive family history and consanguinity, we have shown a Mendelian inheritance rate of more than 40%, including 3 patients with exon deletions in PRKN, 2 with point mutations in PINK1 and 2 other patients with the founder mutation W258X in ATP13A2 which was responsible for Kufor-Rakeb syndrome, a juvenile-onset atypical Parkinson disease associated with spasticity and dementia (Bouhouche et al. 2017a).
In the present study, after excluding the recurrent LRRK2 p.G2019S mutation, we analyzed the genetic bases of 18 familial PD patients from Morocco, with an approach that included both gene dosage and gene panel next-generation sequencing (NGS) associated with PD and overlapping phenotypes. We also compared clinical characteristics between patients with and without PD gene causal mutations.
Patients and Methods
Patients
The 18 Moroccan index cases, with a familial form of PD, were recruited from the Movement Disorder Unit of the Department of Neurology (Specialties Hospital, Rabat, Morocco). PD was diagnosed according to the United Kingdom Parkinson’s Disease Society Brain Bank criteria (Hughes et al. 1992) and patients were submitted to a structured clinical interview as described in previous literature (Bouhouche et al. 2017a). A pedigree was drawn for all families which were divided into ADPD with parent-child transmission, ARPD with consanguineous healthy parents, and unspecified inheritance (UIPD) when parents were consanguineous and one of them was with PD.
The Biomedical Research Ethics Committee of the Medical School of Rabat (CERB) approved this study and written informed consent was obtained from all subjects in accordance with the Declaration of Helsinki.
Genetic Analysis Procedure
Genomic DNA was extracted from peripheral blood leukocytes using the Isolate II Genomic DNA kit from Bioline. All patients were first screened by Sanger sequencing for the LRRK2 p.G2019S mutation as described in prior literature (Bouhouche et al. 2017b). Negative patients for the p.G2019S mutation were then screened for exon-rearrangements in the PD genes SNCA, PRKN, PINK1, DJ-1, ATP13A2, and LRRK2 through the multiplex ligation-dependent probe amplification (MLPA) using the P-051 kit, according to the manufacturer’s protocol. Data analysis was performed using the Coffalyser software (MRC- Holland, Amsterdam, The Netherlands).
Afterward, patients whose pedigree suggested an ADPD were analyzed for point mutations at the ICM of Paris, by NGS illumina technology, using a panel of 22 genes as also described previous literature (Bouhouche et al. 2017a). Those suggestive of ARPD and UIPD were screened at UATRS of Rabat, by a Thermo Fisher technology, using a panel of 25 PD-related genes. Briefly; 697 amplicons, separated into two pools, were targeted with a coverage of 99.06%. Library preparation, bead templating (emulsion PCR), and PI Chips v2 loading were performed on an Ion Chef System and the sequencing was done on the Ion Proton machine. The generated VCF files were then imported to the online Server of Ion Reporter Software v5.10, for variant analysis and annotations. The 22- and 25- gene panels used here contained 19 genes in common and their complete gene lists are given in Supplementary Table 1. For the two NGS technologies used, functional variants with MAF < 0.5% in public databases (ExAC, 1000G dbSNP138, GnomAD) and of Caucasian population were considered potential PD variants. Twenty healthy control individuals of Moroccan origin were also screened by the 25-gene panel as a reference for the study population. All the reported variants were confirmed by Sanger sequencing.
For all validated mutations, the interpretation of pathogenicity was based on population data (ExAC, 1000 genomes and GnomAD), computational prediction tools (Sift, Polyphen2, MutationAssessor, MutationTaster and CADD), the amino-acid conservation, and the evolutionary nucleotide conservation. Exon deletions, INDELS causing frameshift, and nonsense mutations were all considered pathogenic, whereas missense mutations were classified as pathogenic, likely pathogenic, likely benign, benign, or variant of unknown significance according to the American College of Medical Genetics and Genomics (ACMG) guidance for pathogenicity classification (Richards et al. 2015).
Results
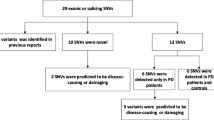
A total of 18 unrelated PD patients with positive family history were recruited in the Department of Neurology of Rabat, of which 11 were male (61%). Based on inheritance information recorded from the pedigrees, the inheritance mode was in favor of ADPD for 9 families (50%), of ARPD for 6 (33%), and UIPD for 3 (17%) by the simultaneous presence of parent-child transmission and consanguinity (Supplementary Fig. 1). The mean age at examination was 62.64 ± 16.68 years (range 29–83) and the mean age at onset was 57.4 ± 18.90 (range 17–81) years. All these 18 index cases were first screened for exon 41 of the LRRK2 gene and were all negative for p.G2019S mutation—the most common PD mutation in North-Africa. They were then investigated for genetic analysis combining MLPA and gene panels by targeted NGS. Analysis of NGS sequence data revealed uniform coverage and high read depths in all samples. The mean coverage was 1475X ± 695X and target base coverage was 98.90% at 20X and 97.12% at 100X. From the variants generated by the two technologies, we selected only functional variants with the criterion of the frequency of the minor allele being less than < 0.5% in public databases. All rare variants identified in our PD patients were confirmed by Sanger sequencing and are summarized in Table 1 and their classification of pathogenicity according to the ACMG guidance is reported in Table 2. Rare genetic variants that could be related to the disease were identified in 72% patients (13 out of 18). Although most index cases came from families whose inheritance was suspected to be dominant, the mutations were frequently found in genes with recessive transmission (9/18, 50%) and this was in a homozygous or compound heterozygous state.
Mutations Identified in Genes with Autosomal Recessive Inheritance
MLPA assays identified PRKN deletions in two patients, a homozygous exon 3–5 deletion in patient 3983, while a heterozygous exon 4 deletion was identified in patient 3565 (Table 1). In addition to exon deletions in the PRKN gene, three other point mutations were identified in three patients by NGS analysis. The mutation c.458C>G (p. Pro153Arg) in exon 4 (Fig. 1b), identified at homozygous state in patient 3929, was rare in public databases but never found at homozygous state, absent in Moroccan control individuals and predicted likely pathogenic by bioinformatics tools. This mutation was found also in patient 3875 at compound heterozygous state, with the already reported pathogenic mutation c.1204C>T (p.Arg402Cys). The third mutation c.818A>G (p.Asn273Ser) was identified in compound heterozygous state in patient 3565, having a heterozygous deletion of the PRKN exon 4 (Fig. 1c). The PRKN p.Asn273Ser mutation was absent in ExAc, 1000 genomes, and the in-house individual controls and was predicted likely pathogenic.

Sanger validation of novel and rare mutations predicted pathogenic. a Mutation c.328A>C in DJ-1. b Mutation c.458C>G in PARK2. c Mutation c.818A>G in PARK2. d Mutation c.5926C>T in VPS13C. e Mutation c.2297G>A in DCTN1. (i) Normal sequences, (ii) Mutated sequences, (iii) Mutation conservation in different species
The PINK1 nonsense mutation p.Gln456* was identified at homozygous state in two cases (3889 and 3989) and classified pathogenic according to the ACHG criteria. The mode of inheritance of the patient’s family 3889 has been classified as UIPD since the patient comes from a consanguineous marriage and his mother was also affected. The Sanger sequencing of exon 7 of the PINK1 gene shows that the mother is also homozygous for this mutation. A novel missense mutation c.328A>C in the DJ-1 gene was found at homozygous state in patient 3949 (Fig. 1a). This mutation results in the replacement of the Threonine amino acid by another non-conservative one, Proline at position 110 (p.Thr110Pro). This was not found in all public databases and only in one out of 232 Moroccan control chromosome; it concerns a highly conservative nucleotide and predicted pathogenic by all in silico prediction tools used, including the highly CADD score of 28.4 obtained.
In patient 3849, we identified two mutations in a compound heterozygous state in the VPS13C gene, a frameshift pathogenic mutation c.6238_6239del leading to a stop codon 17 amino acids after the Asp at position 2080 (p.Gln2080Aspfs*17) and a novel mutation c.5926C>T (p.Leu1976Phe) predicted to be likely pathogenic (Fig. 1d). In patient 3865, we found three heterozygous rare mutations, the c.1147G>A (p.Ala383Thr) and the c.4757C>T (p.Pro1586Leu) in SYNJ1 and one mutation c.3961A>G (p.Ile1321Val) in the VPS13C gene. All these 3 rare mutations were predicted to be likely benign by bioinformatics tools.
Mutations Identified in Genes with Autosomal Dominant Inheritance
Rare mutations in genes causing autosomal dominant inheritance were found only in 4 out of 18 patients (22%). Patient 3795 has a predicted pathogenic mutation in the DCTN1 gene, c.2297G>A (p.Arg766Gln) (Fig. 1e) and also a novel heterozygous mutation in the TH gene implicated in the autosomal recessive Segawa syndrome. In patient 3637 with UIPD, we identified a rare mutation in exon 5 of the UCHL1 gene, c.374C>T (p.Ser125Phe) found only in one individual among 251,351 of gnomAD exome database and predicted pathogenic. Two patients showed a rare variant of the LRRK2 gene; patient 3846 has the c.356T>C (p.Leu119Pro), which was found in 1/40 control chromosomes but predicted as damaging by 3/4 in silico prediction tools and a CADD score of 26.9 was classified a variant of unknown significance (VUS). The other patient 3848 showed a novel mutation in exon 29 of the LRRK2 gene, c4136A>G (p.Gln1379Arg) absent in 192 Moroccan control chromosomes. However, this mutation, predicted benign by all in silico pathogenicity prediction tools, has a CADD score of 15.58 and was classified as VUS. This patient has another mutation in the PINK1 gene in heterozygous state, c.851C>T (p.Ser284Phe) that was predicted to be likely pathogenic.
Clinical Data
Clinical characteristics of the PD probands with mutations in genes causing autosomal recessive genes are summarized in Table 3. All patients with pathogenic mutations in the DJ-1, PINK1, and PRKN genes presenting with early-onset PD were under 50 years of age. The two patients with the PINK1 mutation (3889 and 3949) showed a typical PD phenotype with the absence of non-motor symptoms or levodopa-induced dyskinesia even after a long disease duration. Patients with PRKN pathogenic mutations (3929, 3983, and 3875) started the disease with bradykinesia or dystonia and manifested an akinetic-rigid form of parkinsonism. Only patient 3929, with a long disease duration of 19 years, showed a severe phenotype with motor fluctuation and dyskinesia for a moderate levodopa dose and non-motor symptoms such as pain, constipation, sleep disorder, and autonomic symptoms. Patient 3565 with compound heterozygous mutations, the pathogenic exon 4 deletion and the p.Asn273Ser predicted to be likely pathogenic, manifested Parkinsonism at 81 years; this was improved by very low doses of antiparkinsonian drugs and this patient showed only a mild degree of cognitive impairment. Furthermore, patients with likely benign or likely pathogenic mutations in SYNJ1and VPS13C presented with a late-onset PD over 60 years of age with mixed phenotype, various non-motor symptoms and motor fluctuation with dyskinesia under very low-dose levodopa therapy.
Clinical characteristics of patients with mutations in genes causing autosomal dominant PD are summarized in Table 4. All these patients presented with late-onset PD (over 60 years of age) and tremor as the initial symptom; they had mixed phenotypes and non-motor symptoms. To note, urinary dysfunction and constipation were the most frequent signs. The most severe phenotype was seen in patient 3848 who had also psychiatric symptoms and cognitive decline after a disease duration of 8 years and a levodopa equivalent dose of 600 mg per day.
Patient 3795, with DCTN1 mutation, started the disease at 70 years and was diagnosed as PD after 2 years of disease evolution. At age 78, she developed frightening visual hallucinations with low doses of dopaminergic agonists that were otherwise ineffective. The hallucinations initially decreased but persisted with levodopa. She also had depressive symptoms associated with apathy, rapid eye movement (REM) behavioral disorder, and sleep apnea syndrome. She complained about autonomic symptoms as urgency, orthostatic hypotension, gastroparesis, and constipation. She also had exercise dyspnea without cardiac or respiratory diseases. Dyspnea affects her daily life activities as she had to get regular rest periods during housework and to split her meal times. Otherwise, she did not have dysexecutive syndrome, but she reported intermittent dysphasia. Of note, her father presented Parkinsonism and her brother suffered from respiratory dyspnea. The patient’s cerebral MRI showed fronto-parietal atrophy.
Finally, the five patients without any causal mutation identified all showed tremor as the initial symptom but were variable for disease age of onset, clinical phenotype, and non-motor symptoms (Table 4).
Discussion
In this study, we describe the genetic and phenotypic features of 18 Moroccan patients with familial form of PD. Based on the pedigree structure, the inheritance mode was in favor of autosomal dominant for 9, of autosomal recessive for 6, and unspecified for 3 by the simultaneous presence of parent-child transmission and consanguinity. After excluding the most frequent LRRK2 p.G2019S mutation in the Maghreb, which accounts for app. 40% of all PD cases (Bouhouche et al. 2017b), the genetic analysis strategy adopted here was one of searching for CNV in PD genes by MLPA analysis and gene-panel NGS. Overall, 16 mutations were identified in the 18 index patients studied. According to the ACMG guidelines to classify their pathogenicity, seven of them were categorized as pathogenic, 5 as likely pathogenic, one to be of uncertain significance, and 3 of them were predicted to be likely benign.
The most common mutated gene was PRKN, with two exon deletions and three missense mutations found in four patients. The exon rearrangements are located between exons 2 and 8 of the PRKN gene, as reported previously in autosomal recessive Moroccan patients (Bouhouche et al. 2017a), further confirming this region as a mutational hotspot. The three missense mutations were predicted pathogenic or likely pathogenic. The p.Pro153Arg was found in homozygous state in one patient and at compound heterozygous with the p.Arg402Cys mutation. In addition, the mutation p.Asn273Ser was found at compound heterozygous state in association with the pathogenic exon 4 deletion. These different combinations thus confirm the pathogenicity of these three rare missense mutations.
The second most frequent mutated gene was PINK1 in which the same nonsense and pathogenic mutation p.Gln456* was found in two patients. This pathogenic mutation was already reported in another patient of Moroccan origin (Bouhouche et al. 2017a) suggesting that it probably resulted from a common founder.
The clinical phenotype of patients with PRKN and PIsNK1 mutations is indistinguishable from each other and is mainly characterized by early-onset manifestation, good levodopa response, and slow disease progression as reported earlier (Kasten et al. 2018). This was not the case for patient 3565 who started the disease at 81 years old probably due to weaker pathogenic effect of the PRKN p.Asn273Ser mutation, resulting in the replacement of the asparagine by the conservative amino acid serine that could predispose to a late and less severe form of the disease. Similarly, while the SYNJ1 and VPS13C genes are known to cause an early-onset PD generally between 20 and 40 years (Lunati et al. 2018), patients 3889 and 3865, with mutations in these two genes, were aged 73 and 65 at onset respectively. The mutation p.Leu1976Phe in VPS13C was predicted likely pathogenic but gives rise to a conservative amino acid, whereas the 3 mutations in the SYNJ1 and VPS13C genes of patients 3865 were predicted likely benign but gave a non-conservative amino acid. All these missense mutations could induce a weaker pathogenic effect and could act as a genetic susceptibility factor for late-onset PD.
For rare mutations found in genes known to cause autosomal dominant inheritance, patient 3795, with DCTN1 p.Arg766Gln mutation, presented a defined Perry syndrome based on diagnostic criteria published by Mishima et al. (2018). Some clinical features are worth mentioning especially the late disease onset and slow progression. Indeed, the mean age of onset reported in the literature was 49.1 ± 6.6 and an age at death of 55 ± 7.4 years (Mishima et al. 2018). Dopamine agonists were ineffective in our patient as reported in the literature (Perry et al. 1975; Lechevalier et al. 1992; Bhatia et al. 1993; Tsuboi et al. 2002; Mishima et al. 2015) with visual hallucinations that resolved after drug withdrawal but persisted even with levodopa, which supposed a cortical spread of the pathogenic process. Contrary to known co-occurrence of visual hallucination with cognitive impairment (Aarsland et al. 2014), our patient did not report cognitive complaints except for some intermittent dysphasia. This symptom was reported to precede respiratory failure (Perry et al. 1975). The patient presented exercise dyspnea limiting her activity of daily living. Sleep disorders were reported in 20.6% including insomnia and sleep apnea syndrome (SAS) (Mishima et al. 2015). We described in our case rapid eye movement behavioral disorders too. SAS probably participated in the respiratory failure but polysomnography cannot be performed to confirm this. Depressive symptoms and apathy are well-defined psychiatric features of Perry syndrome. We prescribed Escitalopram but the outcome is under investigation; antidepressant therapy resistance was reported by several authors (Wider and Wszolek 2008). Furthermore, our patient was considered and treated as PD based on age at onset, asymmetric phenotype, sensitivity to levodopa and the occurrence of non-motor symptoms that fitted with recent criteria for idiopathic PD (Postuma et al. 2015; Postuma and Berg 2017). Respiratory symptoms were occulted as the patient reported familial history of asthma. Indeed, Perry syndrome may be misdiagnosed as Parkinson’s disease (PD) especially familial early PD. Weight loss, respiratory symptoms, and rapid progression allow distinguishing it from EOPD but it is more difficult in the early stage (Mishima et al. 2018). In our case, the age of onset was late, the progression was slow, and there was no weight loss, which could explain our misdiagnosis.
In patient 3637, we identified a heterozygous c.374C>T mutation in exon 5 of the UCHL1 gene resulting in a p.Ser125Phe substitution in a highly conserved motif. This mutation was reported in only one individual in all the public databases and was classified pathogenic. To date, only one mutation in the UHCL1 gene was reported in a patient, from German origin, with the familial form of PD (Leroy et al. 1998), but this finding has not been replicated and so far the gene is considered only as a risk factor for PD. Recent data increasingly involve the UCHL1 gene in the development of PD. Indeed, it has been shown that S-nitrosylation of UCHL1 promotes α-synuclein aggregation, the major component of Lewy bodies (Kumar et al. 2017) and the UCHL1 gene was upregulated in PD patients when compared with healthy controls (Wang et al. 2017). Moreover, the knockdown of the Drosophila ortholog of UCHL1 led to the degeneration of dopaminergic neurons which resulted in locomotor dysfunction that mimics the PD phenotype (Tran et al. 2018).
Two rare variants in the LRRK2 gene, the p.Leu119Pro and the p.Gln1379Arg predicted as VUS, were identified in two patients with a late-onset PD, rapid disease progression, and non-motor symptoms.
The causal gene remained unidentified for 5 families all apparently ADPD, with variable age at onset and variable severity. This suggests that mutations of known genes may have been missed such as in gene-regulatory elements or that other new genes were involved; these may be potentially identifiable by whole-exome or whole-genome sequencing. Interestingly, no rare or pathogenic mutation was discovered in the SNCA gene in this Moroccan series of 18 patients with familial PD, as well as in a previous study on patients from the same population (Bouhouche et al. 2017a). Indeed, SNCA mutations were reported to be the cause of a minority of familial PD; most of them were from Caucasian and Asian origin (Trinh et al. 2018; Riederer et al. 2019).
Twelve out of the 16 rare mutations found were in genes with autosomal recessive inheritance and only five in genes with autosomal dominant inheritance. Therefore, the inheritance mode of the causal gene found in these families does not correspond, in the majority of cases, to what has been assumed from the pedigree information. Indeed, it is rare to have several living patients over several generations because Parkinson’s disease occurs at a late age, and this in addition to the fact that the ancestors of the index case carrying pathogenic mutations could die before declaring the disease and count as healthy individuals. Moreover, the status of deceased patients was based only on interrogatories of family members and could be false, inducing errors of judgment on the mode of inheritance. In these conditions, the pedigree is not always relevant in the orientation of the genetic analysis to be performed and the real mode of transmission can only be determined from the inheritance of the gene in which the pathogenic mutation is identified. The most efficient diagnostic strategy should consider the frequency of gene mutations in the population studied and the screening of the known PD genes by the most appropriate new techniques.
One of the limitations of our study was the small sample size. This is mainly due to the low proportion of familial forms of PD and to the fact that the majority of them in the Moroccan population are due to the recurrent LRRK2 p.G2019S mutation. On the other hand, in some patients, mutations in the compound heterozygous state have not been verified if they are in cis or trans due to unavailability of DNA samples from parents mostly deceased. Finally, the possible pathogenicity of some of the missense variants was based on the frequency and bioinformatics analysis, and therefore, other functional studies would be desirable to ascertain their pathogenicity.
Conclusion
In conclusion, 16 rare variants in PD genes were identified in this study, 7 of them were classified as pathogenic, 5 as likely pathogenic, and 4 were categorized as likely benign or of uncertain significance. These last variants were seen particularly in late-onset PD indicating that they also contribute to the risk of PD. Otherwise, the disease course of patients with mutations causing ADPD is more severe and of rapid progression in comparison with AR genes. It should be noted that the disease, when it occurs late in age (over 60 years), is often associated with non-motor symptoms regardless of the mutated gene.
Data Availability
The datasets used and analyzed during the current study are available from the corresponding author upon request.
References
Aarsland D, Taylor JP, Weintraub D (2014) Psychiatric issues in cognitive impairment. Mov Disord 29:651–662
Bhatia KP, Daniel SE, Marsden CD (1993) Familial parkinsonism with depression: a clinicopathological study. Ann Neurol 34:842–847
Bouhouche A, Tesson C, Regragui W, Rahmani M, Drouet V, Tibar H, Souirti Z, Ben El Haj R, Bouslam N, Yahyaoui M, Brice A, Benomar A, Lesage S (2017a) Mutation analysis of consanguineous Moroccan patients with Parkinson’s disease combining microarray and gene panel. Front Neurol l8:567
Bouhouche A, Tibar H, Ben El Haj R, El Bayad K, Razine R, Tazrout S, Skalli A, Bouslam N, Elouardi L, Benomar A, Yahyaoui M, Regragui W (2017b) LRRK2 G2019S mutation: prevalence and clinical features in Moroccans with Parkinson’s disease. Park Dis 2017:2412486
De Virgilio A, Greco A, Fabbrini G, Inghilleri M, Rizzo MI, Gallo A, Conte M, Rosato C, Ciniglio Appiani M, de Vincentiis M (2016) Parkinson’s disease: autoimmunity and neuroinflammation. Autoimmun Rev 15:1005–1011
Deng H, Wang P, Jankovic J (2018) The genetics of Parkinson disease. Ageing Res Rev 42:72–85
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55:181–184
Kasten M, Hartmann C, Hampf J, Schaake S, Westenberger A, Vollstedt EJ, Balck A, Domingo A, Vulinovic F, Dulovic M, Zorn I, Madoev H, Zehnle H, Lembeck CM, Schawe L, Reginold J, Huang J, König IR, Bertram L, Marras C, Lohmann K, Lill CM, Klein C (2018) Genotype-phenotype relations for the Parkinson’s disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov Disord 33:730–741
Kumar R, Jangir DK, Verma G, Shekhar S, Hanpude P, Kumar S, Kumari R, Singh N, Sarovar Bhavesh N, Ranjan Jana N, Kanti Maiti T (2017) S-nitrosylation of UCHL1 induces its structural instability and promotes α-synuclein aggregation. Sci Rep 7:44558
Lechevalier B, Schupp C, Fallet-Bianco C, Viader F, Eustache F, Chapon F, Morin P (1992) Familial parkinsonian syndrome with athymhormia and hypoventilation. Rev Neurol (Paris) 148:39–46
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH (1998) The ubiquitin pathway in Parkinson's disease. Nature 395:451–452
Lesage S, Dürr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, Pollak P, Brice A (2006) LRRK2 G2019S as a cause of Parkinson’s disease in north African Arabs. N Engl J Med 354:422–423
Lunati A, Lesage S, Brice A (2018) The genetic landscape of Parkinson's disease. Rev Neurol (Paris) 174:628–643
Mishima T, Fujioka S, Kurisaki R, Yanamoto S, Higuchi MA, Tsugawa J, Fukae J, Neshige R, Tsuboi Y (2015) Impulse control disorders and punding in Perry syndrome. Parkinsonism Relat Disord 21:1381–1382
Mishima T, Fujioka S, Tomiyama H, Yabe I, Kurisaki R, Fujii N, Neshige R, Ross OA, Farrer MJ, Dickson DW, Wszolek ZK, Hattori N, Tsuboi Y (2018) Establishing diagnostic criteria for Perry syndrome. J Neurol Neurosurg Psychiatry 89:482–487
Monfrini E, Di Fonzo A (2017) Leucine-rich repeat kinase (LRRK2) genetics and Parkinson’s disease. Adv Neurobiol 14:3–30
Perry TL, Bratty PJ, Hansen S, Kennedy J, Urquhart N, Dolman CL (1975) Hereditary mental depression and parkinsonism with taurine deficiency. Arch Neurol 32:108–113
Postuma RB, Berg D (2017) New diagnostic criteria for Parkinson’s disease. Int Rev Neurobiol 132:55–78
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, Obeso J, Marek K, Litvan I, Lang AE, Halliday G, Goetz CG, Gasser T, Dubois B, Chan P, Bloem BR, Adler CH, Deuschl G (2015) MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 30:1591–1601
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Riederer P, Berg D, Casadei N, Cheng F, Classen J, Dresel C, Jost W, Krüger R, Müller T, Reichmann H, Rieß O, Storch A, Strobel S, van Eimeren T, Völker HU, Winkler J, Winklhofer KF, Wüllner U, Zunke F, Monoranu CM (2019) α-Synuclein in Parkinson’s disease: causal or bystander? J Neural Transm (Vienna) 126:815–840
Tran HH, Dang SNA, Nguyen TT, Huynh AM, Dao LM, Kamei K, Yamaguchi M, Dang TTP (2018) Drosophila ubiquitin C-terminal hydrolase knockdown model of Parkinson’s disease. Sci Rep 8:4468
Trinh J, Zeldenrust FMJ, Huang J, Kasten M, Schaake S, Petkovic S, Madoev H, Grünewald A, Almuammar S, König IR, Lill CM, Lohmann K, Klein C, Marras C (2018) Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord 33:1857–1870
Tsuboi Y, Wszolek ZK, Kusuhara T, Doh-ura K, Yamada T (2002) Japanese family with parkinsonism, depression, weight loss, and central hypoventilation. Neurology 58:1025–1030
Tysnes OB, Storstein A (2017) Epidemiology of Parkinson’s disease. J Neural Transm (Vienna) 124:901–905
Wang J, Liu Y, Chen T (2017) Identification of key genes and pathways in Parkinson’s disease through integrated analysis. Mol Med Rep 16:3769–3776
Wider C, Wszolek ZK (2008) Rapidly progressive familial parkinsonism with central hypoventilation, depression and weight loss (Perry syndrome)-a literature review. Parkinsonism Relat Disord 14:1–7
Acknowledgments
We are grateful to all patients for their participation in this study and to Dr. Leslie Hunter for improving the English language of the manuscript.
Funding
The study was supported by the “Ministère de l’Enseignement Supérieur, de la Recherche Scientifique et de la Formation des Cadres” (MESRSFC) of Morocco and the “Centre National de Recherche Scientifique et Technique” (CNRST).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Approval
All research was approved by the ethics committee of biomedical research of Medical School and Pharmacy of Rabat (CERB).
Consent to Participate
Written informed consent was obtained from all of the participants in the study.
Consent for Publication
Written informed consent for publication of clinical details and clinical images was obtained from all of the participants.
Code Availability
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Supplementary Fig. 1
Family pedigree of the eighteen index PD cases studied. a Families with probably autosomal dominant inheritance; b families with probably autosomal recessive inheritance; c families with unspecified inheritance. (PPT 925 kb)
Supplementary Table 1
Comparison between the two NGS gene panels used in this study. (DOCX 13 kb)
Rights and permissions
About this article

Cite this article
Smaili, I., Tesson, C., Regragui, W. et al. Gene Panel Sequencing Identifies Novel Pathogenic Mutations in Moroccan Patients with Familial Parkinson Disease. J Mol Neurosci 71, 142–152 (2021). https://doi.org/10.1007/s12031-020-01635-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-020-01635-3