
Abstract
Ferroportin plays an essential role for iron transport through the blood-brain barrier (BBB), which is formed by brain capillary endothelial cells (BCECs). To maintain the integrity of the BBB, the BCECs gain support from pericytes and astrocytes, which together with neurons form the neurovascular unit (NVU). The objectives of the present study were to investigate ferroportin expression in primary cells of the NVU and to determine if ferroportin mRNA (Fpn) expression is epigenetically regulated. Primary rat BCECs, pericytes, astrocytes, and neurons all expressed ferroportin mRNA at varying levels, with BCECs exhibiting the highest expression of Fpn, peaking when co-cultured but examined separately from astrocytes. Conversely, Fpn expression was lowest in isolated astrocytes, which correlated with high DNA methylation in their Slc40a1 promoter. To provide further evidence for epigenetic regulation, mono-cultured BCECs, pericytes, and astrocytes were treated with the histone deacetylase inhibitors valproic acid (VPA) and sodium butyrate (SB), which significantly increased Fpn and ferroportin protein in BCECs and pericytes. Furthermore, 59Fe export from BCECs was elevated after treatment with VPA. In conclusion, we present first time evidence stating that Fpn expression is epigenetically regulated in BCECs, which may have implications for pharmacological induction of iron transport through the BBB.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The main protection against influx of toxins, pathogens, and other harmful molecules into the brain is denoted by the vascular barrier system, generally referred to as the blood-brain barrier (BBB) [1, 2]. The BBB is formed by brain capillary endothelial cells (BCECs) with their intermingling tight junctions, which regulate the transport of molecules into the parenchyma [3, 4]. The BCECs gain support from the basement membrane, pericytes, and astrocytes that together with neurons form the neurovascular unit (NVU) [4,5,6].
The transport of iron through the BBB remains a source of uncertainty with various hypotheses for iron transport prevailing, including transport via endocytotic or transcytotic pathways [7]. In both pathways, iron import into the BCECs from the blood plasma is initiated by binding of transferrin to transferrin receptor 1 (TfR1), located on the apical membrane of the BCECs. The endocytotic pathway predicts that transferrin-TfR1 complex is internalized at the luminal membrane to the endosome, followed by reduction of ferric iron with release from the endosome through DMT1. The iron will thereafter be exported through the abluminal membrane via ferroportin followed by oxidation of ferrous iron by ceruloplasmin [2]. This pathway is favored by the expression of both TfR1, the ferric reductases Steap 2 and 3, DMT1, ferroportin, and the ferrous oxidase ceruloplasmin in BCECs [7]. Within the brain, iron participates as a co-factor for enzymes in multiple physiological functions, including mitochondrial respiration, myelination of axons, synthesis of neurotransmitters, and maintenance of iron homeostasis inside the brain, which is crucial as both iron deficiency and iron overload have serious damaging effects on the brain [8, 9].
Ferroportin (Fpn) is the only known iron exporter [10,11,12] and regulates iron transport into the systemic circulation through the basolateral membrane of intestinal enterocytes [13, 14], as well as the transfer of iron from cells to plasma [15,16,17]. Ferroportin is encoded by the SLC40A1 gene, located on chromosome 2 at position 2q32 [14, 15]. The ferroportin protein is assumed to be 571 amino acids long, containing 12 transmembrane regions, each thought to comprise 21–23 amino acids [16], but some controversy regarding its structure [17,18,19,20] and distribution remains [21,22,23]. The control of ferroportin expression is multilayered and varies between cell types and tissues. It can be controlled at transcriptional, post-transcriptional, and post-translational levels [24]. At transcriptional level, Fpn can be induced by hypoxia and heme [18] due to a so-called antioxidant response element (ARE) in the promoter sequence located 7 kb upstream of the transcription start. AREs are bound by either BACH1 which leads to gene repression or nuclear factor erythroid 2-related factor 2 (NRF2) which leads to gene activation. Heme causes BACH1 degradation, allowing NRF2 to activate the transcription of ferroportin [25, 26]. Iron status regulates ferroportin at a translational level by controlling the stability of Fpn through interaction between iron regulatory proteins (IRP) and iron responsive elements (IRE) [27]. Fpn has an IRE at the 5’untranslated region meaning that its translation is reduced when cells are deprived of iron [9, 25]. However, an isoform of ferroportin mRNA (Fpn1b), detected in mouse duodenum and erythroblasts, lacks the 5’IRE enabling it to escape regulation by IRP/IRE and will therefore export iron even under conditions with cellular iron deficiency [28]. This isoform was however not detectable in brain tissue [28]. Lastly, ferroportin is regulated at a post-translational level by the hormone hepcidin, which has a high affinity for ferroportin and binds to a domain on its extracellular loop [29,30,31,32]. This binding results in phosphorylation, internalization, and degradation of the ferroportin-hepcidin complexes in lysosomes [14].
Epigenetic regulation of ferroportin was previously reported in breast cancer tissue [33], but to the best of our knowledge, no published work has investigated the possible epigenetic regulation of ferroportin in the brain. DNA methylation is the best characterized covalent modification of DNA and is a stable epigenetic mark in differentiated cells, which ensures cell integrity and transcriptional gene silencing [34, 35]. DNA methylation occurs when a methyl group is added in the 5-carbon position of cytosine, generating a 5-methylcytosine (5mC) catalyzed by methyltransferases DNMT1, DNMT3A, and DNMT3B [36, 37]. The regions of interest are those that contain a high frequency of cytosine-guanine (CG) dinucleotides, called CpG islands, which are usually found near or within the gene promoters [36,37,38]. The DNA methylation status of CpG islands is known to influence gene expression, where actively transcribed genes usually contain low DNA methylation percentage in the promoter region and vice versa [37]. DNA methylation is a rigid form of transcriptional silencing resulting in long-term alteration of the gene expression. It has become apparent that DNA methylation and chromatin configuration are connected and that alterations on histones precede DNA methylation through e.g., histone deacetylase (HDAC) [39, 40]. Acetylation of histones determines chromatin folding and thus the accessibility of transcription factors. Histone acetylation is regulated by two counteracting protein families, HDACs and histone acetyltransferases (HATs) [41,42,43]. HDAC is a negative regulator of gene expression and is able to repress transcription by decreasing acetylation on histones, thereby increasing chromatin compaction [44, 45]. Hypoacetylation has been linked to adverse effects in multiple neurodegenerative models [46].
With the complicated nature of Fpn expression in mind, the objective of the present work was to examine the transcriptional regulation of Fpn in the major cell types of the NVU, namely BCECs, pericytes, astrocytes, and neurons isolated as primary cells from intact rat brain. The cells were studied either in mono-cultures or as reconstructs of the BBB using co-cultures grown in a transwell system under polarized conditions. To elaborate on the epigenetic regulation of Fpn, two well-known HDAC inhibitors (HDACi), i.e., valproic acid (VPA) and sodium butyrate (SB), were applied. Our data signify, for the first time, a profound epigenetic regulation of Fpn in BCECs adding significant novelty to the potential for pharmacological handling of iron transport across the BBB.
Methods
Primary Cell Cultures
Brain Capillary Endothelial Cells and Pericytes
Primary cultures of rat pericytes and BCECs were derived from 2 to 3-week-old Sprague-Dawley rats as described previously [47]. The rats were deeply anesthetized using isoflurane, decapitated, and their brain extracted under sterile conditions. The meninges were removed, and the cerebral cortices cut into smaller pieces before being incubated for 75 min at 37 °C in Dulbecco’s modified Eagle’s medium/nutrient F-12 Ham (DMEM-F12) (Gibco, cat#31331) supplemented with DNAse I (Sigma-Aldrich cat#10104159001 Roche) and collagenase II (Gibco cat# 17101015). The digested brain material was resuspended in 20% bovine serum albumin (BSA) (Europa Bioproducts, cat#EQBAH62) in DMEM-F12, and centrifuged at 1000g for 20 min to obtain vessel fragments. The vessel fragments were further digested for 50 min at 37 °C in DMEM-F12 containing collagenase/dispase (Sigma-Aldrich cat#11097113001 Roche) and DNase I, before being loaded on to a 33% percoll gradient (Sigma-Aldrich cat# P1644) to obtain microvessels. The isolated microvessels were seeded directly onto collagen IV (Sigma-Aldrich cat#C5533) and fibronectin (Sigma-Aldrich cat#F1141)-coated dishes. BCECs were maintained in DMEM-F12 supplemented with 10% plasma-derived bovine serum (First Link cat# 60–00-810), 1% insulin transferrin sodium selenite (Sigma-Aldrich, cat# 11074547001 Roche), 10 μg/ml gentamicin sulfate (Lonza Copenhagen, cat#17-518Z), and 1 ng/ml freshly added basic fibroblast growth factor (Roche, cat#F1141). To obtain pure BCEC cultures, 4 μg/ml puromycin (Sigma-Aldrich, cat#P8833) was added to the culture media for the first 3 days of culture. Pericytes were acquired by prolonged culture of the isolated microvessel fragments containing BCECs and pericytes. By culturing these fragments for 10 days in uncoated dishes in DMEM (Gibco, cat#21885) with 10% FCS and 10 μg/ml gentamicin sulfate, the proliferation of pericytes were favored and revealed a pure culture. The pericytes were subsequently seeded directly in uncoated wells.
Astrocytes
Primary cultures of astrocytes were obtained from neonatal (2–3 days old) Sprague-Dawley rats as described previously [48]. Isolated astrocytes were cultured in poly-L-lysine (Sigma-Aldrich, cat#P6282)-coated flasks for 2–3 weeks in DMEM supplemented with 10% FCS and 10 μg/ml gentamicin sulfate. The astrocytes were seeded directly in poly-L-Lysine-coated 12-well dishes for co-culture experiments, or on to poly-L-lysine-coated coverslips added to 24-well plates for 14 days before being used in experiments.
Neurons
Primary neurons were derived from embryos (E17–19) of Sprague Dawley rats obtained from the Animal Facility, Aalborg University. Animals were housed with humidity 40–60%, 12 h light/dark cycle, constant temperature (20–24 °C), and diet (altromin 1324 Roogaarden) and water ad libitum. The experiments were performed in accordance with relevant guidelines (EU Directive 2010/63/EU) and the handling of animals was approved by The Danish Animal Experiment Inspectorate (license no. 2013-15-2934-00776). Pregnant female rats were anesthetized with a subcutaneous injection of hypnorm/dormicum (0.3 ml/100 g, 0.315 mg/ml fentanyl and 10 mg/ml fluanisone mixed with 5 mg/ml midazolam and sterile water in a ratio 1:1:2). The temperature of the female rat was kept at 37 °C and blood supply to the fetuses was ensured, while embryos were taken individually by cesarean section. The dissection was initiated by decapitation of fetuses. The brain was placed on a Sylgard platform containing cold phosphate-buffered saline (PBS) under a dissection microscope. The meninges were removed, and hippocampi isolated. The isolated hippocampal structures were pelleted by centrifugation at 130×g for 1 min, the supernatant was aspirated, and papain solution (Leibovitz’s L-15 Medium (Thermo Fisher Scientific, cat#11415) consisting of ethylenediaminetetraacetic acid (EDTA), 20 U/ml papain (Worthington), and NaOH was added to the hippocampcal extracts, which were incubated at 37 °C for 30 min.
DMEM media supplemented with 10% FCS and DNase I (Roche, cat#10104159001) was added to stop digestion of the tissue followed by centrifugation at 130g for 1 min. To dissociate the tissue, cells were resuspended in DMEM/FCS and DNase I. Subsequently, cells were centrifuged at 130×g for 5 min and resuspended in DMEM/FCS. Finally, the cells were seeded onto pre-coated coverslips or in culture wells with a density of 50,000–60,000 cells/cm2 and incubated at 37 °C. Coverslips or well plates were pre-coated with poly-L-lysine for 2 h at 37 °C, rinsed in ddH2O, and then coated with laminin and incubated for 1 h at 37 °C. The cells were cultured in DMEM/FCS for 24 h, after which the medium was replaced by neuronal medium supplemented with glial inhibitor, cytosine β-D-arabinofuranoside (Sigma-Aldrich, C6645) (AraC, 0.5 μM). The neuronal medium consisted of Neurobasal Medium (Thermo Fisher Scientific, cat#21103) supplemented with 1X B27 supplement (Thermo Fisher Scientific, cat#17504044), 20 μM 5-fluoro-2′-deoxyuridine (Sigma-Aldrich, cat#F0503), 20 μM uridine (Sigma-Aldrich, cat#U3750), 2 mM glutamine-L, and 100 μg/ml primocin (InvivoGen, cat#ant-pm). After 3 days, half of the medium was replaced by neuronal medium without AraC. The medium was changed every other day. Experiments were terminated after 7 days of culture.
In Vitro Model of the Blood-Brain Barrier
Pure cultures of BCECs were used in either mono-culture or non-contact, co-culture setting together with astrocytes. For mono-cultures, BCECs were seeded onto collagen IV fibronectin-coated coverslips added to 24-well plates at a cell density of 100,000 cells/cm2. For co-culture experiments, BCECs were seeded onto collagen IV fibronectin double-coated 12 well Thincert hanging filter inserts (1 μm pore diameter) (in vitro, cat#665610) at a cell density of 100,000 cells/cm2. The cells were left to adhere for 24 h to reveal 100% confluence. To induce barrier integrity, the filter inserts were moved to a 12-well plate containing astrocytes at the bottom of the wells, and the media was supplemented with hydrocortisone (550 nM) (Sigma-Aldrich, cat# H4001), CTP-cAMP, (250 μM) (Sigma-Aldrich, cat# C3912), and RO-201724 (17.5 μM) (Sigma-Aldrich, cat# B8279) in the upper chamber, and hydrocortisone in the lower chamber. The media composition in the lower chamber was a 1:1 mixture of endothelial media and astrocyte media, while the upper chamber only contained endothelial media [47]. Co-culturing the BCECs with astrocytes for 24 h resulted in increased barrier integrity measured as the trans-endothelial electrical resistance (TEER) using a Millicell ERS-2-epithelial volt-ohm meter and a STX01 chopstick electrode. TEER values of double-coated cell-free filter inserts were extracted from the TEER values of the co-cultured BCECs containing filter inserts and multiplied with the filter area (1.12 cm2).
Treatment with HDACi
In order to investigate whether ferroportin mRNA and protein expression was epigenetically regulated, primary BCECs, pericytes, and astrocytes were treated with 2 or 4 mM sodium butyrate (SB) (Sigma-Aldrich) or valproic acid (VPA) (Sigma-Aldrich) for 6 or 24 h. Cells were incubated at 37 °C with 5% CO2 and 95% O2.
Probe-Based qPCR
The purification of DNA and RNA from BCECs, astrocytes, pericytes, and neurons was carried out using an AllPrep DNA/RNA Mini Kit (QIAGEN, cat#80204) according to the manufacturer’s instructions. For the co-culture, BCECs from the upper chamber were purified separately from astrocytes of the lower chamber. Accordingly, BCEC transcripts obtained in the qPCR were not mixed with those of astrocytes. The concentration and purity of RNA were assessed using a NanoDrop spectrophotometer (ND-1000, Fisher Scientific). A total of 500 ng RNA was treated with DNase I enzyme (Thermo Scientific, cat#EN0521) to remove potential genomic DNA contamination. Complementary DNA (cDNA) was synthesized using a Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Thermo Scientific, cat#K1672). Subsequently, 2.5 ng of each sample was used in the PCR reaction. Taq-man probe-based qPCR was carried out for analysis of Fpn using a Brilliant III Probe Mastermix (Agilent Technologies, cat#600880), primer, and probe from TAG Copenhagen (see Table 1). The optimal concentration of probe was 150 nM and the optimal concentration of each primer was individually determined (see Table 2). The thermal profile was as follows: hot start at 95 °C for 3 min followed by 40 amplification cycles with 95 °C for 5 s and 60 °C for 10 s. The Fpn expression was run in duplicates and normalized to the geometric mean of Actb and Cops8 expression. All qPCR reactions were performed using AriaMx real-time PCR system (Agilent Technologies). Non-reverse transcribed RNA and water served as negative controls.
DNA Methylation Analysis
Three CpG islands were found in the Slc40a1 gene promoter. The first consists of 95 base pairs (bp) and contains five CpGs, the second is 113 bp long and contains six CpGs, while the third CpG is 303 bp long and contains 34 CpGs (Fig. 3a). Primers targeting the first two regions were designed, as well as primers targeting the third region (Table 3). The forward primers were designed to bind 100 bp prior to the start of CpG regions 1 and 3 to ensure the whole island was captured. The region 1 and region 2 products were 383 bp, and the region 3 product was 460 bp. A total of 100 ng DNA from astrocytes, mono-cultured BCECs, and co-cultured BCECs were bisulfite converted using the EZ DNA Methylation-Gold™ Kit (Zymo Research, cat#D5006) following the manufacturer’s protocol. The three regions upstream of the Slc40A1 transcription start were included for bisulfite sequencing. These regions were amplified with HotStartTaq DNA polymerase (Qiagen cat#203203) along with the primers stated in the previous section. The protocol belonging to the HotStartTaq DNA polymerase kit was followed except for the reaction volume, which was reduced to 50 μl. For each region, a 50-μl reaction was prepared to contain 10 ng bisulfite-converted DNA and a final concentration of 0.5 μM of each primer for region 1 and 0.3 μM of each primer for region 2. The optimal annealing temperature was determined to be 53 °C by a gradient PCR. The thermal profile for the PCR reaction was as follows: 1 cycle for 15 min at 95 °C, 40 cycles with 30 s at 94 °C, 2 min at 53 °C, and 1 min at 72 °C, and 1 cycle for 10 min at 72 °C.
Before sequencing and purification, the PCR products were analyzed on a 1.3% agarose gel stained with GelRed™ nucleic acid gel stain (Biotium, cat#41003) to ensure the right products had been amplified. The PCR products were then purified using QIAquick® PCR purification kit (Qiagen, cat#28106) and sequenced using an Ion Personal Genome Machine (Life Technologies). The percentage of cytosine methylation was calculated as % C/C+T based on minimum 10 sequencing reads.
Immunocytochemistry
The cells were fixed in 4% paraformaldehyde for 10 min at room temperature. They were then blocked with 3% BSA and 0.3% Triton X in PBS for 30 min at room temperature. The primary antibodies, rabbit anti-Slc40a1 (Alpha Diagnostics cat#MTP11-S), mouse anti α-smooth muscle actin (α-SMA) (Sigma, cat#A5228–2), mouse anti-zonula occludens protein 1 (ZO-1) (Invitrogen, cat# 339100), mouse anti-glial fibrillary acidic protein (GFAP) (Millipore, cat# MAB360), and mouse anti-tubulin β3 (TU-20) (Millipore, cat#MAB1637), were diluted in 1% BSA (1:200), added to the cells, and incubated for 1 h at room temperature. The secondary antibodies, Alexa Fluor 594 goat anti-rabbit IgG (Invitrogen, cat# A11037), Alexa Fluor 488 donkey anti-mouse (Invitrogen, cat# A21202), and goat anti-mouse IgG (Invitrogen, cat#A11032) conjugated with Alexa Fluor 594 were diluted in 1% BSA (1:200) and incubated for 1 h at room temperature. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma, cat#D9542) diluted 1:1000 in PBS for 10 min. The cells were mounted on glass slides in fluorescent mounting medium (DAKO, cat#S3023) and examined under a × 40 oil objective (Zeiss Axio Observer Z1 microscope). All cells were stained on the same day. Images were analyzed by the Image J software [49].
Protein Analysis
Enzyme-linked immunosorbent assay (ELISA) was utilized to measure the amount of ferroportin protein before and after treatment with 2 mM VPA in pericytes and BCECs. The media was discarded, and the cells rinsed twice in PBS. The pericytes were lysed with neuronal protein extraction reagent (Thermo-Fischer Scientific, #87792), kept on ice for 5 min, and harvested and centrifuged at 14,000g for 10 min at 4 °C. The supernatant was collected. BCECs were lysed with of mammalian protein extraction reagent (Thermo-Fischer Scientific, #78501) and shaken gently for 5 min. The cells were harvested using cell scraper (VWR International), collected and centrifuged at 10,000g for 10 min at 4 °C. Supernatants were collected and analyzed using Sandwich-ELISA specific for rat Slc40a1 (BioSite, #EKR1138), carried out following the manufacturer’s instructions. In brief, a twofold standard curve ranging from 0.312 to 20 ng/ml was prepared. Plates were pre-coated with anti-Slc40a1 antibody and incubated with the cell culture samples for 90 min at 37 °C. Plates were washed before adding biotin-labeled anti-Slc40a1 detection antibody and incubated for 60 min at 37 °C. The wells were washed and incubated with HRP-streptavidin conjugate (SABC) working solution and incubated for 30 min at 37 °C, washed and incubated in a dark room with TMB substrate for 20 min at 37 °C. The reaction was terminated, and the absorbance read at 450 nm using plate reader (Enspire Plate Reader, Perkin Elmer). The concentration of Slc40a1 was calculated using a standard curve.
In order to account for the slightly varying number of cells in each flask, the total protein concentration was investigated in all samples using BCA protein assay (Pierce BCA Protein Assay kit, #23225). Standards were prepared using diluted albumin standards with final concentrations ranging from 0 to 2 mg/ml. A standard curve was generated, and the unknown protein concentration of the samples calculated. The results from the ELISA were normalized to the total protein content of each individual sample.
59Fe Export Through BBB In Vitro
To investigate whether the upregulation of ferroportin mRNA and protein resulted in increased iron export at the BBB, BCECs were mono-cultured in a 12-well plate until they reached confluence. Half of the wells were treated with 2 mM VPA for 24 h to induce FPN expression. A total of 4 μCi 59Fe (Perkin Elmer) was added to the media for 4 h where after the cells were rinsed twice with PBS and the media replaced to remove 59Fe from the extracellular environment. A total of 100 μl media samples were collected 1 h, 2 h, and 4 h after media change, and replaced with 100 μl fresh media. All samples were added to 2 ml Ultima Gold scintillations fluid (PerkinElmer) and counted in a Hidex 300SL automatic TDCR liquid scintillation counter with MicroWin 300 SL software for 5 min. Iron uptake was measured as counts per minute and quantified in pmol/l using a standard curve with known 59Fe concentrations. The experiment was repeated three times.
Statistics
The relative gene expression of Fpn mRNA was calculated according to Pfaffl [50] using the neuronal expression as a calibrator for the expression in different cell types, and the control group of a given cell type in the VPA and SB treatment groups. Fpn mRNA expression in cells of the NVU was analyzed using GraphPad Prism 6 by one-way ANOVA with Tukey’s multiple comparisons post hoc test. Fpn expression after VPA treatment was analyzed by one-way ANOVA with Dunnett’s multiple comparisons post hoc test. The FPN protein concentration before and after VPA treatment in BCECs and pericytes was analyzed using unpaired t test. Significance levels were *p = 0.01–0.05, **p = 0.001–0.01, ***p = 0.0001–0.001, and ****p < 0.0001.
Results
Expression of Ferroportin in Primary Brain Cells
The expression of Fpn was investigated in all cell types of the neurovascular unit, which all express Fpn. However, the Fpn expression in primary astrocytes and pericytes was very low (Fig. 1). In comparison, neurons had × 27 and × 3 higher expression of Fpn than astrocytes and pericytes, respectively. BCECs had the highest expression of Fpn; thus, mono-cultured BCECs displayed 151-fold higher expression than astrocytes, 19-fold higher than pericytes, and 5-fold higher than neurons. Interestingly, we observed a further significant increase in the Fpn expression in BCECs co-cultured with astrocytes (Fig. 1) compared with mono-cultured BCECs. The expression of Fpn was much higher in co-cultured BCECs when compared with astrocytes (239-fold) and pericytes (31-fold).

The relative gene expression of ferroportin mRNA (Fpn) in primary rat brain cells. The Fpn expression is significantly higher in brain capillary endothelial cells (BCECs) cultured as mono-cultures or in non-contact, co-cultures with astrocytes, compared with astrocytes, pericytes, or neurons. The highest expression of Fpn is seen in co-cultured BCECs analyzed after being separated from astrocytes. The results are analyzed with one-way ANOVA with a Tukey’s multiple comparisons post hoc test (n = 5). Data are presented as mean ± SEM. Significance (****p < 0.0001)
In order to confirm the cellular content of FPN, we analyzed the cells by immunocytochemistry. GFAP-positive astrocytes displayed very low FPN immunoreactivity (Fig. 2a–c). Compared with astrocytes, FPN was more visible in pericytes (Fig. 2d–f), although still quite weak. Neurons displayed prominent FPN immunoreactivity with marked labeling of both the perinuclear soma and peripheral processes (Fig. 2 n and o). In accordance with the mRNA expression, both mono-cultured BCECs (Fig. 2 h and i) and BCECs co-cultured with astrocytes (Fig. 2 k and l) revealed high FPN expression both intracellularly and on the abluminal membrane.

Ferroportin protein in primary rat brain cells. Left column, cell-specific hallmark proteins (labeled in green) identifying: a astrocytes immunolabeled with glial fibrillary acidic protein (GFAP), d pericytes with α-smooth muscle actin (α-SMA), g Neurons with tubulin beta III (TU-20), and j, m BCECs with zonula occludens protein 1 (ZO-1) when cultured as j mono-cultures or in co-cultures with m astrocytes. Middle column, ferroportin immunolabeling. All cell types express ferroportin (red). The highest immunolabeling is seen in n neurons, h mono-cultured BCECs, and k co-cultured BCECs, while the weakest immunolabeling is seen in b astrocytes and e pericytes. Pericytes consistently display ferroportin immunoreactivity, but they vary in their content of α-SMA with their stage of differentiation. The right column displays the merging of the two illustrations shown in the same row. Cellular nuclei are counterstained with DAPI (blue). Scale bar = 20 μM
DNA Methylation Analysis
In order to investigate if the varying ferroportin expressions in the cell types of the neurovascular unit were due to epigenetic regulation, we analyzed DNA methylation status of CpG islands, which is known to influence gene expression, where actively transcribed genes often contain low DNA methylation percentage in their promoter region and vice versa [37]. We identified three CpG islands in the Slc40a1 promoter region. CpG islands 1 and 2 (region 1 and 2) were located upstream of the transcription start site (TSS), whereas CpG island 3 (region 3) encompassed the TSS and a part of exon 1. The CpG sites are displayed in Fig. 3a and the methylation percentage is graphically presented in Fig. 3b. DNA methylation analysis was carried out for all three identified CpG islands present in the Slc40a1 promotor in primary astrocytes, mono-cultured BCECs, and BCECs co-cultured with astrocytes, which were selected because they represented the lowest and highest expression of Fpn. Noteworthy, region 1, which includes CpG sites 45–70 (Fig. 3b, blue bars), showed higher DNA methylation on average in astrocytes compared with mono-cultured BCECs and co-cultured BCECs. All five CpG sites in the region 1 were methylated in astrocytes and mono-cultured BCECs, whereas only three CpG sites were methylated in region 1 in co-cultured BCECs (Fig. 3b). Region 2, which includes CpG sites 150–220 (Fig.3b, green bars), displayed lower degree of methylation than region 1 in astrocytes and co-cultured BCECs, whereas region 2 was more methylated than region 1 in mono-cultured BCECs. There was 2-times lower degree of DNA methylation in region 2 in astrocytes compared with region 1. When looking at the CpG positions, the most methylated position was 70 in astrocytes with 8%, but only 3.6% and 3.45% in co-cultured and mono-cultured BCECs, respectively. Position 50 was more methylated in co-cultured BCECs compared with astrocytes and mono-cultured BCECs. DNA methylation was lowest in region 3 in both cell types (data not shown). These results correlate well with the low Fpn expression in astrocytes and high expression in BCECs, suggesting that Fpn is epigenetically regulated.

DNA methylation analysis. a Schematic overview of the Slc40a1 gene. Included are the three CpG islands presented as regions 1, 2, and 3. The region within the two red vertical lines contains the CpG regions that were methylated (regions 1 and 2) when sequenced. b DNA methylation in regions 1 and 2. DNA methylation proportions as determined by pyrosequencing of regions 1 and 2 in the Slc40a1 promotor of primary astrocytes and primary brain capillary endothelial cells (BCECs) cultured alone (mono-culture) or in the presence of astrocytes (co-culture). The CpG sites 45–70, located in region 1, are presented in different shades of blue, while the CpG site 117 between regions 1 and 2 is presented in white, and the CpG sites 150–220 of region 2 are presented in different shades of green. Data are presented as a percentage of C-methylation at each of the CpG sites 45–220. The data shown here is from a single sample of each cell type (n = 1)
Expression of Fpn After Treatment with HDAC Inhibitors
The analysis of DNA methylation on the Slc40a1 promoter raised the question whether it would be possible to manipulate Fpn gene expression pharmacologically. Two well-known and broadly tested HDAC inhibitors (HDACi) VPA and SB were used in order to answer this question. In astrocytes, no indication of a significant expressional change was seen (not shown).
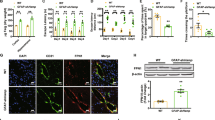
Even though Fpn mRNA expression was generally high in BCECs compared with other cell types cultured under normal conditions (Fig. 1), it was significantly increased by the HDACi in BCECs compared with untreated cells (Fig. 4a). The Fpn expression in BCECs after stimulation with 4 mM SB for 6 h resulted in a significantly higher expression (3.3-fold), and with 2 and 4 mM VPA for 6 h, the increases were 3.6-fold and 2.5-fold, respectively. The increase was even higher after 24 h, reaching 5.1-fold increase with 2 mM VPA and 6.2-fold increase with 4 mM VPA, compared with the controls. In contrast to that of pericytes, BCECs did not increase their expression of the Fpn mRNA following treatment with SB for 24 h.

Valproic acid (VPA) increases expression of ferroportin in brain capillary endothelial cells (BCECs) and pericytes. To investigate whether the expression of Fpn could be modified epigenetically, mono-cultured brain capillary endothelial cells (BCECs) and pericytes were treated with sodium butyrate (SB) or valproic acid (VPA) in different time intervals and concentrations. In the a BCECs, VPA induces a robust increase in Fpn at both 2 and 4 mM for 6 h and 24 h. SB increases expression of Fpn at 4 mM for 6 h. SB and VPA did increase the expression of Fpn in c pericytes when VPA was added in concentration 2 and 4 mM for 24 h and 4 mM for 6 h. SB increased Fpn expression at 2 mM for 6 h, as well as 4 mM for 24 h. The results are analyzed with one-way ANOVA with a Dunnett multiple comparisons post hoc test (n = 6). Number of asterisks (*) indicates the level of statistical significance (*p < 0.05, **p < 0.01, ****p < 0.0001). b Mono-cultured BCECs and d pericytes were treated with 2 mM VPA for 24 h and examined for FPN using immunocytochemistry. BCECs (identified by ZO-1 immunoreactivity) display strong FPN immunolabeling signal after treatment with b VPA. Conversely, pericytes display unaltered FPN immunoreactivity after treatment with d VPA. Scale bar = 20 μM
Contrary to the very low expression of ferroportin in untreated pericytes (Fig. 1), the HDACi, VPA and SB, were both able to induce a significant increase in pericytes (Fig. 4c). Hence, there was an increase in Fpn expression after treatment with 2 mM SB and 4 mM VPA after 6 h (12-fold), after 24 h of treatment with 4 mM SB (21-fold) and 2 (21-fold) and 4 mM VPA (19-fold) compared with untreated cells (Fig. 4c). These results indicated that inducers of epigenetic regulators allow modification of Fpn expression in BCECs and pericytes (Fig. 4), but not in astrocytes (not shown). Thus, only BCECs and pericytes had a regulatory epigenetic layer of control for Fpn expression following histone acetylation.
Ferroportin Protein Expression After Treatment with VPA
To investigate whether the increased mRNA expression of Fpn translated to protein, BCECs and pericytes were treated with VPA. Based on the results from mRNA analysis, the cells were treated with 2 mM VPA for 6 and 24 h and examined by immunocytochemistry. We did not find any visible difference in FPN expression after treatment with 2 mM VPA for 6 h compared with the untreated cells (data not shown). However, there was a strong increase in FPN expression in the BCECs after treatment with 2 mM VPA after 24 h compared with controls (Fig. 4b). This correlated well with the increased Fpn mRNA expression after 24 h treatment with VPA. In contrast, untreated pericytes displayed low expression of FPN even after treatment with VPA for 24 h (Fig. 4d).
In order to quantify the FPN expression after treatment with VPA in BCECs and pericytes, the ELISA analysis revealed that in control culture situations, FPN was approximately two-fold higher in BCECs compared with that of pericytes. For BCECs, we observed a significant raise in FPN expression after VPA treatment compared with controls leveling the content of FPN almost two-fold higher in the treated cells (0.357 ± 0.0468 ng/ml vs. 0.5698 ± 0.0831 ng/ml) (Fig. 5a). In spite of the significant increase in pericytic Fpn after treatment with VPA for 24 h, we were not able to detect a difference in FPN expression (Fig. 5b).

Ferroportin (FPN) protein analysis and functionality in brain capillary endothelial cells (BCECs) and pericytes after valproic acid (VPA) treatment. a, b ELISA analysis of the ferroportin content in cultured BCECs and pericytes. a BCECs cultured as mono-cultures display higher content of FPN after treatment with 2 mM VPA for 24 h being almost two-fold higher compared with controls (n = 4). b The content of FPN in pericytes does not change after treatment with 2 mM VPA for 24 h (n = 3). Of note, the content of FPN in the control wells of the BCECs is more than twice that of the pericytes. Statistical analysis was carried out using an unpaired t test. Number of asterisks (*) indicates the level of statistical significance (**p = 0.001–0.01). c59Fe export in mono-cultured BCEC after treatment with VPA. As BCECs display higher expression of ferroportin protein after epigenetic challenge (see above), we compared iron efflux from BCECs following treatment with 2 mM VPA for 24 h to the control situation (n = 4). Iron export was measured after 1 h, 2 h, and 4 h. BCECs receiving VPA treatment prior to iron loading (blue) export more iron than the untreated cells (gray) at all-time points
59Fe Export Through BBB Model
To investigate whether the epigenetic induction of the ferroportin mRNA and protein also translated functionally to higher activity of FPN, a transport study was conducted in BCECs, which were treated with 2 mM VPA for 24 h prior to exposure for radiolabeled iron. The cells were added with 4 μCi 59Fe for 4 h followed by removal of extracellular radiolabeled iron by change of the culture medium. After 1 h, the controls, which had not received VPA treatment, had exported less iron than cells receiving VPA (Fig. 5c). After 2 and 4 h, the pattern of the results was very similar to that observed after 1 h with more iron being exported from BCECs of the treatment group than the control group (Fig. 5c).
Discussion
Ferroportin is a crucial regulator of iron homeostasis and has a unique function in the BBB as it controls the amount of iron transported into the brain parenchyma [23, 51]. It is thus highly relevant to investigate the expression level of Fpn in the cells associated with the BBB. The purpose of this study was to investigate how ferroportin is regulated in BCECs and other cell types of the neurovascular unit. We report that astrocytes, pericytes, neurons, and BCECs all express Fpn but that BCECs, cultured as mono- or co-cultures, express significantly more Fpn compared with astrocytes, pericytes, and neurons.
Pericytes are crucial for maintaining the integrity of the BBB by supporting angiogenesis and ensuring microvascular stability [52, 53]. Although another study did not find expression of ferroportin in pericytes using immunostaining or Western blotting [54], we show here that pericytes express ferroportin mRNA and protein in vitro. This is a noteworthy finding as the expression of Fpn in pericytes has not been documented before, and the presence of ferroportin in pericytes suggests that they are also involved in maintaining iron homeostasis in the NVU.
Astrocytes are thought to have a pivotal role in regulating iron transport to the brain. Although they are unlikely to store iron due to their low expression of ferritin, or export iron due to low expression of Fpn [55], astrocytes are known to secrete various neurotrophic factors and can have protective properties when cultured with other cells [56]. For instance, the primary astrocytes in this study seem to positively regulate Fpn expression in the BCECs, as co-cultured BCECs presented the greatest expression of Fpn. Additionally, astrocyte-conditioned medium has been shown to positively influence the expression of Fpn in BCECs [7], which further indicates that astrocytes are able to upregulate the expression of Fpn in adjacent cells possibly through secretion of various molecules. Such molecules include ceruloplasmin, which provides ferroxidase activity needed for iron efflux through FPN [7, 57].
Primary rat neurons displayed a higher expression of Fpn than astrocytes and pericytes, as well as strong staining for FPN. The high expression of FPN is in line with the results of Burdo et al. [51]. Neurons utilize iron for metabolic purposes, store iron in ferritin, and release the remainder through ferroportin [23]. The presence of ferroportin is of great importance, as downregulation of Fpn in neurons has been shown to cause iron accumulation, inflammation, and compromised integrity of the BBB [58]. High expression of Fpn can, therefore, be expected in neurons due to their low iron storage ability and low tolerance for unbound iron.
In order to investigate the possibility of epigenetic regulation through DNA methylation, the Fpn promotor was analyzed by bisulfite conversion and deep sequencing. The addition of methyl groups to CG dinucleotides in CpG islands within the promotor can lead to transcriptional silencing [59, 60]. Supporting this notion, the Slc40A1 promotor displayed a higher degree of methylation in astrocytes compared with BCECs, which correlates with lower Fpn mRNA expression in astrocytes. The biggest difference in DNA methylation observed between astrocytes and BCECs was in region 1, where astrocytes displayed more DNA methylation. On the other hand, region 3 was surprisingly free of DNA methylation in both cell types despite being located closest to the TSS. The overall levels of DNA methylation on the Slc40A1 promoter seem to be very low. However, it needs to be kept in mind that methylation in the mammalian genome is rare, occurring only at about 1% of the DNA [60]. Thus, we suggest that DNA methylation is a regulator of Fpn expression in BCECs despite subtle overall differences in promoter methylation.
Since DNA methylation regulates Fpn gene expression, it was appealing to manipulate the epigenetic landscape with HDACi in the interest of possible future therapy options. Hemochromatosis is a genetic disorder that causes systemic iron overload due to reduced hepcidin or hepcidin-ferroportin binding. With reduced activity of hepcidin, more ferroportin is present on the cell surface, enabling ferroportin to transport more iron into plasma. The higher iron concentration in plasma increases iron uptake, particularly by hepatocytes and cardiomyocytes, which can lead to hepatic and cardiac iron overload [61]. The main therapy for hemochromatosis is iron chelation [61], which binds and neutralizes the iron that has been transported to plasma. Theoretically, a pharmacological epigenetic induction of ferroportin could aid the cellular export of iron from organs compromised in function by iron loading.
It has previously been shown that HDACi have enhanced cognition in a transgenic mouse model of Alzheimer’s disease [62] and that VPA is able to upregulate different efflux transporters in mice in vivo [63] and in immortalized human endothelial cells in vitro [64]. In the current study, both VPA and SB caused an increase in the Fpn gene expression in BCECs and pericytes, but not in the astrocytes. This suggests differential regulatory pathways for Fpn gene expression among cell types and the higher DNA methylation found in astrocytes may be the reason why there is no alteration in Fpn expression after treatment with HDACi. Theoretically, if VPA and SB increased Fpn expression within the brain, the therapeutic effect would most likely be higher iron transport across the BBB, which could be utilized in iron deficiency, or increased iron transport from neurons in age-related neurodegeneration. It would be interesting to investigate whether ferroportin expression in neurons can be manipulated by VPA or SB, as it has been documented that ferroportin levels progressively decrease in transgenic Alzheimer’s mice compared with wild-type littermates [65]. This could possibly be due to the blocked activity of the ferroxidase amyloid precursor protein that stabilizes ferroportin, thus resulting in reduced neuronal iron accumulation [66].
In the last few years, it has become apparent that DNA methylation and chromatin configuration are connected and that alterations on histones precede DNA methylation through e.g., HDACs [39, 40]. Both VPA and SB are short-chain fatty acids that cross the BBB and have been used clinically to treat epilepsy [67]. The main difference lies in the type of HDACs these substances can inhibit. SB inhibits class I HDACs [68], whereas VPA is able to inhibit both class I and II HDACs [69]. VPA can upregulate many different genes and is not specific for Fpn. VPA’s effect on the brain is versatile as it can alter activity of the inhibitory neurotransmitter GABA through multiple mechanisms, block Na+, Ca2+, and K+ channels and decrease NMDA excitation [70]. In addition, VPA affects the BBB in a positive way by improving the integrity of the barrier after traumatic brain injury [71], preventing barrier disruption after subarachnoid hemorrhage [72] and attenuating BBB disruption in transient focal cerebral ischemia [73]. VPA has gained popularity in traumatic brain injury research where its neuroprotective properties are credited upregulation of several genes within certain pathways [71, 74, 75]. The upregulation in ferroportin mRNA and protein displayed here could therefore also be a secondary effect to alterations in expression of other genes that induces iron regulatory proteins or prevent their degradation, as this in turn leads to stabilization of the FPN transcript [76].
In conclusion, we find that the cell types denoting the NVU of the brain, i.e., BCECs, pericytes, astrocytes, and neurons, all express Fpn with BCEC expression being highest. The regulation of Fpn is complex with epigenetic regulation occurring in BCECs and pericytes, adding to the well-established mechanisms of transcriptional, post-transcription, and post-translational regulatory mechanisms of ferroportin [9, 24, 25, 29]. As VPA is a commonly used anti-epileptic drug that easily crosses the BBB, it could theoretically also lead to increase in ferroportin at the BBB in vivo, hence providing the substrate for elevated transport of iron into the brain.
Abbreviations
- 5mC:
-
5-Methylcytosine
- α-SMA:
-
α-Smooth actin
- Actb:
-
β-Actin
- ARE:
-
Antioxidant responsive element
- BACH1:
-
BTB and CNC homology 1
- BBB:
-
Blood-brain barrier
- BCECs:
-
Brain capillary endothelial cells
- BSA:
-
Bovine serum albumin
- cDNA:
-
Complementary DNA
- CG:
-
Cytosine-guanine
- DAPI:
-
4′,6-diamidino-2-phenylindole
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- DMSO:
-
Dimethyl sulfoxide
- DMT1:
-
Divalent metal transporter 1
- EDTA:
-
Ethylenediaminetetraacetic acid
- ELISA:
-
Enzyme-linked immunosorbent assay
- FCS:
-
Fetal calf serum
- Fe:
-
Iron
- Fpn:
-
Ferroportin
- GFAP:
-
Glial fibrillary acidic protein
- HDAC:
-
Histone deacetylase
- HDACi:
-
Histone deacetylase inhibitor
- IRE:
-
Iron responsive element
- IRP:
-
Iron regulatory protein
- NRF2:
-
Nuclear factor (erythroid-derived 2)-like-2
- NVU:
-
Neurovascular unit
- PBS:
-
Phosphate-buffered saline
- SAM:
-
SAdenosylmethionine
- SB:
-
Sodium butyrate
- TfR1:
-
Transferrin receptor 1
- VPA:
-
Valproic acid
- TEER:
-
Trans-endothelial electrical resistance
- TSS:
-
Transcription start site
- TU-20:
-
β3 tubulin
- ZO-1:
-
Zonula occludens protein 1
References
Abbott NJ (2013) Blood–brain barrier structure and function and the challenges for CNS drug delivery. J Inherit Metab Dis 36:437–449. https://doi.org/10.1007/s10545-013-9608-0
Mills E, Dong X-P, Wang F, Xu H (2010) Mechanisms of brain iron transport: insight into neurodegeneration and CNS disorders. Future Med Chem 2:51–64. https://doi.org/10.1021/ac901991x
Duck KA, Connor JR (2016) Iron uptake and transport across physiological barriers. BioMetals 29:573–591. https://doi.org/10.1007/s10534-016-9952-2
Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37:13–25. https://doi.org/10.1016/j.nbd.2009.07.030
McConnell HL, Kersch CN, Woltjer RL et al (2016) The translational significance of the neurovascular unit. JBC Pap. https://doi.org/10.1074/jbc.R116.760215
Thomsen MS, Routhe LJ, Moos T (2017) The vascular basement membrane in the healthy and pathological brain. J Cereb Blood Flow Metab 37:3300–3317. https://doi.org/10.1177/0271678X17722436
Burkhart A, Skjørringe T, Johnsen KB, Siupka P, Thomsen LB, Nielsen MS, Thomsen LL, Moos T (2016) Expression of iron-related proteins at the neurovascular unit supports reduction and reoxidation of iron for transport through the blood-brain barrier. Mol Neurobiol 53:7237–7253. https://doi.org/10.1007/s12035-015-9582-7
Moos T, Rosengren Nielsen T, Skjørringe T, Morgan EH (2007) Iron trafficking inside the brain. J Neurochem 103:1730–1740. https://doi.org/10.1111/j.1471-4159.2007.04976.x
Belaidi AA, Bush AI (2016) Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J Neurochem 139:179–197. https://doi.org/10.1111/jnc.13425
Abboud S, Haile DJ (2000) A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 275:19906–19912. https://doi.org/10.1074/jbc.M000713200
Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A et al (2000) Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403:776–781. https://doi.org/10.1038/35001596
McKie AT, Marciani P, Rolfs A et al (2000) A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 5:299–309
Wallace DF (2016) The regulation of iron absorption and homeostasis. Clin Biochem Rev 37:51–62
Nemeth E, Tuttle MS, Powelson J et al (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science (80-) 306:2090–2093. https://doi.org/10.1126/science.1104742
Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC (2005) The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 1:191–200. https://doi.org/10.1016/j.cmet.2005.01.003
Rochette L, Gudjoncik A, Guenancia C, Zeller M, Cottin Y, Vergely C (2015) The iron-regulatory hormone hepcidin: a possible therapeutic target? Pharmacol Ther 146:35–52. https://doi.org/10.1016/j.pharmthera.2014.09.004
Le Gac G, Ka C, Joubrel R et al (2013) Structure-function analysis of the human ferroportin iron exporter (SLC40A1): effect of hemochromatosis type 4 disease mutations and identification of critical residues. Hum Mutat 34:1371–1380. https://doi.org/10.1002/humu.22369
Ward DM, Kaplan J (2012) Ferroportin-mediated iron transport: expression and regulation. Biochim Biophys Acta, Mol Cell Res 1823:1426–1433. https://doi.org/10.1016/j.bbamcr.2012.03.004
Bonaccorsi di Patti MC, Polticelli F, Cece G, Cutone A, Felici F, Persichini T, Musci G (2014) A structural model of human ferroportin and of its iron binding site. FEBS J 281:2851–2860. https://doi.org/10.1111/febs.12825
Andersen HH, Johnsen KB, Moos T (2014) Iron deposits in the chronically inflamed central nervous system and contributes to neurodegeneration. Cell Mol Life Sci 71:1607–1622. https://doi.org/10.1007/s00018-013-1509-8
Clardy SL, Wang X, Boyer PJ, Earley CJ, Allen RP, Connor JR (2006) Is ferroportin–hepcidin signaling altered in restless legs syndrome? J Neurol Sci 247:173–179. https://doi.org/10.1016/J.JNS.2006.04.008
Boserup MW, Lichota J, Haile D, Moos T (2011) Heterogenous distribution of ferroportin-containing neurons in mouse brain. BioMetals 24:357–375. https://doi.org/10.1007/s10534-010-9405-2
Wu LJC, Leenders AGM, Cooperman S, Meyron-Holtz E, Smith S, Land W, Tsai RYL, Berger UV et al (2004) Expression of the iron transporter ferroportin in synaptic vesicles and the blood-brain barrier. Brain Res 1001:108–117. https://doi.org/10.1016/j.brainres.2003.10.066
Muckenthaler MU, Rivella S, Hentze MW, Galy B (2017) A red carpet for iron metabolism. Cell 3:1–18. https://doi.org/10.1016/j.cell.2016.12.034
Drakesmith H, Nemeth E, Ganz T (2015) Ironing out ferroportin. Cell Metab 22:777–787. https://doi.org/10.1016/j.cmet.2015.09.006
Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, Muckenthaler MU (2010) Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica 95:1261–1268. https://doi.org/10.3324/haematol.2009.020123
Silva B, Faustino P (2015) An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim Biophys Acta 1852:1347–1359. https://doi.org/10.1016/j.bbadis.2015.03.011
Zhang D-L, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA (2009) A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab 9:461–473. https://doi.org/10.1016/j.cmet.2009.03.006
Gulec S, Anderson GJ, Collins JF (2014) Mechanistic and regulatory aspects of intestinal iron absorption. AJP Gastrointest Liver Physiol 307:G397–G409. https://doi.org/10.1152/ajpgi.00348.2013
Helgudottir SS, Lichota J, Burkhart A, Moos T (2018) Hepcidin mediates transcriptional changes in ferroportin mRNA in differentiated neuronal-like PC12 cells subjected to iron challenge. Mol Neurobiol 56:2362–2374 3–5
Rishi G, Wallace DF, Subramaniam VN (2015) Hepcidin: regulation of the master iron regulator. Biosci Rep 35:e00192. https://doi.org/10.1042/BSR20150014
Wang L, Liu X, You L-H, Ci YZ, Chang S, Yu P, Gao G, Chang YZ (2019) Hepcidin and iron regulatory proteins coordinately regulate ferroportin 1 expression in the brain of mice. J Cell Physiol 234:7600–7607. https://doi.org/10.1002/jcp.27522
Chen Y, Zhang S, Wang X, Guo W, Wang L, Zhang D, Yuan L, Zhang Z et al (2015) Disordered signaling governing ferroportin transcription favors breast cancer growth. Cell Signal 27:168–176. https://doi.org/10.1016/j.cellsig.2014.11.002
Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Balazer JA, Eaves HL et al (2011) Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci 14:1345–1351. https://doi.org/10.1038/nn.2900
Kohli RM, Zhang Y (2013) TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502:472–479. https://doi.org/10.1038/nature12750
Yao B, Christian KM, He C, Jin P, Ming GL, Song H (2016) Epigenetic mechanisms in neurogenesis. Nat Rev Neurosci 17:537–549. https://doi.org/10.1038/nrn.2016.70
Tough DF, Tak PP, Tarakhovsky A, Prinjha RK (2016) Epigenetic drug discovery: breaking through the immune barrier. Nat Rev Drug Discov 15:835–853. https://doi.org/10.1038/nrd.2016.185
Benayoun BA, Pollina EA, Brunet A (2015) Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol 16:593–610. https://doi.org/10.1038/nrm4048
Milutinovic S, Detich N, Szyf M (2007) Valproate induces widespread epigenetic reprogramming which involves demethylation of specific genes. Carcinogenesis 28:560–571. https://doi.org/10.1093/carcin/bgl167
Li E, Zhang Y (2014) DNA methylation in mammals. Cold Spring Harb Perspect Biol 6:a019133. https://doi.org/10.1101/cshperspect.a019133
Rossaert E, Pollari E, Jaspers T, van Helleputte L, Jarpe M, van Damme P, de Bock K, Moisse M et al (2019) Restoration of histone acetylation ameliorates disease and metabolic abnormalities in a FUS mouse model. Acta Neuropathol Commun 7:107. https://doi.org/10.1186/s40478-019-0750-2
Konsoula Z, Barile FA (2012) Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J Pharmacol Toxicol Methods 66:215–220. https://doi.org/10.1016/j.vascn.2012.08.001
Saha RN, Pahan K (2006) HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ 13:539–550. https://doi.org/10.1038/sj.cdd.4401769
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS (2001) Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 276:36734–36741. https://doi.org/10.1074/jbc.M101287200
Zhu M-M, Li H-L, Shi L-H, Chen XP, Luo J, Zhang ZL (2017) The pharmacogenomics of valproic acid. J Hum Genet 62:1009–1014. https://doi.org/10.1038/jhg.2017.91
Bennett SA, Tanaz R, Cobos SN, Torrente MP (2019) Epigenetics in amyotrophic lateral sclerosis: a role for histone post-translational modifications in neurodegenerative disease. Transl Res 204:19–30. https://doi.org/10.1016/J.TRSL.2018.10.002
Burkhart A, Skjørringe T, Andresen TL et al (2017) Transfection of primary brain capillary endothelial cells for protein synthesis and secretion of recombinant erythropoietin: a strategy to enable protein delivery to the brain. Cell Mol Life Sci 74:2467–2485. https://doi.org/10.1007/s00018-017-2501-5
Burkhart A, Azizi M, Thomsen MSS et al (2014) Accessing targeted nanoparticles to the brain: the vascular route. Curr Med Chem 21:4092–4099
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. https://doi.org/10.1038/nmeth.2019
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Burdo JR, Menzies SL, Simpson IA, Garrick LM, Garrick MD, Dolan KG, Haile DJ, Beard JL et al (2001) Distribution of divalent metal transporter 1 and metal transport protein 1 in the normal and Belgrade rat. J Neurosci Res 66:1198–1207. https://doi.org/10.1002/jnr.1256
Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV (2015) Establishment and dysfunction of the blood-brain barrier. Cell 163:1064–1078. https://doi.org/10.1016/j.cell.2015.10.067
Winkler EA, Sagare AP, Zlokovic BV (2014) The pericyte: a forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol 24:371–386. https://doi.org/10.1111/bpa.12152
Yang WM, Jung KJ, Lee MO, Lee YS, Lee YH, Nakagawa S, Niwa M, Cho SS et al (2011) Transient expression of iron transport proteins in the capillary of the developing rat brain. Cell Mol Neurobiol 31:93–99. https://doi.org/10.1007/s10571-010-9558-0
Dringen R, Bishop GM, Koeppe M, Dang TN, Robinson SR (2007) The pivotal role of astrocytes in the metabolism of iron in the brain. Neurochem Res 32:1884–1890. https://doi.org/10.1007/s11064-007-9375-0
Schipper HM, Vininsky R, Brull R, Small L, Brawer JR (1998) Astrocyte mitochondria: a substrate for iron deposition in the aging rat substantia nigra. Exp Neurol 152:188–196. https://doi.org/10.1006/exnr.1998.6854
McCarthy RC, Kosman DJ (2014) Glial cell ceruloplasmin and hepcidin differentially regulate iron efflux from brain microvascular endothelial cells. PLoS One 9:e89003. https://doi.org/10.1371/journal.pone.0089003
Urrutia P, Aguirre P, Esparza A, Tapia V, Mena NP, Arredondo M, González-Billault C, Núñez MT (2013) Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J Neurochem 126:541–549. https://doi.org/10.1111/jnc.12244
Browne EP, Dinc SE, Punska EC, Agus S, Vitrinel A, Erdag GC, Anderton DL, Arcaro KF et al (2014) Promoter methylation in epithelial-enriched and epithelial-depleted cell populations isolated from breast milk. J Hum Lact 30:450–457. https://doi.org/10.1177/0890334414548224
Yong-Quan Ng G, Yun-An L, Sobey C et al (2018) Epigenetic regulation of inflammation in stroke. Ther Adv Neurol Disord 11:1–30
Brissot P, Pietrangelo A, Adams PC, de Graaff B, McLaren CE, Loréal O (2018) Haemochromatosis. Nat Rev Dis Prim 4:399–408. https://doi.org/10.1038/nrdp.2018.16
Qing H, He G, Ly PTT, Fox CJ, Staufenbiel M, Cai F, Zhang Z, Wei S et al (2008) Valproic acid inhibits Aβ production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J Exp Med 205:2781–2789. https://doi.org/10.1084/JEM.20081588
You D, Shin HM, Mosaad F, Richardson JR, Aleksunes LM (2019) Brain region-specific regulation of histone acetylation and efflux transporters in mice. J Biochem Mol Toxicol e22318. https://doi.org/10.1002/jbt.22318
You D, Wen X, Gorczyca L, Morris A, Richardson JR, Aleksunes LM (2019) Increased MDR1 transporter expression in human brain endothelial cells through enhanced histone acetylation and activation of aryl hydrocarbon receptor signaling. Mol Neurobiol. https://doi.org/10.1007/s12035-019-1565-7
Belaidi AA, Gunn AP, Wong BX, Ayton S, Appukuttan AT, Roberts BR, Duce JA, Bush AI (2018) Marked age-related changes in brain Iron homeostasis in amyloid protein precursor knockout mice. Neurotherapeutics 15:1055–1062. https://doi.org/10.1007/s13311-018-0656-x
Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BXW, Adlard PA et al (2012) Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med 18:291–295. https://doi.org/10.1038/nm.2613
Biermann J, Boyle J, Pielen A, Lagrèze WA (2011) Histone deacetylase inhibitors sodium butyrate and valproic acid delay spontaneous cell death in purified rat retinal ganglion cells. Mol Vis 17:395–403
Candido EPM, Reeves R, Davie JR (1978) Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 14:105–113. https://doi.org/10.1016/0092-8674(78)90305-7
Minucci S, Zhu P, Kra OH et al (2001) Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 20:6969–6978. https://doi.org/10.1093/emboj/20.24.6969
Chateauvieux S, Morceau F, Dicato M, Diederich M (2010, 2010) Molecular and therapeutic potential and toxicity of valproic acid. J Biomed Biotechnol:1–18. https://doi.org/10.1155/2010/479364
Nikolian VC, Dekker SE, Bambakidis T, Higgins GA, Dennahy IS, Georgoff PE, Williams AM, Andjelkovic AV et al (2018) Improvement of blood-brain barrier integrity in traumatic brain injury and hemorrhagic shock following treatment with valproic acid and fresh frozen plasma. Crit Care Med 46:e59–e66. https://doi.org/10.1097/CCM.0000000000002800
Ying G, Jing C, Li J, Wu C, Yan F, Chen JY, Wang L, Dixon BJ et al (2016) Neuroprotective effects of valproic acid on blood-brain barrier disruption and apoptosis-related early brain injury in rats subjected to subarachnoid hemorrhage are modulated by heat shock protein 70/matrix metalloproteinases and heat shock protein 70/AKT pathways. Neurosurgery 79:286–295. https://doi.org/10.1227/NEU.0000000000001264
Wang Z, Leng Y, Tsai L-K, Leeds P, Chuang DM (2011) Valproic acid attenuates blood–brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP-9 inhibition. J Cereb Blood Flow Metab 31:52–57. https://doi.org/10.1038/jcbfm.2010.195
Chang P, Williams AM, Bhatti UF, Biesterveld BE, Liu B, Nikolian VC, Dennahy IS, Lee J et al (2019) Valproic acid and neural apoptosis, inflammation, and degeneration 30 days after traumatic brain injury, hemorrhagic shock, and polytrauma in a swine model. J Am Coll Surg 228:265–275. https://doi.org/10.1016/j.jamcollsurg.2018.12.026
Nikolian VC, Dennahy IS, Higgins GA, et al (2018) Transcriptomic changes following valproic acid treatment promote neurogenesis and minimize secondary brain injury. In: Journal of Trauma and Acute Care Surgery. Lippincott Williams and Wilkins, pp 459–465
Zhang DL, Ghosh MC, Rouault TA (2014) The physiological functions of iron regulatory proteins in iron homeostasis-an update. Front Pharmacol 5(JUN):124
Acknowledgments
We thank Merete Fredsgaard and Hanne Krone Nielsen, Aalborg University, Denmark, for their excellent technical assistance. We also thank Lykke Christina Grubach, Clinical Diagnostics Department at Aalborg University Hospital, Denmark for providing technical instruments.
Funding
The present work has been supported by the Danish Multiple Sclerosis Society, “Kong Christian den Tiendes fond,” “Åse og Ejner Danielsens Fond,” and the Lundbeck Foundation Research Initiative on Brain Barriers and Drug Delivery (Grant no. R155-2013-14113).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Helgudottir, S.S., Routhe, L.J., Burkhart, A. et al. Epigenetic Regulation of Ferroportin in Primary Cultures of the Rat Blood-Brain Barrier. Mol Neurobiol 57, 3526–3539 (2020). https://doi.org/10.1007/s12035-020-01953-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-01953-y