
Abstract
Treatment of chronic disorders affecting the central nervous system (CNS) is complicated by the inability of drugs to cross the blood–brain barrier (BBB). Non-viral gene therapy applied to brain capillary endothelial cells (BCECs) denotes a novel approach to overcome the restraints in this passage, as turning BCECs into recombinant protein factories by transfection could result in protein secretion further into the brain. The present study aims to investigate the possibility of transfecting primary rat brain endothelial cells (RBECs) for recombinant protein synthesis and secretion of the neuroprotective protein erythropoietin (EPO). We previously showed that 4% of RBECs with BBB properties can be transfected without disrupting the BBB integrity in vitro, but it can be questioned whether this is sufficient to enable protein secretion at therapeutic levels. The present study examined various transfection vectors, with regard to increasing the transfection efficiency without disrupting the BBB integrity. Lipofectamine 3000™ was the most potent vector compared to polyethylenimine (PEI) and Turbofect. When co-cultured with astrocytes, the genetically modified RBECs secreted recombinant EPO into the cell culture medium both luminally and abluminally, and despite lower levels of EPO reaching the abluminal chamber, the amount of recombinant EPO was sufficient to evolve a biological effect on astrocytes cultured at the abluminal side in terms of upregulated gene expression of brain-derived neurotropic factor (BDNF). In conclusion, non-viral gene therapy to RBECs leads to protein secretion and signifies a method for therapeutic proteins to target cells inside the CNS otherwise omitted due to the BBB.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Disorders affecting the central nervous system (CNS) like neurodegenerative disorders, tumors and infections are becoming more prevalent with great demands for development of new pharmaceutics. The treatment is, however, complicated by the inability of potential drugs to cross the blood–brain barrier (BBB) [1], which explains why more than 98% of drug candidates for treatment of CNS disorders never make it to the clinic [2–5]. The BBB is a dynamic physical and biological barrier located between the bloodstream and the CNS, more specifically at the capillary level. The BBB is formed by brain capillary endothelial cells (BCECs) lining the cerebral microvasculature. These cells are in close contact with pericytes and astrocytic endfeet, which are important for the formation and maintenance of the BBB properties [6–8]. The BBB is important in controlling the passage of endogenous and exogenous substances in and out of the CNS [9].
Neurotrophins and growth factors are implicated in a variety of neurological disorders [10]. A general feature of many neurological disorders is the loss of specific populations of neurons. Several studies have reported on the potential of neurotrophins and growth factors like growth hormone 1 (GH1), glia-derived neurotrophic factor (GDNF), brain-derived neurotropic factor (BDNF), and erythropoietin (EPO), to protect diseased or injured neurons from dying, induce neuronal sprouting, and increase neuronal metabolism and function [11–14]. Therefore, they are believed to be useful as therapeutic agents in a variety of neurologic disorders. These neuroprotective peptides are however not able to enter the CNS due to the BBB, and must therefore be transported across the BBB by use of different drug delivery strategies, e.g., gene therapy. Over the past decade, many therapeutic agents have moved from tissue culture studies and animal models into clinical trials using the gene therapy delivery approach to treat neurodegenerative diseases like Parkinson’s and Alzheimer’s disease [15–19]. One of these therapeutic agents is EPO [20].
EPO is a 165-aminoacid glycoprotein and member of the type I cytokine superfamily, known to increase red cell mass, and thereby increase tissue oxygenation [21, 22]. Its main site of production is in the kidneys in response to hypoxia. However, EPO and its receptor (EPO-R) are also weakly expressed in other tissues including the CNS [23]. Brain EPO is of lower molecular weight (30.3 vs. 30.5 kD) than systemic EPO, and the effects of brain EPO in vitro is higher in lower concentrations compared to the effects of systemic EPO [24]. Its main production within the brain is in neurons, neuronal progenitor cells, glial, and cerebrovascular endothelial cells, but is particularly highly expressed in brain regions containing neurons vulnerable to ischemic insult, such as the hippocampus and cerebral cortex [25–27]. EPO-R is expressed in BCECs, neurons, astrocytes, and microglial cells [22, 27–29]. The main neuroprotective function of EPO is believed to be inhibition of apoptosis in cells, which express the EPO-R [30]. Several in vitro [31–35] and in vivo [22, 36–39] studies have shown that EPO is able to protect neurons during hypoxia, indicating that EPO has beneficial effects in the treatment of stroke patients, which has also been confirmed in a clinical trial [40]. Additionally, these neuroprotective effects have also been seen in an animal model of Parkinson disease, by decreasing the loss of dopaminergic neurons, and consequently significantly increase the locomotor activity. Furthermore, the protective effects of EPO have been observed in animal models of schizophrenia, epilepsy, multiple sclerosis, and amyotrophic lateral sclerosis [22, 41–44]. In addition to its direct neuroprotective effect, EPO has also been shown to have a protective effect on the BBB by decreasing its permeability, and might thereby also contribute indirectly to neuroprotection [23, 28].
The use of gene therapy as a drug delivery strategy is dependent on the use of a vector that can protect the genetic material against degradation and enable efficient gene delivery to the target cells. Both viral and non-viral vectors have been widely explored. The viral vectors are the most efficient in delivering the genetic material, but the non-viral gene carriers exhibit lower immunogenicity, and are, therefore, considered safer to use [45, 46]. Non-viral vectors include cationic lipids, polymers, and peptides [47, 48]. Efficient gene therapy is very dependent on the selection of an appropriate transfection vector, which should have the following properties. The vector should be able to bind and condensate the genetic material, e.g., plasmid DNA. This can be achieved by an excess positive charge, which both will condensate the negatively charged DNA, but also interact with the negatively charged cell membrane. Secondly, it should be able to protect the DNA against degradation from blood components, like enzymes and endonucleases on its way to the target site. Upon arrival, it must endorse internalization of the genetic material into the target cell by endocytosis or another biological process. Within the endosome, the DNA:vector complex should bypass degradation by the endosomal/lysosomal system and release from the endosome into the cytoplasm, a process referred to as endosomal escape [47, 49]. The final task of the vector is to facilitate transport of the genetic material into the cell nucleus. This process is believed to either involve transport of DNA through pores in the nuclear membrane or by cell division, during which the nuclear envelope is temporally disassembled, allowing the genetic material to enter the nucleus [50, 51]. The major limitation of the non-viral gene vector in regards to the viral vectors is, despite a lot of advantage in the area, still low transfection efficiency. Their advantages over the viral vectors are, however, numerous and include larger loading size of genetic material, lower immunogenicity, ease of production and upscaling, reproducibility, low cytotoxicity, and a general safer use. They, therefore, still holds the potential to supplant viral vectors in the future [47, 49].
Delivery of genetic material to the brain is complicated by the BBB, and is dependent on the ability of the vector to bypass the BBB [1]. Other strategies for gene delivery to the CNS involves risky and invasive procedures like temporary opening of the BBB and intraparenchymal injections [47, 49]. Therefore, it is highly relevant to develop methodologies that allow for noninvasive routes for delivering genetic material into the brain. In this study, we explore the strategy of using the BCECs as a target for gene therapy. Gene therapy to the BCECs involves a two-step delivery strategy, which includes the uptake and process of genetic material by the cells, and subsequently secretion of the recombinant protein into the brain by the genetically modified BCECs [52, 53]. Several obstacles must be overcome for this strategy to be successful. First of all, BCECs in vivo are not in a mitotic state (referred to as non-dividing), so the non-viral carrier must be able to deliver the DNA through pores in the nuclear envelope. Secondly, the transfection should not cause an opening of the BBB, thereby allowing the entrance of unwanted substances into the brain parenchyma. Thirdly, genetically modified BCECs will need to secrete the recombinant protein towards the brain parenchyma, not only towards the blood stream. Finally, this drug delivery strategy should result in a high percentage of genetically modified BCECs secreting the recombinant protein, in order to ensure a high bioavailability of the therapeutic product in the diseased tissue. We have recently shown that non-viral gene therapy to non-diving primary rat brain capillary endothelial cells (RBECs) cultured in an in vitro BBB model together with astrocytes could be transfected without the disruption of the cells barrier properties [54]. We now report on higher transfection efficiency using a different vector and secretion of the neuroprotective protein EPO towards the brain tissue, with a measurable biological effect on the gene expression of BDNF in astrocytes.
Materials and methods
The following reagents were purchased from Sigma-Aldrich (Brondby, Denmark, DK): Heparin (Cat. No. H3149), Insulin transferrin sodium selenite (Cat. No. 11074547001), puromycin (Cat. No. P8833), collagen type IV (Cat. No. C5533), fibronectin (Cat. No. F1141), poly-L-lysine (Cat. No. P6282), hydrocortisone (Cat. No. H4001), Dimethyl Sulfoxide (DMSO) (Cat. No. D2650), CTP-cAMP (Cat. No. C3912), 4-(3-Butoxy-4-methoxybenzyl)imidazolidin-2-one (RO-201724) (Cat. No. B8279), Competent CG5 Escherichia coli strain (Cat. No. G3169), Paraformaldehyde (Cat.No. 441244), Triton™-X-100 (Cat. No. X100), and 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) (Cat. No. D9542). The following reagents were purchased from Life Technology (Naerum, Denmark, DK): Fetal calf serum (Cat. No. 10270), Dulbecco’s Modified Eagle Medium consisting of nutrient Mixture F-12 (DMEM/F-12) (Cat. No. 31331), DMEM (Cat. No. 21885), Trypsin (Cat. No 15090-46), Turbofect™ (Cat. No. R0531), Lipofectamine 3000™ (Cat. No. L3000), phosphate-buffered saline (PBS) (Cat. No SH3025802), GeneJet plasmid Midiprep kit (Cat. No. K0481), Rabbit Anti-Zonula Occludens (ZO1) (Cat. No. 61-7300), Alexa Fluor 594-conjugated goat anti-rabbit IgG (Cat. No. A11037), GeneJet RNA purification kit (Cat. No. K0731), DNaseI enzyme (Cat. NO. EN0523), Thermo Scientific Maxima H Minus first strand cDNA synthesis kit (Cat. NO. K1651), Maxima SYBR Green Master mix (Cat.No. K0252), TAG Copenhagen (Frederiksberg, Denmark, DK) synthesized primers. Basic fibroblast growth factor (Cat. No. 100-18B) was purchased from PeproTech Nordic (Stockholm, Sweden, SE). Plasma-derived bovine serum (Cat. No. 60-00-810) was purchased from First Link (Wolverhampton, United Kingdom, UK). Gentamicin Sulfate (Cat.No. 17-518Z) was purchased from Lonza Copenhagen (Vallensbaek Strand, Denmark, DK). Greiner bio-one Thincert cell culture insert for 12-well plates, with a transparent polyethylene terephthalate (PET) membrane and a pore diameter of 1 µm (Cat No. 665610), was purchased from In Vitro (Fredensborg, Denmark, DK). CytoFLEX Daily QC Fluorospheres (Cat. No. B53230) were purchased from Beckman Coulter (Copenhagen, Denmark, DK). Bovine serum albumin (BSA) (Cat. No. EQBAH62) was purchased from Europa Bioproducts (Cambridge, United Kingdom, UK). Rabbit anti-EPO antibody (Cat. No sc-7956) was purchased from Santa Cruz (Heidelberg, Germany). Promokine Cell proliferation kit I (CFDA SE) (PK-CA707-30050) was from BioNordika Denmark A/S (Herlev, Denmark, DK) and Fluorescence mounting media (Cat. No S3023) were purchased from DAKO (Glostrup, Denmark, DK). Rat EPO Gene ORF cDNA expression pCMV3-C-green fluorescent protein (GFP) Spark plasmid, from Sino Biological Inc. (Cat.No. RG80055-ACG) and LEGEND MAX™ Rat EPO ELISA Kit with pre-coated plates from BioLegend (Cat.No 442807) were purchased from Nordic BioSite (Copenhagen, Denmark, DK).
Isolation of primary rat brain capillary endothelial cells (RBECs) and astrocytes
Primary cultures of RBECs were prepared from two-to-three-week-old Sprague Dawley rats as described previously [54]. RBECs were maintained in DMEM/F12 supplemented with 10% plasma-derived bovine serum, heparin, insulin, transferrin, sodium, selenite, 10 µg/mL gentamicin sulfate, and 1 ng/µl basic fibroblast growth factor (referred to as RBEC media). Puromycin (4 µg/mL) was additionally added to the culturing medium for the first three days of culturing. All surface areas for culturing RBECs were coated twice with collagen IV/fibronectin. RBECs were isolated three days prior to the experiment and either frozen (RBEC media supplemented with 30% plasma-derived bovine serum and 7.5% DMSO) for later use or used directly in the transfection experiments.
Primary astrocytes were prepared from two-day old Sprague Dawley rats as described previously [54, 55]. Astrocytes were maintained in DMEM supplemented with 10% fetal calf serum and 10 µg/mL gentamicin sulfate. All surface areas for astrocytes were coated with poly-L-Lysine. Astrocytes were isolated and cultured for three weeks, after which they were either frozen (DMEM supplemented with 30% Fetal calf serum and 7.5% DMSO) or seeded directly into 12-well plates at a density of approximately 30,000 cells/cm2, and maintained for a minimum of two weeks prior to co-culture experiments. The cell culture medium was changed every fourth day. All cells were cultured in an incubator with humidified 5% CO2/95% air at 37 °C.
Cell cultures
For monocultures, RBECs were seeded in 24-well plates at a density of 100.000 cells/cm2, and left to adhere for 24 h resulting in a confluent monolayer. The RBECs were maintained in RBEC media and cultured for additionally 24 h to ensure the cells were 100% confluent and non-dividing. Monocultures of the cervix cancer cell line HeLa were likewise prepared in 24-well plates and maintained in DMEM supplemented with 10% fetal calf serum and 10 µg/mL gentamicin sulfate. HeLa cells were seeded at a density of 15.000 cells/cm2, left to adhere for 24 h, and maintained for an additionally 24 h to ensure a confluency of 80–90%. The media of both monocultures were changed prior to the addition of the transfection complexes.
RBECs in co-culture were seeded on hanging culture inserts at a cell density of 100.000 cells and left to adhere for 24 h, after which the cells were confluent. Induction of the RBECs barrier integrity was stimulated by culturing the RBECs together with astrocytes (non-contact co-culture), and by adding hydrocortisone (550nM), CTP-cAMP (250 µM), and RO (17.5 µM) to the upper chamber, and hydrocortisone to the lower chamber. The media composition of the lower chamber was a combination of RBEC media and astrocyte-conditioned media, while only RBEC medium was used in the upper chamber. Barrier integrity of RBECs in co-culture with astrocyte was present after 24 h measured as trans-endothelial electrical resistance (TEER). TEER was measured using a Millicell ERS-2 epithelial Volt-Ohm meter and a STX01 chopstick electrode (Millipore, Hellerup Denmark, DK) as described previously [54]. TEER values of coated but cell-free inserts were subtracted the measured TEER values, and the difference was multiplied the size of the insert (1.12 cm2). TEER values are given as Ω*cm2 or as percentage TEER, where the difference in TEER between 0 and 24 h was calculated for each culture insert and multiplied with 100. Data were analyzed in GraphPad Prism 6.0 (GraphPad Software, Inc., CA, USA) using one-way ANOVA with Dunnett’s post hoc test. Only RBECs co-cultures with TEER values above 150 Ω*cm2 were used for the transfection studies [54, 56].
In vitro transfection
Transfection was analyzed both in monocultures, in co-cultures of RBECs, and in monocultures of HeLa cells. The cells were transfected using Turbofect™, PEI, and Lipofectamine 3000™. The transfection efficacies were analyzed using monocultures of RBECs and HeLa. In addition, different combinations of DNA vs transfection agents were analyzed in the monocultures out of which the two most potent combinations of each transfection agent were used for further studies. Co-cultures of RBECs were used to assess the toxicity of the transfection agents with regards to the barrier integrity of the RBECs. The most potent transfection agents with respect to high transfection efficacy and minimum disruption of the barrier integrity were used for the remaining part of the study, which were all carried out in the co-culture setup.
Rat EPO Gene ORF cDNA expression pCMV3-C-GFPSpark plasmid was propagated in a competent CG5 Escherichia coli (E-coli) strain by heat shock and purified with ion exchange chromatography with the plasmid DNA purification kit prepared according to the manufacture’s protocol. DNA was complexed with three different transfection agents as described in the following section. The concentrations all applies to a single well in a 24-well plate (1.9 cm2) or a single 12-well hanging culture insert (1.12 cm2). Turbofect™: 0.5 or 1 µg plasmid DNA was mixed in 100 µl DMEM-F12. 2 µl Turbofect™ were added and immediately mixed by vortexing. Turbofect™:DNA complexes were left to form for 20 min at room temperature. A low molecular weight Polyethylenimine (PEI F25-LMW) was generated as described previously [57]. One or 2 µg plasmid DNA was diluted in 25 µl HN buffer (10 mM HEPES, 150mM NaCl in DEPC-treated water, pH 7.4). In another tube, PEI was diluted to a concentration corresponding to five times the mass ratio of DNA in 25 µl HN buffer. The two tubes were incubated separately for five min before being mixed together by vortexing. PEI:DNA complexes were left to form for 30 min at room temperature. 0.5 µg plasmid DNA was diluted in DMEM-F12 together with a P3000 reagent (2 µl/µg DNA). Either 0.75 or 1.5 µl Lipofectamine 3000™ was diluted in DMEM-F12, and both solutions were mixed well separately before DNA was added to the tubes containing Lipofectamine 3000™ in a 1:1 ratio. The DNA:Lipofectamine 3000™ complexes were left to form for 15 min at room temperature. The cells were incubated with the complexes for 24 h.
Flow cytometry
The transfection efficiency was analyzed using flow cytometry. The cells were transfected for 24 h, then washed twice in 0.1 M PBS, pH 7.4, detached from the culture support by trypsin, and washed twice in PBS by spinning the sample at 300xG for 5 min. Cells from three wells in a 24-well plate or three hanging culture insert were pooled together. Transfection efficiency was analyzed using CytoFLEX S (Beckman Coulter Copenhagen, Denmark, DK)), which prior to the flow cytometic analysis was calibrated using CytoFLEX Daily QC Fluorospheres. The cells were gated using forward and side scatters to eliminate cell debris and doublets. Approximately, 50.000 cells were analyzed in each sample. Non-transfected cells were used as a control for auto fluorescence. EPO-GFP-positive cells were gated based on auto fluorescence from the non-transfected cells to ensure less than 0.5% false positive events occurred. The results were analyzed using the CytExpert software (Beckman Coulter, Copenhagen, DK), and GraphPad Prism 6.0 software using a one-way ANOVA with Dunnett’s multiple comparisons post hoc test.
CFSE-DA assay
The cell division activity of RBECs in monoculture was analyzed using a CFDA SE assay [54], which is a dye that binds to intracellular proteins. The dye becomes fluorescent (495/519), when hydrolyzed by intracellular esterases, causing a long-term labeling of the cells. The label is inherited each time the cell divides, resulting in each daughter cell receiving half the label [58]. In brief, the cells were seeded in 24-well plates, and left to adhere for 24 h. The cells were labeled with 1 µM CFSA SE in PBS for 10 min. The CFDA SE solution was then removed, and the cells incubated in RBEC media for additionally 30 min to ensure hydrolysis of the CFDA SE label. The cells were subsequently cultured for 0–72 h to analyze their cell division activity. Every 24 h, a sample was terminated by detaching the cells of three wells in the 24-well plate by trypsin and washed in PBS by spinning the sample for 5 min at 300xG. The cells were subsequently fixed in 4% paraformaldehyde for 5 min, followed by two PBS washing steps. The cells were analyzed using the flow cytometer Cytoflex S. The cells were then gated using forward and side scatters to eliminate cell debris, and approximately 40.000 cells were analyzed in each sample. Non-labeled cells served as control for auto fluorescence. The results were analyzed using the CytExpert software.
Immunocytochemistry
Cells were washed in 0.1 M PBS, pH 7.4 and fixed for 10 min in 4% paraformaldehyde at room temperature. The cells were permeabilized and blocked for unspecific binding of primary antibody in PBS supplemented with 3% BSA and 0.2% Triton-X-100 for 30 min. All incubations were performed at room temperature with mild agitation. The RBECs were stained for ZO1 (1:500) and EPO (1:250). The cells were incubated with primary antibodies diluted in PBS supplemented with 3% BSA and 0.2% Triton-X-100 for one hour at room temperature. Alexa Fluor 594-conjugated goat anti-rabbit IgG antibodies was used in a dilution of 1:250 in PBS supplemented with 3% BSA and 0.2% Triton-X-100 and incubated for 30 min. Non-transfected cells were used as control for non-specific binding of primary antibodies. Nuclei were counterstained with DAPI. All cells were mounted on glass slides with fluorescent mounting media and examined in a fluorescence Observer Z1 microscope with ApoTome 2 under a Plan-Apochromat 40x/1.3 Oil DIC objective (Carl Zeiss, Germany). Captured images were corrected for brightness and contrast with ImageJ.
Gene expression analysis
RBECs cultured in co-culture and astrocytes with and without the exposure to recombinant EPO were used for RT-qPCR analysis. RNA was extracted using the GeneJet RNA purification kit according to the manufacture’s protocol. Four to seven RNA samples were obtained for each situation. Each RNA sample corresponded to three hanging culture inserts (RBECs) or three wells in a 12-well culture plate (astrocytes). RNA samples were treated with DNaseI enzyme to remove genomic DNA contamination. 100 ng of each DNA-free RNA sample was used as template for RT-qPCR. cDNA synthesis was carried out using the Thermo Scientific Maxima H Minus First Strand cDNA Synthesis Kit. To assess the expression profile of the transfected cells, qPCR was performed using primers specific for rat EPO, rat EPO-R, rat glial fibrillary acidic protein (GFAP), and rat BDNF (for additional information on primers see Table 1). The primers specifically designed to amplify rat EPO did not distinguish between the endogenous and recombinant forms of EPO. Rat β-Actin and rat hypoxanthine phosphoribosyltransferase 1 (HPRT1) were used as a housekeeping control genes for normalization purpose. 2.4 ng cDNA and 10 pmol of each primer were used together with Maxima™ SYBR Green qPCR Master Mix. Non-transfected cells, non-reverse transcribed RNA, and water served as negative controls. RT-qPCR was performed using the Stratagene Mx3000P™ QPCR system (Agilent Technologies, Horsholm, Denmark, DK). PCR conditions were 95 °C for 10 min, 40 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s. The relative expression of mRNA was calculated according to Pfaffl [59] and analyzed in the GraphPad Prism 6.0 software using a one-way ANOVA with Dunnett’s or Tukey’s multiple comparisons post hoc test, or an unpaired t-test.
Protein secretion analysis
The amount of recombinant EPO secreted from the transfected RBECs was analyzed using ELISA specific for rat EPO. The direction of recombinant EPO secretion from the RBECs was determined in the co-culture setup by sampling the cell culture media from both the upper and lower chambers 24 h after transfection. Additionally, the transport of rat EPO across the BBB was studied in the co-culture setup by adding 250 pg/mL rat EPO standard (provided in the ELISA kit) to either the upper or the lower chamber of the hanging culture insert. After incubation for 24 h, media from the upper and lower chamber was sampled and analyzed with ELISA. Media samples were stored at −80 °C, prior to the ELISA analysis. A twofold standard curve ranging from 1 ng/mL to 16 pg/mL rat EPO standard were prepared for each assay, according to the manufacture’s instruction. In brief, the plates were pre-coated with a rat monoclonal anti-EPO antibody, and incubated with the cell culture samples for two hours, after which the plate was washed several times before being incubated with anti-rat EPO antibody for one hour. The wells were again washed and incubated with avidin-HRP solution for 30 min, followed by a thorough washing step prior to the addition of the substrate solution, which were incubated in the dark for 10 min. The reaction was terminated, and the absorbance measured at 450 nm and subtracted the background absorbance at 570 nm using Tecan Sunrise Microplate Reader (Tecan, Switzerland). All samples were analyzed in duplicates and all incubation steps were performed at room temperature during shaking. Data were analyzed with a computer-based curve-fitting software using a 5-parameter logistics curve-fitting algorithm (Elisaanalysis.com). The mean absorbance for each set of duplicate was calculated, and in diluted samples concentrations were multiplied the by the appropriate dilution factor. The data were analyzed using the GraphPad Prism 6.0 software using a two-way ANOVA with Sidak´s multiple comparisons post hoc test.
Results
Transfection of BCECs to secrete proteins into the brain has been recognized as a novel drug delivery strategy [52, 53]. We have recently shown that non-diving RBECs cultured in co-culture with astrocytes are able to take up genetic material and process it into proteins without disrupting the barrier integrity [54]. The percentage of transfectable cells was 4%, which might raise questions to whether the amount of protein getting into the brain might be enough to have an effect. We have therefore in the present study compared three different transfection agents with the aim of increasing the transfection efficiency.
Screening of transfection agents using monocultures
The three different transfection agents studied were Turbofect™, PEI, and Lipofectamine 3000™. Turbofect™ and Lipofectamine 3000™ are both commercially available transfection agents, both claimed to have high transfection potential and low cytotoxicity. The RBECs were cultured in monoculture and transfected with a plasmid encoding rat EPO, which is coupled to a GFP tag, making EPO-GFP positive cells easily detectable in a fluorescent microscope, as they appear green. A series of DNA:transfection agents ratios were investigated in order to find the most potent combination, and based on fluorescent microscopic analyses, the two most potent combinations were subsequently analyzed in flow cytometric analyses (Fig. 1). Turbofect™ was complexed with either 0.5 or 1 µg DNA, while PEI was complexed with either 1 or 2 µg DNA. PEI is known to be somewhat toxic to the cells [47], albeit lower molecular weight PEIs, like the PEI F25-LMW used here, have been found to be more biocompatible in vitro and in vivo [57, 60, 61]. Still, to address possible adverse effects on in vitro BBB integrity, different exposure times (24 h and three hours) of the cells to the DNA:PEI complexes were additionally analyzed. Either 0.75 or 1.5 µl Lipofectamine 3000™ was complexed to 0.5 µg DNA. The transfection efficacies of the RBECs were compared to that of the easily transfectable cervix cancer cell line HeLa (Fig. 1).

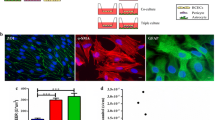
Transfection efficacies in monocultures of RBECs and HeLa cells. a Transfection efficacies were analyzed using three different transfection agents and different DNA:transfection agents ratios. The cells were transfected with a plasmid encoding rat EPO coupled to the green fluorescent tag (EPO-GFP). Positively transfected cells are therefore recognized as green fluorescent cells. Flow cytometric analysis was used to count the percentage of transfected cells, and thereby determine the most potent transfection agent. Lipofectamine 3000™ was the most potent transfection agent resulting in 27.74 ± 1.0% EPO-GFP expressing RBECs (dark purple). b HeLa cells were used as positive controls and compared to the RBECs transfection resulted in more than twice as many positive cells. The percentage of positive cells was statistically compared to the non-transfected cells using one-way ANOVA with a Dunnett´s multiple comparison post hoc test. Data are presented as mean ± SEM (n = 4–8). *P < 0.05, **P < 0.01, ***P < 0.001. c To evaluate if this high transfection efficiency was due to high cell division activity of the endothelial cells, their cell proliferation activity was analyzed using a CFDA SE assay. Here no or very little mitotic activity was observed between 24 and 48 h (red and purple respectively) after seeding, which corresponded to the timeframe during which the transfection studies were performed. d Immunocytochemical staining of RBECs transfected with Turbofect™ (1 µg), PEI (2 µg) and Lipofectamine 3000™ (0.75 µl). EPO-GFP-positive cells are seen in green, while ZO1 is stained red. Double-labeling EPO-GFP-positive cells (green) with an anti-EPO antibody (red), co-localization is observed, indicating recombinant protein expression is EPO. More EPO expressing cells are observed, when the cells are transfected with Lipofectamine. Nuclei are stained with DAPI (blue). Scale bar 20 µm
Transfection with Turbofect™ resulted in 2.76 ± 0.51% (light green) to 5.3 ± 0.33% (dark green) EPO-GFP-positive RBECs (Fig. 1a). Lipofectamine 3000™ was the most promising transfection agent ranging from 21.78 ± 2.9% (light purple) to 27.74 ± 1.0% (dark purple) positive EPO-GFP RBECs. RBECs transfected with PEI showed transfection efficacies of 6.28 ± 2.97% (red), while decreasing the cells’ exposure to PEI to only three hours was found insufficient for transfection (1.70 ± 0.62% (dark red)) (Fig. 1a). When transfecting HeLa cells with the same combinations of DNA:transfection agents ratios, more than three times as many cells were transfected. Turbofect (0.5 µg) and Lipofectamine 3000™ (1.5 µl) were the most potent transfection agents, resulting in approximately 65% EPO-GFP expressing HeLa cells (green and purple). When transfecting the HeLa cells with PEI, an ~40% transfection rate was observed upon 24 h exposure, with again a decrease in percentage of EPO-GFP expressing cells, when reducing the exposure time of PEI to three hours (blue and red) (Fig. 1b).
EPO-GFP-positive RBECs were seen as green fluorescent cells, when examined under the fluorescent microscope, and when immunolabeled with ZO1 (red), which is a tight junction protein highly expressed by BCECs, the EPO-GFP-positive cells were recognized as BCECs (Fig. 1d). RBECs were cultured in monoculture without the induction of any barrier properties, which is also observed by the rifled lines in the ZO1 protein expressions at the cell–cell borders. When immunolabeling the RBECs using anti-EPO, co-localization between EPO-GFP and EPO (red) was obvious. More EPO-GFP-positive cells were observed, when the cells were transfected with Lipofectamine 3000™ (0.75 µl) compared to cells transfected with Turbofect™ and PEI (Fig. 1d).
The transfected RBECs show low mitotic activity
Dividing cells were reported to be more prone to non-viral transfection than non-dividing cells, due to the easy accessibility of the plasmid to the nucleus during cell division [62, 63]. RBECs in monoculture were seeded at a high cell density two days prior to the transfection studies to ensure, the cells had reached a confluent monolayer. However, to ensure that the relative high transfection efficacy of RBECs in monoculture was not caused by a high mitotic activity, cells were incubated with CFDA SE, which is a fluorescent tracer that binds and labels intracellular proteins (Fig. 1c). When cells divide, the two daughter cells inherit approximately half the label. RBECs were seeded in 24-well plates and left to adhere for 24 h, before being incubated with the CFDA SE tracer for 0–72 h. Non-labeled cells were used to assess the cells auto fluorescence (black). The lowest cell division activity was seen between 24 and 48 h (red and purple, respectively) after labeling, which corresponds to the timeframe, where the transfection experiments were carried out. Between 48 and 72 h (purple and blue, respectively), some of the cells probably started to die (Fig. 1c), which caused an increased cell proliferation to ensure the monolayer remained confluent, which was also confirmed, when the cells were examined under a light microscope (data not shown). However, increasing the time of the transfection experiments from 24 to 72 h did not increase the number of transfected cells (data not shown).
Effect of transfection on the barrier integrity of RBECs using a non-contact co-culture setup
To investigate the transfection agent’s effect on the RBECs barrier properties, RBECs were cultured in a non-contact co-culture setup together with astrocytes. RBECs were seeded at high cell density on hanging culture inserts and left to adhere for 24 h, after which they formed a confluent cell layer. The RBECs were then stimulated to exhibit BBB properties by co-culturing them with astrocytes and by the addition of BBB inducing factors (Hydrocortisone, cAMP, and RO). After another 24 h, the RBECs exhibited TEER values above 150 Ω*cm2, at which time point the transfection complexes were added. RBECs cultured on hanging culture inserts were previously shown to be non-diving [54]. The cells were incubated with the complexes for 24 h, and TEER was measured before and after the experiment. In all situations, TEER decreased during the 24 h of transfection. However, the most noticeable decrease was seen in cells transfected with PEI for 24 h (Fig. 2a). By decreasing the cells’ exposure to PEI to only three hours, a less dramatic decrease was observed. RBECs exhibit a tight monolayer to small tracers like mannitol and sodium fluorescein as long as TEER remains above 131–150 Ω*cm2 (stifled line) [54, 56]. Only RBECs transfected with Turbofect™ (0.5 and 1 µg) and Lipofectamine 3000™ (0.75 µl) remained above this point. To evaluate the decrease compared to the decrease also observed in the non-transfected cells, the decrease in percentage was calculated for each culture insert (Fig. 2b). Non-transfected cells decreased to 87.87 ± 2.48% after 24 h, while the decrease was significantly lower in all the transfected cells. Turbofect™ (1 µg) caused the lowest decrease (to 70.36 ± 1.95%), while Lipofectamine 3000™ decreased to 57.57 ± 2.8% after 24 h.

Effect on barrier integrity and transfection efficacies in RBECs in co-culture with astrocytes. a RBECs, cultured on hanging culture inserts and induced to exhibit BBB properties, were transfected with three different transfection agents and different DNA:transfection agent ratios for 24 h. The barrier integrity (TEER) was monitored and compared to that of non-transfected cells. In all setups TEER decreased. The most dramatic drop in TEER occurred, when the cells were exposed to PEI for 24 h. However, by decreasing the exposure for PEI to three hours, the decrease in TEER was less dramatic. TEER values above 150 Ω*cm2 (stifled line) indicate a tight endothelial cell layer. Only Turbofect™ and Lipofectamine 3000™ remained above this limit. b The difference in %TEER was calculated to evaluate the decrease of each situation compared to the control. All transfection setups were significantly lower than non-transfected cells. Statistical significances compared to non-transfected cells were analyzed using one-way ANOVA with a Dunnett´s multiple comparison post hoc test. Data are presented as mean ± SEM (n = 12–51). c The transfection efficiency using Lipofectamine 3000™ (0.75 µl) on RBECs in co-culture were additionally analyzed using flow cytometric analysis and found to be 8.41 ± 1.21% for Lipofectamine 3000™, which is lower than that observed, when the RBECs were in monoculture. The percentage of positive cells was statistically compared to the non-transfected cells using an unpaired t-test. Data are presented as mean ± SEM (n = 5–8). d The expression of EPO was furthermore validated using gene expression analysis. It was significantly higher in RBECs transfected with Lipofectamine 3000™. The gene expression was statistically analyzed using an unpaired t-test. Data are presented as mean ± SEM (n = 4). ***P < 0.001
Transfection of RBECs in co-cultures
Lipofectamine 3000™ (0.75 µl) showed a high transfection potential and even though it caused a decrease of 42%, it did not cause a total disruption of the BBB properties, we decided to move forward using only this transfection agent. To ensure that the transfection efficacy was comparable between the RBECs in monoculture and RBECs in co-culture with astrocytes, the transfection efficiency was likewise investigated in co-cultures (Fig. 2c). Unfortunately, the transfection efficacies of Lipofectamine 3000™ decreased to 8.41 ± 1.21%. The expression of EPO was additionally investigated at the gene level, and here the relative gene expression level of EPO was significantly higher in transfected cells compared to non-transfected cells (Fig. 2d).
Immunolabeling of RBECs in co-culture revealed a more continuous expression of ZO1 at the cell–cell borders of RBECs in both the transfected and non-transfected cells (Fig. 3) compared to those cultured in monoculture (Fig. 1d). The expression of the ZO1 was, however, more continuous in non-transfected cells compared to the transfected cells. Also in the co-culture setup, the EPO-GFP-positive cells expressed ZO1, and co-localization was seen between EPO-GFP and immunolabeled EPO. The co-localization was especially evident in close proximity to the cell nucleus possibly corresponding to the site of the rough endoplasmic reticulum and the Golgi apparatus, i.e., the site where translation and packing of proteins for secretion are located. No EPO-GFP-positive cells or unspecific binding of the anti-EPO antibody were seen in the non-transfected cells (Fig. 3).

Immunocytochemical staining of RBECs in co-culture with astrocytes. The tight junction protein ZO1 (red) was determined in cells transfected with Lipofectamine 3000™ and compared to that of non-transfected cells. The latter exhibited higher TEER values than the transfected cells, which is seen as a continued expression of ZO1 at the cell borders, while this expression pattern was a little less evident in the transfected cells. EPO-GFP-positive cells are observed as green fluorescent cells, and immunocytochemical staining for EPO reveals co-localization between EPO and the GFP tag in the cell cytoplasm, especially close to the nucleus, probably corresponding to the site of the Golgi apparatus and the rough endoplasmic reticulum. Nuclei are stained with DAPI (blue). Scale bar 20 µm
Directional secretion of EPO from genetically modified RBECs
Using the in vitro BBB model, it is possible to define a blood and a brain side of the endothelial cells, since they become polarized, when cultured on hanging culture inserts [54]. EPO is a naturally occurring protein that exhibits specific physiological functions following secretion from kidney cells to promote proliferation of red blood cells [23]. EPO-positive cells would, therefore, theoretically secrete this recombinant protein into their surroundings, in this case the cell culture medium. Using the in vitro BBB model, it should therefore be possible to assess in which direction transfected RBECs will secrete recombinant EPO. Whether EPO is able to cross the BBB is site of conflict [22, 26, 64–66]. If EPO can cross the BBB, it will not be possible to determine the direction of EPO secretion using the in vitro BBB model. It was therefore investigated, whether EPO was able to cross the BBB using our in vitro BBB setup. RBECs in co-culture with astrocytes were incubated with rat EPO (250 pg/mL) added to either the upper compartment (blood) or lower compartment (brain). Media from both chambers were collected after 24 h and analyzed using ELISA specifically detecting rat EPO. The ELISA analysis revealed no significant transport of EPO across the BBB, irrespective to which side of the RBEC layer EPO was added (Fig. 4a). Approximately the same concentration as added was recovered in the media after 24 h (EPO added to upper chamber: 236.9 ± 32.9 pg/mL; EPO added to lower chamber: 265.9 ± 37.01 pg/mL). The sensitivity of the ELISA has some limitations, which is also why approximately 6 pg/mL EPO was found in the chambers in where no EPO was added. This corresponds almost to the same amount as that observed in the opposite chambers to where EPO was added (EPO added to upper chamber, lower chamber: 9.9 ± 9.38 pg/mL; EPO added to lower chamber, upper chamber: 4.7 ± 3.32 pg/mL). TEER remained at approximately the same level during the study, indicating that the exposure of RBECs to EPO did not have any effect on the RBECs barrier properties (Fig. 4b). BCECs in vitro express EPO-R [28, 29, 66], but since we did not see any transport of EPO, it was investigated whether the RBECs expressed EPO-R. Gene expression analysis revealed that RBECs express EPO-R, and this expression pattern was not significantly different, when the cells were transfected with EPO-GFP (Fig. 4c).

Transport of EPO across the in vitro BBB model. (a + b) Possible transport of EPO from the simulated blood side to the simulated brain side and reverse was examined using RBECs in co-culture with astrocytes. Rat EPO was added to either the upper chamber (blood side) or lower chamber (brain side) for 24 h, during which timeframe the barrier integrity was monitored using TEER measurements. a Cell culture media from upper and lower chambers was collected, and the concentration of EPO determined using ELISA specifically detecting rat EPO. No transport of EPO was seen after 24 h indicating that EPO is not able to cross the BBB from either side of the barrier. Significant differences were analyzed using a two-way ANOVA with Sidak’s multiple comparisons post hoc test. Significant differences were not found between the lower chamber with EPO added to upper chamber and the lower chamber with no EPO added, or between upper chamber with EPO added to lower chamber and upper chamber of no EPO added, indicating non-significant transport of EPO across the BBB (n = 6–9). b TEER values remained high, and no difference was seen between the cells not exposed to EPO and cells exposed to EPO at the brain- or blood site (n = 6). c The gene expression of EPO-receptors (EPO-R) by endothelial cells were additionally examined by gene expression analysis and found to be relative low (n = 4). All data are represented as mean ± SEM, and statistically significant differences compared to non-transfected cells were analyzed using a one-way ANOVA with Dunnett’s multiple post hoc test. ***P < 0.001
Since EPO was unable to cross the barrier of this in vitro BBB, the setup was useful in determining the amount and direction of recombinant EPO secretion. RBECs co-cultured with astrocytes were transfected with Lipofectamine 3000™. After 24 h, the media was collected and analyzed for the presence of recombinant EPO using ELISA specific against rat EPO. Media from non-transfected cells were collected and used in the analysis as negative controls (Fig. 5). The primary direction of EPO secretion by the RBEC was towards the upper chamber, corresponding to the blood side. Transfection with Lipofectamine 3000™ resulted in 1067.6 ± 108 pg recombinant EPO being secreted towards the blood side (upper chamber), while 198.74 ± 47.7 pg recombinant EPO was secreted towards the brain side (lower chamber) (Fig. 5a). Again, to evaluate the tightness of the endothelial cell layer during the experiment to ensure EPO was unable to pass though due to a leaky cell layer, TEER was monitored during the experiment. A decrease in TEER was observed, however not below 150 Ω*cm2 (Fig. 5b).

Secretion of recombinant EPO from transfected RBECs in co-culture with astrocytes. a RBECs were transfected using Lipofectamine 3000™ (0.75 µl) for 24 h, after which the media from the upper and lower chambers were collected, and the concentration of recombinant EPO analyzed using an ELISA specifically detecting rat EPO. RBECs secreted significantly higher amounts of recombinant EPO both towards the luminal and abluminal chambers, compared to non-transfected cells. The highest amount of recombinant EPO was secreted towards the luminal chamber (blood side). b TEER values were monitored during transfection and did not decrease below 150 Ω*cm2, meaning the RBEC cell layer remained intact. Significant differences were calculated using a one-way ANOVA with Sidaks multiple comparison post hoc test. All data are represented as mean ± SEM (n = 10–19) **P < 0.001, ***P < 0.001
Recombinant EPO secreted towards the abluminal chamber regulates BDNF expression in the astrocytes
Cells transfected with Lipofectamine 3000™ resulted in both secretion of recombinant EPO towards the blood and the brain side. Therefore, we examined whether the exposure to EPO had any effect on the astrocytes, cultured at the brain side in the in vitro BBB setup (Fig. 6). First, the presence of GFAP expressing astrocytes was confirmed in both situations. We additionally analyzed whether astrocytes expressed EPO-R at gene level, which would be an indication that astrocytes are able to take up EPO. We found expression of EPO-R, and the expression of EPO-R was not affected, when the astrocytes were exposed to recombinant EPO. EPO has previously been shown to regulate the expression of BDNF in neurons [21, 67, 68]. We therefore examined whether the exposure to recombinant EPO would have any biological effect on the expression of BDNF in the astrocytes, and here BDNF was significantly upregulated in astrocytes exposed to recombinant EPO, compared to astrocytes that had been co-cultured with non-transfected RBECs (Fig. 6).

Effect of recombinant EPO expression by RBECs on astrocytes. GFAP-positive astrocytes were cultured at the brain side (lower chamber) in the in vitro BBB model and therefore exposed to the recombinant EPO secreted by the RBECs transfected with Lipofectamine 3000™. Astrocytes were able to take up secreted recombinant EPO due to their expression of EPO-receptors (EPO-R). The astrocytes exposed to recombinant EPO had a higher gene expression level of BDNF compared to astrocytes, which were cultured together with non-transfected RBECs and not exposed to EPO. Significant differences were calculated using an unpaired t-test. All data are represented as mean ± SEM (n = 6–8). **P < 0.001
Discussion
Screening of different transfection agents to increase transfection efficiency of RBECs without disrupting BBB properties of the RBECs
We have for the first time successfully genetically modified primary isolated RBECs in vitro into protein factories that express the recombinant protein EPO, which is a glycoprotein showing great neuroprotective potential in a variety of neurodegenerative diseases. We have previously reported a transfection efficiency of RBECs expressing a red fluorescent protein to be 4% using the transfection agent Turbofect™ [54]. We questioned if this transfection efficiency was high enough to observe a significant biological effect within the diseased brain. We therefore set out to find a more potent non-viral transfection agent, which was able to increase this transfection efficiency without disrupting the barrier integrity of the RBECs. When using monocultures of RBECs, Lipofectamine 3000™ seemed to be a very promising vector with transfection efficacies over 20%. Lipofectamine 3000™ was also able to increase the transfection efficiency of RBECs in co-culture with astrocytes to approximately 8% compared to the 4% seen with the use of Turbofect™. Compared to Turbofect™, Lipofectamine 3000™ was somehow more compromising to the BBB properties of the RBECs, although not to an extent that allowed the cells to become leaky. We have previously reported on low cytotoxicity, when transfecting RBECs with Turbofect™ [54], which is confirmed in the present study.
PEI is one of the most widely used cationic polymers for transfection both in vitro and in vivo and is therefore also believed to be the golden standard among the non-viral vectors [61]. Previously, different PEI modifications have been investigated in order to decrease the cytotoxicity without decreasing the transfection potential and PEI with linear structures and low molecular weight are believed to be less cytotoxic than the branched PEI with higher molecular weights [61]. The PEI F25-LMW, used in this study has previously been widely examined in vivo in several organs and is generally found to be well tolerated [60, 61], possibly because of an excessive buffering by plasma proteins, when present in the circulation. The cytotoxicity observed in this in vitro study using PEI-F25 (LMW) has therefore not been confirmed, when used in vivo.
BCECs in vivo are characterized as having low endocytotic activity compared to endothelial cells of other organs [69]. This indicates that RBECs cultured under in vivo-like conditions, like in the in vitro BBB model, are less likely to take up material by endocytosis than RBECs monoculture without BBB properties. The presence of astrocytes might also cause a decreased rate of endocytosis in the RBECs [70, 71], which might explain the lower transfection efficiency of RBECs in co-culture compared to RBECs in monoculture. The highest transfection potential was not surprisingly found in HeLa cells. These cells have been extensively used for gene transfection experiments [72–75], since they have high endocytic activity, are easy to transfect and highly susceptible for expressing the newly introduced genes, which were also confirmed in this study. HeLa cells were primarily included in this study as a positive control for the assay, but also as a comparison, in order to better analyze the transfection potential of the primary isolated endothelial cells.
Directional secretion of recombinant EPO from genetically modified RBECs
Gene therapy to BCECs with the purpose of turning the BCECs into protein factories for protein secretion into the brain parenchyma was previously only studied using immortalized brain endothelial cell lines [14, 52]. Thomsen et al. [52] studied protein secretion of human GH1 from monocultures of immortalized human brain microvascular endothelial cell line (HBMEC) and the rat brain endothelial cell line (RBE4) using a similar strategy as reported in the present study. They used two different transfection agents for nuclear delivery of the plasmid, namely Pullulan–Spermine and Turbofect™. They did not report on the transfection efficiency, but they were able to obtain a transfection efficiency high enough to detect the presence of the recombinant GH1 secreted into the cell culture medium [52]. Jiang et al. [14] were also able to detect secretion of a neuroprotective protein after gene transfection. They transfected a mouse brain endothelial cell line (MBEC4) with a plasmid encoding the mouse GDNF using the commercially available transfection agent Lipofectamine 2000™ [14]. By culturing the MBEC4 cells on hanging culture inserts, they observed a significantly higher secretion of the recombinant GDNF protein towards the abluminal side (brain) as opposed to the luminal (blood) side. This observation was important; since it emphasizes that distribution of proteins to the brain after luminal transfection might not be an unrealistic drug delivery approach. In our study, however, we were not able to report on similar findings. We observed a secretion pattern, which was mainly luminal, with a significantly lower amount reaching the abluminal compartment. One might, however, question the integrity of the monolayer used in the study by Jiang and colleagues, since the use of immortalized cell lines are, generally, not considered the best cell type for obtaining a sufficiently tight in vitro BBB model [76, 77]. Additionally, the use of astrocytes, astrocyte-conditioned medium, or other tight junction inducing agents is often necessary to stimulate the integrity of the endothelial cell layer [76], of which they used neither [14]. We observed a high amount of recombinant EPO in the abluminal compartment, when the RBECs barrier integrity had dropped below approximately 130 Ω*cm2, indicating that RBEC layer was leaky even to larger proteins like EPO of 30.4 kDa [GFP tag (27 kDa)], when the barrier integrity was compromised (data not shown). Jiang and colleagues [14] evaluated the tightness of the polarized MBEC4 cell layer prior to their transfection experiments by Evans blue dye-binding bovine serum albumin, but they did not evaluate the impact of the transfection on this tightness. It might therefore had become leaky because of transfection. For their transfection studies, they used Lipofectamine 2000™, which according to the manufacture’s are more toxic than the newer version, Lipofectamine 3000™, which we observed had a significant impact on the barrier integrity. Jiang and colleagues also attempted the strategy in vivo in rats by intra carotid artery injection of Hemagglutination virus of Japan (HVJ)-mGDNF-liposomes, and observed a marked increase in brain levels of GDNF for up to 12 days. When they attempted the same in rats with a retrograde 6-hydroxydopamine (6-OHDA) lesion, GDNF showed protective effects on neurons [14], indicating that in vivo BCECs secrete therapeutic levels of the recombinant proteins into the brain tissue.
We speculated whether the lower concentration of EPO in the abluminal compartments could be caused by the presence of astrocytes in the abluminal compartments. Astrocytes express EPO-R [27] and could therefore take up the secreted recombinant EPO and clear it from the abluminal compartments. We therefore repeated the study using astrocyte-conditioned media to induce BBB properties instead of astrocytes. We did, however, not detect any change in the amount of recombinant EPO present in the abluminal compartment, indicating that the astrocytes did not clear EPO from the media. This was additionally confirmed by the fact that it was possible to recover the added concentration of EPO, which was added to the abluminal chamber in the EPO transport study (Fig. 4a). We then wondered whether EPO was trapped in the filter support of the hanging culture inserts. However, when the BBB properties of the cell layers were compromised (TEER >130 Ω*cm2), EPO could readily pass through the culture inserts, indicating that this was not the case.
Gene therapy as a drug delivery strategy for transport through the BBB
An important aspect to discuss is the possible side effects of this strategy. Transfection of BCECs in vivo will also result in a higher amount of the recombinant protein circulating in the blood. Unfortunately, in the previously mentioned study by Jiang et al., they did not analyze the impact of increasing circulatory levels of GDNF on other organs of the body than the brain [14]. However, a more recent study attempted to deliver GDNF across the BBB of Parkinson’s monkey by means of a Trojan horse technology, to which GDNF where coupled to a monoclonal antibody against the insulin receptor [78]. They did not observe any measurable improvements in the animals, but when assessing other organs than the brain, they found that the animals had developed pancreatic cancer. This study clearly emphasizes the risk of increased systemic amount of recombinant proteins of therapeutic potential within the brain on other organs of the body. The strategy of turning BCECs into protein factories might therefore not be a feasible strategy for all proteins. In our study, we have attempted to increase the availability of EPO within the brain tissue. EPO is a natural blood circulating protein; however, prolonged increased amounts of circulating EPO might cause side effects like seizures, hypertension, stroke, myocardial infarction, congestive heart failure, and tumor progression [79]. Studies have, however, shown that high levels of recombinant EPO are well tolerated [65, 80–82]. Other diseases, in which this strategy for delivery of genes into the BCECs for protein secretion to the brain might be useful, could be in the treatment of lysosomal storage diseases. More than 50 different lysosomal storage diseases exist with the majority of them characterized by the accumulation of storage material in somatic and nervous system cells, due to lack of certain enzymes involved in metabolic degradation of waste products in the lysosomes [83, 84]. Normally, cells of the brain will release lysosomal enzymes, which will be taken up by adjacent cells [45]. Therefore, by genetically modifying the BCECs to express and secrete the missing enzyme into the brain tissue, the missing enzyme becomes available for degradation of metabolic waste products in the diseased cells, a process known as cross-correction. The fact that the BCECs also secretes the recombinant protein into the circulation might in this case only be beneficial, as the enzyme is missing in all organs and tissues.
The large neuroprotective potential of EPO within the brain has resulted in several attempts to increase the presence of blood circulating recombinant EPO within the brain. Several study’s claim that EPO is able to cross the BBB [22, 64, 65], while other studies were not able to obtain transport across the BBB [66], unless the BBB was compromised [85]. Several in vivo studies have shown that less than 1% of injected dose are able to cross the BBB, and in order to measure this transport of EPO, very high levels of recombinant EPO must be systemically administered [79, 86]. It is a consensus that brain capillaries express EPO-R [22, 28, 29], which was also confirmed in this study. Despite this, several studies fail to show that EPO enters the brain via a receptor mediated transport system [23, 64, 65], but instead points to an extracellular pathway similar to that of albumin [64], which might also explain why high systemic injection doses are necessary for BBB transport. In accordance with many of the studies on EPO transport across the BBB, we did not observe any significant transport of EPO across the in vitro BBB model, despite the presence of EPO-R on the RBECs. In another in vitro BBB study using bovine BCECs co-cultured with rat astrocytes, it was shown that EPO was transported across the BCEC layer and that this transport could be decreased in the presence of an anti-EPO-R antibody, indicating that the transport of EPO was EPO-R dependent [28]. The transport of EPO was additionally observed both in the luminal to abluminal and abluminal to luminal direction, with the highest transport being luminal to abluminal [28]. We did not observe any significant transport of EPO in any direction; however, it cannot be excluded that this is due to lower concentrations of EPO added to the cell culture medium. The concentrations used in our assay were determined by the sensitivity of the ELISA used. The study using bovine BCECs in co-culture with astrocytes additionally showed that EPO was able to protect the BBB model against vascular endothelial growth factor-induced permeability. EPO added alone were likewise able to significantly decrease the BBB permeability [28]. In contrast, we did not observe any difference in TEER measurement between cultures incubated with EPO either at the luminal or abluminal side, compared to RBECs not incubated with EPO. TEER and permeability measures are not directly comparable, and it can therefore not be excluded that EPO might have an effect on the RBECs permeability.
Recombinant EPO secreted towards the abluminal chamber exert a biologic effect in the astrocytes
EPO has been postulated to be a potential target in the treatment of depression and has been shown to exert neurotrophic and neuroprotective properties by binding to EPO-R in the brain, and it is also believed to have antidepressants effects [21]. Depression is often associated with a lower level of BDNF within the brain [87, 88] and studies have shown that EPO is capable of altering the expression of BDNF [21, 67, 68]. With EPO being a cytokine expressed by several brain cells, including astrocytes, which express the EPO-R receptor, EPO might have a paracrine and autocrine effect on cells-like astrocytes [21]. Even though we observed lower levels of recombinant EPO secreted to the abluminal chamber than the luminal chamber, we wondered whether it was enough to measure any biological function on the astrocytes located in the abluminal compartment. We therefore confirmed the expression of EPO-R in astrocytes and subsequently examined the gene expression level of BDNF in astrocytes exposed to the secreted recombinant EPO, and compared it to the level in astrocytes, which had not been exposed to recombinant EPO. BDNF was significantly increased in astrocytes exposed to secreted amounts of recombinant EPO, indicating that the levels of secreted recombinant EPO exerted a biological change in the cultured astrocytes. This observation is in consensus with other studies showing that EPO is able to increase or restore the gene expression levels of BDNF in hippocampus [21, 67] and in primary cultured hippocampal neurons [68].
Translation from in vitro to in vivo
In the present study, we have explored the possibility of using gene therapy to the BBB in vitro, since there are less variables to consider than when examining the BBB in vivo. For the purpose, we have used an in vitro BBB model that mimics the in vivo situation as closely as possible. The results presented in this paper does therefore not directly translate to the in vivo situation in its current state. When BCECs are cultured on hanging culture inserts, they become polarized and have a defined apical, lateral, and basal membrane. This made it possible to evaluate the direction of secretion of the recombinant proteins. Whether the secretion of recombinant proteins will be different when studied in vivo is unknown, and of course, it needs to be evaluated. Additionally, it could be highly relevant to examine the mechanisms that directs secretion of recombinant proteins towards either the brain or the blood side, and to investigate whether this could be manipulated to increase the degree of recombinant proteins reaching the CNS. Furthermore, in vivo the BBB carries a dense negative charged glycocalyx on its luminal surface [89] that might negatively influence the uptake of the DNA:vector complex into the BCECs. We did not examine whether the glycocalyx is present, when the RBECs are cultured in vitro; however, the presence of a glycocalyx layer has previously been confirmed in an in vitro BBB model using immortalized mouse brain endothelial cells (bEnd3) [90]. For future in vivo studies, the choice of transfection agent and BBB targeting also need consideration. Intravenous injection of Lipofectamine 3000™ will not result in BBB specific transfection; however, intracarotid injection might enhance the bioavailability of the vector complex to the BBB endothelium. Several modifications to this protein delivery strategy need to be modified in order to examine the strategy in vivo. First of all, the vector should be designed to specifically target the BBB endothelium, e.g., by the use of a nanocarrier that has BBB specific targeting moieties on its surface, and the vector should be protected from blood-borne molecules. An alternative approach to specifically target the CNS could be through direct intraparenchymal injection, directly into the ventricles, or into the cerebrospinal fluid (CSF) to target the blood–CSF barrier. Even though this is a highly invasive route for administration, several studies have exploited these pathways with success primarily using viral vectors [91, 92].
Conclusions
We have established a method to deliver secretory neuroprotective proteins across the BBB and into the brain parenchyma by transferring genetic material into BCECs. Using primary isolated RBECs in co-culture with primary rat astrocytes, we show that BCECs take up and process the genetic material into recombinant proteins, which they subsequently secrete in the direction of both blood and brain. Turning BCECs into protein factories could be done without compromising the barrier properties of the BCECs, and the BCECs could be transfected with a sufficiently high transfection efficacy to yield recombinant protein reaching the brain side to exert a biological function in astrocytes. We therefore show that gene therapy to the BBB is a feasible drug delivery strategy, which result in protein expression and secretion of recombinant protein towards the brain tissue.
Abbreviations
- BBB:
-
Blood–brain barrier
- BCECs:
-
Brain capillary endothelial cells
- BDNF:
-
Brain-derived neurotrophic factor
- BSA:
-
Bovine serum albumin
- CSF:
-
Cerebrospinal fluid
- CNS:
-
Central nervous system
- DAPI:
-
4′,6-diamidino-2-phenylindole dihydrochloride
- DMEM:
-
Dulbecco’s Modified Eagle Medium
- DMSO:
-
Dimethyl sulfoxide
- EPO:
-
Erythropoietin
- EPO-R:
-
EPO-receptor
- GDNF:
-
Glial cell-derived neurotrophic factor
- GFAP:
-
Glial fibrillary acidic protein
- GFP:
-
Green fluorescent protein
- GH1:
-
Growth hormone 1
- HBMEC:
-
Human brain microvascular endothelial cells
- MBEC4:
-
Mouse brain endothelial cell line
- PEI:
-
Polyethylenimine
- PBS:
-
Phosphate-buffered saline
- RBECs:
-
Rat brain endothelial cells
- TEER:
-
Trans-endothelial electrical resistance
- ZO1:
-
Zonula occludens
References
Georgieva JV, Hoekstra D, Zuhorn IS (2014) Smuggling drugs into the brain: an overview of ligands targeting transcytosis for drug delivery across the blood-brain barrier. Pharmaceutics 6:557–583
Patel MM, Goyal BR, Bhadada SV, Bhatt JS, Amin AF (2009) Getting into the brain: approaches to enhance brain drug delivery. CNS Drugs 23:35–58
Pardridge WM (2005) The blood-brain barrier: bottleneck in brain drug development. NeuroRx 2:3–14
Pardridge WM (2012) Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab 32:1959–1972
Pardridge WM, Boado RJ (2012) Reengineering biopharmaceuticals for targeted delivery across the blood-brain barrier. Methods Enzymol 503:269–292
Abbott NJ (2002) Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat 200:629–638
Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C (2010) Pericytes regulate the blood-brain barrier. Nature 468:557–561
Daneman R, Zhou L, Kebede AA, Barres BA (2010) Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468:562–566
Abbott NJ (2013) Blood-brain barrier structure and function and the challenges for CNS drug delivery. J Inherit Metab Dis 36:437–449
Price RD, Milne SA, Sharkey J, Matsuoka N (2007) Advances in small molecules promoting neurotrophic function. Pharmacol Ther 115:292–306
Neumann JT, Thompson JW, Raval AP, Cohan CH, Koronowski KB, Perez-Pinzon MA (2015) Increased BDNF protein expression after ischemic or PKC epsilon preconditioning promotes electrophysiologic changes that lead to neuroprotection. J Cereb Blood Flow Metab 35:121–130
Wu D (2005) Neuroprotection in experimental stroke with targeted neurotrophins. NeuroRx 2:120–128
Arce VM, Devesa P, Devesa J (2013) Role of growth hormone (GH) in the treatment on neural diseases: from neuroprotection to neural repair. Neurosci Res 76:179–186
Jiang C, Koyabu N, Yonemitsu Y, Shimazoe T, Watanabe S, Naito M, Tsuruo T, Ohtani H, Sawada Y (2003) In vivo delivery of glial cell-derived neurotrophic factor across the blood-brain barrier by gene transfer into brain capillary endothelial cells. Hum Gene Ther 14:1181–1191
Marks WJ Jr, Ostrem JL, Verhagen L, Starr PA, Larson PS, Bakay RA, Taylor R, Cahn-Weiner DA, Stoessl AJ, Olanow CW, Bartus RT (2008) Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: an open-label, phase I trial. Lancet Neurol 7:400–408
Marks WJ Jr, Bartus RT, Siffert J, Davis CS, Lozano A, Boulis N, Vitek J, Stacy M, Turner D, Verhagen L, Bakay R, Watts R, Guthrie B, Jankovic J, Simpson R, Tagliati M, Alterman R, Stern M, Baltuch G, Starr PA, Larson PS, Ostrem JL, Nutt J, Kieburtz K, Kordower JH, Olanow CW (2010) Gene delivery of AAV2-neurturin for Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol 9:1164–1172
Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R, Patel P, Blesch A, Vahlsing HL, Ho G, Tong G, Potkin SG, Fallon J, Hansen L, Mufson EJ, Kordower JH, Gall C, Conner J (2005) A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med 11:551–555
Feigin A, Kaplitt MG, Tang C, Lin T, Mattis P, Dhawan V, During MJ, Eidelberg D (2007) Modulation of metabolic brain networks after subthalamic gene therapy for Parkinson’s disease. Proc Natl Acad Sci USA 104:19559–19564
Gasmi M, Herzog CD, Brandon EP, Cunningham JJ, Ramirez GA, Ketchum ET, Bartus RT (2007) Striatal delivery of neurturin by CERE-120, an AAV2 vector for the treatment of dopaminergic neuron degeneration in Parkinson’s disease. Mol Ther 15:62–68
Xue YQ, Ma BF, Zhao LR, Tatom JB, Li B, Jiang LX, Klein RL, Duan WM (2010) AAV9-mediated erythropoietin gene delivery into the brain protects nigral dopaminergic neurons in a rat model of Parkinson’s disease. Gene Ther 17:83–94
Ma C, Cheng F, Wang X, Zhai C, Yue W, Lian Y, Wang Q (2016) Erythropoietin pathway: a potential target for the treatment of depression. Int J Mol Sci doi:10.3390/ijms17050677
Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, Itri LM, Cerami A (2000) Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA 97:10526–10531
Noguchi CT, Asavaritikrai P, Teng R, Jia Y (2007) Role of erythropoietin in the brain. Crit Rev Oncol Hematol 64:159–171
Buemi M, Cavallaro E, Floccari F, Sturiale A, Aloisi C, Trimarchi M, Corica F, Frisina N (2003) The pleiotropic effects of erythropoietin in the central nervous system. J Neuropathol Exp Neurol 62:228–236
Masuda S, Okano M, Yamagishi K, Nagao M, Ueda M, Sasaki R (1994) A novel site of erythropoietin production. Oxygen-dependent production in cultured rat astrocytes. J Biol Chem 269:19488–19493
Marti HH, Wenger RH, Rivas LA, Straumann U, Digicaylioglu M, Henn V, Yonekawa Y, Bauer C, Gassmann M (1996) Erythropoietin gene expression in human, monkey and murine brain. Eur J Neurosci 8:666–676
Nagai A, Nakagawa E, Choi HB, Hatori K, Kobayashi S, Kim SU (2001) Erythropoietin and erythropoietin receptors in human CNS neurons, astrocytes, microglia, and oligodendrocytes grown in culture. J Neuropathol Exp Neurol 60:386–392
Martinez-Estrada OM, Rodriguez-Millan E, Gonzalez-De Vicente E, Reina M, Vilaro S, Fabre M (2003) Erythropoietin protects the in vitro blood-brain barrier against VEGF-induced permeability. Eur J Neurosci 18:2538–2544
Yamaji R, Okada T, Moriya M, Naito M, Tsuruo T, Miyatake K, Nakano Y (1996) Brain capillary endothelial cells express two forms of erythropoietin receptor mRNA. Eur J Biochem 239:494–500
Eid T, Brines M (2002) Recombinant human erythropoietin for neuroprotection: what is the evidence? Clin Breast Cancer 3(Suppl 3):S109–S115
Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R (1997) Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 76:105–116
Lewczuk P, Hasselblatt M, Kamrowski-Kruck H, Heyer A, Unzicker C, Siren AL, Ehrenreich H (2000) Survival of hippocampal neurons in culture upon hypoxia: effect of erythropoietin. Neuroreport 11:3485–3488
Ruscher K, Freyer D, Karsch M, Isaev N, Megow D, Sawitzki B, Priller J, Dirnagl U, Meisel A (2002) Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J Neurosci 22:10291–10301
Weber A, Maier RF, Hoffmann U, Grips M, Hoppenz M, Aktas AG, Heinemann U, Obladen M, Schuchmann S (2002) Erythropoietin improves synaptic transmission during and following ischemia in rat hippocampal slice cultures. Brain Res 958:305–311
Xu X, Dai H, Shi Y (2009) Erythropoietin protects primary cultures of rat cortical neurons from hypoxia-induced toxicity through attenuating both glutamate release and NMDA receptor evoked neurotoxicity pathway. Pharmazie 64:210–213
Calapai G, Marciano MC, Corica F, Allegra A, Parisi A, Frisina N, Caputi AP, Buemi M (2000) Erythropoietin protects against brain ischemic injury by inhibition of nitric oxide formation. Eur J Pharmacol 401:349–356
Genc S, Kuralay F, Genc K, Akhisaroglu M, Fadiloglu S, Yorukoglu K, Fadiloglu M, Gure A (2001) Erythropoietin exerts neuroprotection in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated C57/BL mice via increasing nitric oxide production. Neurosci Lett 298:139–141
Diem R, Sattler MB, Merkler D, Demmer I, Maier K, Stadelmann C, Ehrenreich H, Bahr M (2005) Combined therapy with methylprednisolone and erythropoietin in a model of multiple sclerosis. Brain 128:375–385
Osredkar D, Sall JW, Bickler PE, Ferriero DM (2010) Erythropoietin promotes hippocampal neurogenesis in in vitro models of neonatal stroke. Neurobiol Dis 38:259–265
Ehrenreich H, Hasselblatt M, Dembowski C, Cepek L, Lewczuk P, Stiefel M, Rustenbeck HH, Breiter N, Jacob S, Knerlich F, Bohn M, Poser W, Ruther E, Kochen M, Gefeller O, Gleiter C, Wessel TC, De Ryck M, Itri L, Prange H, Cerami A, Brines M, Siren AL (2002) Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med 8:495–505
Wustenberg T, Begemann M, Bartels C, Gefeller O, Stawicki S, Hinze-Selch D, Mohr A, Falkai P, Aldenhoff JB, Knauth M, Nave KA, Ehrenreich H (2011) Recombinant human erythropoietin delays loss of gray matter in chronic schizophrenia. Mol Psychiatry 16(26–36):1
Ehrenreich H, Fischer B, Norra C, Schellenberger F, Stender N, Stiefel M, Siren AL, Paulus W, Nave KA, Gold R, Bartels C (2007) Exploring recombinant human erythropoietin in chronic progressive multiple sclerosis. Brain 130:2577–2588
Grunfeld JF, Barhum Y, Blondheim N, Rabey JM, Melamed E, Offen D (2007) Erythropoietin delays disease onset in an amyotrophic lateral sclerosis model. Exp Neurol 204:260–263
Koh SH, Kim Y, Kim HY, Cho GW, Kim KS, Kim SH (2007) Recombinant human erythropoietin suppresses symptom onset and progression of G93A-SOD1 mouse model of ALS by preventing motor neuron death and inflammation. Eur J Neurosci 25:1923–1930
Simonato M, Bennett J, Boulis NM, Castro MG, Fink DJ, Goins WF, Gray SJ, Lowenstein PR, Vandenberghe LH, Wilson TJ, Wolfe JH, Glorioso JC (2013) Progress in gene therapy for neurological disorders. Nat Rev Neurol 9:277–291
Choong CJ, Baba K, Mochizuki H (2016) Gene therapy for neurological disorders. Expert Opin Biol Ther 16:143–159
Rezaee M, Oskuee RK, Nassirli H, Malaekeh-Nikouei B (2016) Progress in the development of lipopolyplexes as efficient non-viral gene delivery systems. J Control Release 236:1–14
Georgieva JV, Brinkhuis RP, Stojanov K, Weijers CA, Zuilhof H, Rutjes FP, Hoekstra D, van Hest JC, Zuhorn IS (2012) Peptide-mediated blood-brain barrier transport of polymersomes. Angew Chem Int Ed Engl 51:8339–8342
Jayant RD, Sosa D, Kaushik A, Atluri V, Vashist A, Tomitaka A, Nair M (2016) Current status of non-viral gene therapy for CNS disorders. Expert Opin Drug Deliv 1–13
Lechardeur D, Lukacs GL (2002) Intracellular barriers to non-viral gene transfer. Curr Gene Ther 2:183–194
Lechardeur D, Verkman AS, Lukacs GL (2005) Intracellular routing of plasmid DNA during non-viral gene transfer. Adv Drug Deliv Rev 57:755–767
Thomsen LB, Lichota J, Kim KS, Moos T (2011) Gene delivery by pullulan derivatives in brain capillary endothelial cells for protein secretion. J Control Release 151:45–50
Lichota J, Skjorringe T, Thomsen LB, Moos T (2010) Macromolecular drug transport into the brain using targeted therapy. J Neurochem 113:1–13
Burkhart A, Thomsen LB, Thomsen MS, Lichota J, Fazakas C, Krizbai I, Moos T (2015) Transfection of brain capillary endothelial cells in primary culture with defined blood-brain barrier properties. Fluids Barriers CNS 12:19
Burkhart A, Skjørringe T, Johnsen KB, Siupka P, Thomsen LB, Nielsen MS, Moos T (2015) Expression of iron-related proteins at the neurovascular unit supports reduction and reoxidation of iron for transport through the blood-brain barrier. Mol Neurobiol 53:7237-7253
Gaillard PJ, de Boer AG (2000) Relationship between permeability status of the blood-brain barrier and in vitro permeability coefficient of a drug. Eur J Pharm Sci 12:95–102
Werth S, Urban-Klein B, Dai L, Höbel S, Grzelinski M, Bakowsky U, Czubayko F, Aigner A (2006) A low molecular weight fraction of polyethylenimine (PEI) displays increased transfection efficiency of DNA and siRNA in fresh or lyophilized complexes. J Controll Release 112(257):270
Lyons AB, Parish CR (1994) Determination of lymphocyte division by flow cytometry. J Immunol Methods 171:131–137
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Hobel S, Vornicescu D, Bauer M, Fischer D, Keusgen M, Aigner A (2014) A novel method for the assessment of targeted PEI-based nanoparticle binding based on a static surface plasmon resonance system. Anal Chem 86:6827–6835
Hobel S, Aigner A (2013) Polyethylenimines for siRNA and miRNA delivery in vivo. Wiley Interdiscip Rev Nanomed Nanobiotechnol 5:484–501
Brunner S, Sauer T, Carotta S, Cotten M, Saltik M, Wagner E (2000) Cell cycle dependence of gene transfer by lipoplex, polyplex and recombinant adenovirus. Gene Ther 7:401–407
Laskey J, Webb I, Schulman HM, Ponka P (1988) Evidence that transferrin supports cell proliferation by supplying iron for DNA synthesis. Exp Cell Res 176:87–95
Banks WA, Jumbe NL, Farrell CL, Niehoff ML, Heatherington AC (2004) Passage of erythropoietic agents across the blood-brain barrier: a comparison of human and murine erythropoietin and the analog darbepoetin alfa. Eur J Pharmacol 505:93–101
Juul SE, McPherson RJ, Farrell FX, Jolliffe L, Ness DJ, Gleason CA (2004) Erytropoietin concentrations in cerebrospinal fluid of nonhuman primates and fetal sheep following high-dose recombinant erythropoietin. Biol Neonate 85:138–144
Buemi M, Allegra A, Corica F, Floccari F, D’Avella D, Aloisi C, Calapai G, Iacopino G, Frisina N (2000) Intravenous recombinant erythropoietin does not lead to an increase in cerebrospinal fluid erythropoietin concentration. Nephrol Dial Transpl 15:422–423
Girgenti MJ, Hunsberger J, Duman CH, Sathyanesan M, Terwilliger R, Newton SS (2009) Erythropoietin induction by electroconvulsive seizure, gene regulation, and antidepressant-like behavioral effects. Biol Psychiatry 66:267–274
Viviani B, Bartesaghi S, Corsini E, Villa P, Ghezzi P, Garau A, Galli CL, Marinovich M (2005) Erythropoietin protects primary hippocampal neurons increasing the expression of brain-derived neurotrophic factor. J Neurochem 93:412–421
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37:13–25
Bertossi M, Mancini L, Favia A, Nico B, Ribatti D, Virgintino D, Roncali L (1991) Endothelial vesicles as a transport system in the blood-brain barrier. Morphometric study during development. Boll Soc Ital Biol Sper 67:167–173
Raub TJ, Kuentzel SL, Sawada GA (1992) Permeability of bovine brain microvessel endothelial cells in vitro: barrier tightening by a factor released from astroglioma cells. Exp Cell Res 199:330–340
Guo X, Meng Q, Liu Q, Wang C, Sun H, Kaku T, Liu K (2012) Construction, identification and application of HeLa cells stably transfected with human PEPT1 and PEPT2. Peptides 34:395–403
Jain S, Kumar S, Agrawal AK, Thanki K, Banerjee UC (2013) Enhanced transfection efficiency and reduced cytotoxicity of novel lipid-polymer hybrid nanoplexes. Mol Pharm 10:2416–2425
Yang K, Qin W, Tang H, Tan L, Xie Q, Ma M, Zhang Y, Yao S (2011) Polyamidoamine dendrimer-functionalized carbon nanotubes-mediated GFP gene transfection for HeLa cells: effects of different types of carbon nanotubes. J Biomed Mater Res A 99:231–239
Vankayala R, Chiang CS, Chao JI, Yuan CJ, Lin SY, Hwang KC (2014) A general strategy to achieve ultra-high gene transfection efficiency using lipid-nanoparticle composites. Biomaterials 35:8261–8272
Wilhelm I, Fazakas C, Krizbai IA (2011) In vitro models of the blood-brain barrier. Acta Neurobiol Exp (Wars) 71:113–128
Deli MA, Abraham CS, Kataoka Y, Niwa M (2005) Permeability studies on in vitro blood-brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol 25:59–127
Ohshima-Hosoyama S, Simmons HA, Goecks N, Joers V, Swanson CR, Bondarenko V, Velotta R, Brunner K, Wood LD, Hruban RH, Emborg ME (2012) A monoclonal antibody-GDNF fusion protein is not neuroprotective and is associated with proliferative pancreatic lesions in parkinsonian monkeys. PLoS ONE 7:e39036
Juul S (2012) Neuroprotective role of erythropoietin in neonates. J Matern Fetal Neonatal Med 25(Suppl 4):105–107
Juul SE, McPherson RJ, Bauer LA, Ledbetter KJ, Gleason CA, Mayock DE (2008) A phase I/II trial of high-dose erythropoietin in extremely low birth weight infants: pharmacokinetics and safety. Pediatrics 122:383–391
Kellert BA, McPherson RJ, Juul SE (2007) A comparison of high-dose recombinant erythropoietin treatment regimens in brain-injured neonatal rats. Pediatr Res 61:451–455
Fauchere JC, Koller BM, Tschopp A, Dame C, Ruegger C, Bucher HU, Swiss Erythropoietin Neuroprotection Trial Group (2015) Safety of early high-dose recombinant erythropoietin for neuroprotection in very preterm infants. J Pediatr 167(52–7):e1–e3
Aronovich EL, Hackett PB (2015) Lysosomal storage disease: gene therapy on both sides of the blood-brain barrier. Mol Genet Metab 114:83–93
Boustany RM (2013) Lysosomal storage diseases—the horizon expands. Nat Rev Neurol 9:583–598
Marti HH, Gassmann M, Wenger RH, Kvietikova I, Morganti-Kossmann MC, Kossmann T, Trentz O, Bauer C (1997) Detection of erythropoietin in human liquor: intrinsic erythropoietin production in the brain. Kidney Int 51:416–418
Nguyen AQ, Cherry BH, Scott GF, Ryou MG, Mallet RT (2014) Erythropoietin: powerful protection of ischemic and post-ischemic brain. Exp Biol Med 239:1461-1475
Monteleone P, Serritella C, Martiadis V, Maj M (2008) Decreased levels of serum brain-derived neurotrophic factor in both depressed and euthymic patients with unipolar depression and in euthymic patients with bipolar I and II disorders. Bipolar Disord 10:95–100
Karege F, Perret G, Bondolfi G, Schwald M, Bertschy G, Aubry JM (2002) Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res 109:143–148
Pries AR, Kuebler WM (2006) Normal endothelium. Handb Exp Pharmacol 176(Pt 1):1–40
Yuan W, Li G, Gil ES, Lowe TL, Fu BM (2010) Effect of surface charge of immortalized mouse cerebral endothelial cell monolayer on transport of charged solutes. Ann Biomed Eng 38:1463–1472
Mendez-Gomez HR, Galera-Prat A, Meyers C, Chen W, Singh J, Carrion-Vazquez M, Muzyczka N (2015) Transcytosis in the blood-cerebrospinal fluid barrier of the mouse brain with an engineered receptor/ligand system. Mol Ther Methods Clin Dev 2:15037
Louboutin JP, Reyes BA, Agrawal L, Van Bockstaele E, Strayer DS (2007) Strategies for CNS-directed gene delivery: in vivo gene transfer to the brain using SV40-derived vectors. Gene Ther 14:939–949
Acknowledgements
This work was funded by generous Grants from the Research Initiative on Blood–Brain Barrier and Drug Delivery founded by the Lundbeck foundation (Grant No. 2013–14113), Fonden for Lægevidenskabens Fremme and Lily Benthine Lunds fond af 1.6.1978. We want to thank Merete Fredsgaard, Hanne Krone Nielsen, and Emma Huus, Aalborg University, Denmark for excellent technical assistance. We thank Jacek Lichota for excellent qPCR guidance and for supplying BDNF primers. Furthermore, we would like to thank Brita Holst for expert assistance in operating the Cytoflex flow cytometer system.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Burkhart, A., Andresen, T.L., Aigner, A. et al. Transfection of primary brain capillary endothelial cells for protein synthesis and secretion of recombinant erythropoietin: a strategy to enable protein delivery to the brain. Cell. Mol. Life Sci. 74, 2467–2485 (2017). https://doi.org/10.1007/s00018-017-2501-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2501-5