
Abstract
The peroxisome proliferator-activated receptor (PPAR) family, type II nucleus receptors have been successfully tested for their neuroprotective potential in certain central nervous system diseases. The aim of the present study was to determine if modulation by PPAR-γ could attenuate pilocarpine-induced seizures and decrease neuronal excitability. Adult male C57BL/6 mice were divided into two groups: one group received pretreatment with pioglitazone and the other received dimethyl sulfoxide (DMSO) for a period of 2 weeks. Status epilepticus was then induced in both groups by lithium-pilocarpine, after which seizure susceptibility, severity, and mortality were evaluated. Hippocampal histopathology was carried out on all mice at 24 h post-status epilepticus as well as blood–brain barrier (BBB) damage analysis. With the aid of patch clamp technology, the hippocampal neuronal excitability from mice with PPAR-γ 50% expression (PpargC/C) and PPAR-γ 25% expression (PpargC/−), as well as the effect of pioglitazone on the sodium currents in hippocampal neurons, were evaluated. It was found that pioglitazone, a PPAR-γ agonist, could attenuate pilocarpine-induced seizure severity in mice. Pathological examination showed that pioglitazone significantly attenuated pilocarpine-induced status epilepticus-related hippocampal neuronal loss and BBB damage. Further characterization of neuronal excitability revealed higher excitability in the brain slices from mice with PpargC/− expression, compared with the PpargC/C group. It was also found that pioglitazone could decrease sodium currents in hippocampal neurons. In conclusion, PPAR-γ deficiency aggravated neuronal excitability and excitotoxicity. PPAR-γ attenuated pilocarpine-induced seizure severity, neuronal loss, BBB damage, and sodium currents in hippocampal neurons. Modulation of PPAR-γ could be a potential novel treatment for epileptic seizures.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Epilepsy is a chronic brain disorder that affects about 1% of the worldwide population. It can negatively impact a patient’s safety, relationships, work, and quality of life. Seizures are controllable with anti-epileptic drugs (AEDs) in about 70% of cases, while 30% of patients remain refractory [1]. The development of novel anti-epileptic targets is therefore required.
Peroxisome proliferator-activated receptors (PPARs) are type II nucleus receptors, which play essential roles in the regulation of cellular differentiation, development, and metabolism. There are three isotypes of PPARs, including alpha, gamma, and delta (beta) [2]. PPAR-γ plays an important role in fatty acid storage and glucose metabolism, and is expressed in the endothelium, vascular muscle, and cells of the innate immune system [3]. Evidence has shown that the PPAR-γ agonist has neuroprotective potential in certain central nervous system (CNS) diseases, such as stroke and Parkinson’s [4,5,6]. It has been previously shown that the expression of PPAR-γ increases after kainic acid–induced status epilepticus. Pretreatment with rosiglitazone, a PPAR-γ agonist, can increase uncoupling protein 2 expression and reduce protein oxidation, as well as dysfunction of the mitochondrial complex [7]. It has also been found that rosiglitazone can partially protect hippocampal slices in magnesium-free medium from NMDA excitotoxicity via PPAR-γ activation [8]. Another recent study has suggested that PPAR-γ2 is part of the mechanism by which the ketogenic diet reduces flurothyl-induced seizures [9]. However, the role of PPAR-γ in pilocarpine-induced seizures and overall neuronal excitability is not well understood.
The aim of the present study was to determine if pioglitazone, a PPAR-γ agonist, could reduce the severity of seizures and neuronal damage in pilocarpine-induced seizure modeling. It was also investigated whether PPAR-γ gene deficiency aggravated neuronal excitability, which could serve as a potential gateway to the alteration of epileptogenesis.
Materials and Methods
All experiments were conducted in accordance with the specifications of the Experimental Ethics Committee of National Cheng Kung University. All procedures for animal experimentation were reviewed and approved by the Institutional Animal Care and Use Committee at the university. Efforts were made to reduce the total number of animals used.
Animals
C57BL/6 (controls) and PPAR-γ mutant (PpargP465L/+) mice were housed in the National Cheng Kung University animal center in a 12:12 h light/dark cycle; the humidity and temperature were controlled, and food and water were provided ad libitum. The PpargP465L/+ mice were gifts from Dr. Tsai YS at the Institute of Clinical Medicine, National Chen Kung University (Tainan, Taiwan) [10]. PpargP465L/+ and wild-type mice were bred from C57BL/6 heterozygote mice. The inbred foundation colony was maintained by breeding heterozygous P465L mutant mice with inbred 129/SvEv mice. The colony used for the experiments was maintained by breeding heterozygous P465L knock-in mice on the 129/SvEv genetic background with C57BL/6 mice. This produced both wild-type and heterozygous P465L knock-in mice with the same inbred genetic background (129/SvEvC57LB/6F1). Littermate wild-type mice served as the controls.
To take advantage of message destabilization by AU-rich elements (ARE) in the 3′-untranslated region (3′-UTR) of transcripts, the ARE of the c-fos gene was inserted into the 3′-UTR of the mouse Pparg gene [11]. The Pparg-C allele was made via gene targeting in embryonic stem cells, by inserting a 69-bp DNA fragment containing the ARE of the c-fos gene [12] into the 3′-UTR immediately downstream of the stop codon in the mouse Pparg gene [13]. PpargC/C and PpargC/− mice were littermates of the F2 generation from a cross between 129S6 and C57BL/6JF1 heterozygotes. The Pparg-C allele was subsequently placed on a C57BL/6J background by backcrossing for eight generations. PpargC/− and PpargC/− mice were F1 littermates from the mating of PpargC/− mice on a C57BL/6J background with Pparg−/− mice on a 129S6 background (kindly provided by Dr. Ronald Evans at the Salk Institute) [14]. These PPAR-γ mutant mice have been previously demonstrated to have 50% basal PPAR-γ mRNA levels (PpargC/C) [11]. Transgenic mice with the PpargC/− genotype (25% PPAR-γ expression) were utilized for comparisons of neuronal excitability.
Pilocarpine-Induced Seizure Modeling
At 30 min prior to seizure induction by pilocarpine injection (300 mg/kg in normal saline, intraperitoneal injection (IP)), all mice were injected with methylscopolamine (1 mg/kg in normal saline, IP) to reduce the peripheral effects. During the epileptic seizures, all mice demonstrated behavioral characteristics, which were similar to those previously reported [15,16,17]. Seizures were scored using the Racine scale [18]. Diazepam (5 mg/kg, IP) was administered to reduce the seizure activity if status epilepticus lasted for 60 min; however, if the status epilepticus persisted, then the mice were euthanized. The seizure onset latency was determined by measuring the time interval between pilocarpine injection and the onset of overt seizure behavior (stages 3 and 4). The mice were monitored for 24 h following status epilepticus and supportive care was given, including body temperature maintenance, feeding, and adequate hydration. Status epilepticus–related mortality was calculated during the first 24 h after seizure onset.
Drug Administration
At 14 days before the administration of pilocarpine, the C57BL/6 mice received either pioglitazone (10 mg/kg, orally; Takeda Pharmaceutical Co. Ltd.) dissolved in dimethyl sulfoxide (DMSO), or DMSO only (≥ 99.9%, 10 cc/kg, orally). The animals were randomly allocated to each treatment group. Methylscopolamine and pilocarpine were obtained from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Measuring Glucose Concentrations
Plasma glucose concentrations were measured in all C57BL/6 mice at baseline and 14 days later (before status epilepticus was induced). The mice were fasted before their glucose levels were measured. The tip of each animal’s tail was snipped and a drop of blood (~ 5 μl) was collected on a test strip (Optium blood glucose electrode; Abbott Diabetes Care Inc., Alameda, CA), which was then read using a glucose meter (MediSense Optium Xceed; Abbott). The glucose-level reading was available in 20 s and displayed as milligrams per deciliter.
Genotyping: DNA Preparation and Polymerase Chain Reaction (PCR)
The genotypes of the mice were determined by PCR analysis of DNA extracted from the tails of the P7 mouse pups. Approximately 2 mm of tail was digested in 200 μl buffer (1 M Tris, pH 8.0, 0.5 M EDTA, 5 M NaCl, 10% SDS) with 5 μl 1 mg/ml proteinase-K (Invitrogen) for 30 min at 65 °C. To extract the DNA, 100 μl 5 M sodium chloride was added and the mixture was centrifuged at 13,500 rpm for 20 min at 25 °C. The supernatant (200 μl) was saved and mixed with 400 μl 100% ethanol. To precipitate the DNA, the mixture was centrifuged at 13,500 rpm for 5 min at 25 °C. The pellets were left to dry following removal of the liquid (60 °C for 15 min). The DNA was then dissolved in 50 μl sterile H2O (60 °C for 5 min to aid dissolution).
A total of 1 μl of the extracted DNA was added to a reaction mixture made up of 10× Puri Taq PCR buffer, 20 mM MgCl2, Puri Taq DNA polymerase, 2.5 mM dNTP, and forward and reverse primers, which had a total volume of 20 μl. The primers were as follows: forward 5′-CACGAATCACCAGCAACATG-3′, reverse 5′-CCCATTCTTGTCATGATTCCC-3′. The reaction consisted of 35 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C for 30 s, and elongation at 72 °C for 30 s. The PCR products were analyzed by electrophoresis using Tris–borate–EDTA buffer agarose (2%) labeled with ethidium bromide. The bands were visualized using an LAS-100 cooled CCD camera (Fujifilm, Tokyo, Japan).
Histopathology
After 24 h (for Nissl staining and IgG leakage), the mice were anesthetized with CO2, and then perfused with normal saline followed by 4% paraformaldehyde. Their brains were removed and post-fixed in 4% paraformaldehyde for 24 h, dehydrated in a graded alcohol series and embedded in paraffin wax.
Nissl Staining
Coronal sections of the hippocampus (10-μm thick) were used for crystal violet staining as previously described [19]. The crystal violet stained sections were examined for gross indications of hippocampal damage. Cells were counted in the Nissl-stained sections at × 100 and × 400 magnifications using a computerized image analysis system (ImagePro Plus 4.5) on a Nikon E400 microscope. The hippocampal subfields were defined by an imaginary line connecting the tips of the granule cell layer blades, which separated the Cornu Ammonis (CA) 3 and the CA2 from the CA1 as previously described [15].
The severity of neuronal damage in the different hippocampus subfields was scored semi-quantitatively using the following scale: 0 = no damage; 1 = 1–10% neuron loss; 2 = 11–49% neuron loss; and 3 = greater than or equal to 50% neuron loss [15, 20]. An investigator blind to the study design determined these values for the different groups. The mean values for each group were also determined.
IgG Leakage
As a marker of BBB permeability, extravasation of IgG into the parenchyma was detected by immunohistochemistry 24 h after seizure onset. An immunoPure ABC peroxidase mouse IgG staining kit (Pierce, Rockford) was used with extravascular IgG antibodies (1:200, incubated for 2 h), followed by diamino-benzidine staining. The integrated density of individual optical microscope fields (× 100 and × 400) was examined and graded using ImagePro Plus 6.0 software.
Cell Culture, Maintenance, and Patch-Clamp Technology
The embryonic mouse hippocampal Cell Line-14 (CELLutions Biosystems Inc., Canada) was maintained in Dulbecco’s modified Eagle’s medium with 10% heat-inactivated fetal bovine serum and 1% penicillin–streptomycin. The cultures were incubated at 37 °C in a humidified environment of 5% CO2/95% air.
Shortly before the experiments, the cells were dissociated with 1% trypsin/EDTA solution and an aliquot of cell suspension was transferred to a recording chamber affixed to the stage of a CKX-41 inverted microscope (Olympus, Tokyo, Japan). The cells were bathed at room temperature (20–25 °C) in normal Tyrode’s solution (NaCl 136.5, KCl, 5.4, CaCl2 1.8, MgCl2 0.53, glucose 5.5, HEPES–NaOH buffer 5.5 (pH 7.4)). The patch pipette was filled with CsCl 140 mM, MgCl2 1 mM, Na2ATP 3 mM, Na2GTP 0.1 mM, EGTA 0.1 mM, and CsOH buffer 5 mM (pH 7.2) solution to measure the sodium current; the bath solution contained 10 mM tetraethylammonium.
The electrodes, made from Kimax-51 capillaries (Kimble Glass, Vineland, NJ, USA) in a PP-830 puller (Narishige, Tokyo, Japan), had a resistance of 3–5 MΩ when immersed in normal Tyrode’s solution. They were mounted and controlled by a WR-98 hydraulic micromanipulator (Narishige, Tokyo, Japan). The whole-cell or cell-attached configuration of the patch-clamp technique was performed using an Axopatch-200B amplifier (Molecular Devices, Sunnyvale, CA, USA) [21].
The signals were collected and stored online using a TravelMate-6253 laptop (Acer, Taipei, Taiwan) at 10 kHz through a Digidata-1322A interface (Molecular Devices), which was controlled by pCLAMP 9.2 (Molecular Devices). An Adaptec SlimSCSI card (Milpitas, CA, USA) was used via a PCMCIA slot. Current signals were low-pass filtered at 1 or 3 kHz. The digitized data were analyzed using either pCLAMP 9.2 or Origin 8.0 (OriginLab, Northampton, MA, USA). pCLAMP-generated voltage-step profiles with either rectangular or ramp pulses were used to examine the current–voltage relationships for the ion currents.
Preparation of Brain Slices and Patch Clamp Technology
Hippocampal slices were prepared from 8- to 12-week-old transgenic mice that had either 25% or 50% PPAR-γ expression. The mice were anesthetized using urethane and then decapitated. The brains were removed and placed in ice-cold artificial cerebrospinal fluid (ACSF) containing 126 mM NaCl, 2.5 mM KCl, 2.0 mM MgCl2, 2.0 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM d-glucose. Transverse hemi-sectional slices (350-μm thick) of the hippocampus were then taken and the slices were incubated at room temperature for > 1 h before being transferred to the recording chamber with fresh ACSF (containing the same components as previously). All solutions were saturated with 95% O2/5% CO2.
Whole-cell patch clamp recordings were performed in the CA1 pyramidal neurons using a MultiClamp 700B amplifier. Patch electrodes (3–5 MΩ) were pulled from 1.5 mm outer diameter thin-walled glass capillaries in three stages and were filled with intracellular solutions containing 123 mM K-gluconate, 17 mM KCl, 10 mM HEPES, 1.1 mM EGTA, 0.1 mM CaCl2, and 2 mM Na2-ATP (pH 7.25, osmolarity 290–300). Input resistance was measured before and after each recording, and any recording with > 25% change in input resistance was discarded. Signals were acquired via a Digidata 1440A analog-to-digital interface; they were low-pass filtered at 2 kHz and digitized at 10 kHz [22].
Voltage-gated sodium currents from the neurons were recorded using the voltage-clamp mode with a holding potential of − 60 mV; they were elicited by a 200 ms step depolarization ranging from − 60 to + 60 mV in 5-mV increments. The neuronal firing pattern was recorded by membrane potentials under the current-clamp mode and evoked by 600-ms injection currents ranging from − 150 to + 140 pA. The firing number was calculated from the number of action potentials elicited by the + 130 pA injection current. The rheobase was measured as the lowest current amplitude that led to firing of action potentials from resting potential.
Data Analysis
Values are provided as the mean ± standard error of the mean (SEM) with the sample sizes (n) indicating the number of cells from which the data was taken. Significance was set at p < 0.05. Continuous variables were assessed using t tests or one-way analysis of variance using SPSS version 15.0 (SPSS Institute, Chicago, IL), followed by Fisher’s least significant difference tests. When the Shapiro–Wilk normality test showed that the data was not normally distributed, the Kruskal–Wallis H test and then Dunn’s multiple comparisons tests were used. Categorical data was analyzed using χ2 tests, the Yates χ2 test, or Fisher’s exact test. Continuous data are expressed as the mean ± SEM, unless otherwise indicated.
Results
Pioglitazone Did Not Affect Plasma Glucose
To determine whether pioglitazone affected the plasma glucose and neuronal excitability of experimental mice, the plasma glucose levels were measured at baseline (T1) and after 14 days of daily treatment with either DMSO or pioglitazone + DMSO, but prior to pilocarpine administration (T2). Neither pioglitazone (T1, 168 ± 5 mg/dl; T2, 155 ± 6 mg/dl) nor DMSO (T1, 154 ± 6 mg/dl; T2, 156 ± 5 mg/dl) significantly affected the blood glucose (t test, all p > 0.1; n = 10 in each group; Fig. 1).

Treatment with pioglitazone (10 mg/kg) for 2 weeks did not affect the serum glucose in C57BL/6 mice
Pioglitazone Reduced Seizure Severity and Status Epilepticus–Related Mortality
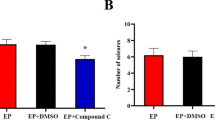
To determine if the activation of PPAR-γ could decrease the severity of seizures, C57BL/6 mice were given daily doses of DMSO (≥ 99.9%, 10 cc/kg, orally) or pioglitazone (10 mg/kg, orally). After 14 days, the mice were injected with pilocarpine (300 mg/kg, IP) to induce seizures and excitotoxicity. In acute seizures, pioglitazone did not significantly alter the latency time to stage 3 seizures (DMSO 17.5 min; pioglitazone 19 min, p > 0.05, n = 12) in each group (Fig. 2a). However, it significantly reduced the sustained time of stage 4–5 seizures (DMSO 140 s; pioglitazone 90 s, p < 0.01 (t = 7.32), n = 12) in each group (Fig. 2b). Pioglitazone also significantly reduced the percentage of status epilepticus (DMSO 10/12 vs. pioglitazone 5/12, p = 0.01, n = 12; Fig. 2c) and mortality (DMSO 7/12 vs. pioglitazone 2/12, p = 0.008, n = 12) in each group (Fig. 2d).

Pilocarpine-induced seizures in the pioglitazone and DMSO groups. (a) There was no significant difference in the latency to stage 3 seizures between the two groups. (b) The pioglitazone group had a significantly lower sustained time (stage 4) compared with the DMSO group. The pioglitazone group had a significantly lower percentage of (c) status epilepticus and (d) status epilepticus–related mortality compared with the DMSO group. The horizontal lines indicate means
Pioglitazone Attenuated Acute Hippocampal Neuronal Loss and BBB Disruption
Pilocarpine-induced hippocampal neuronal loss was significantly reduced by pioglitazone in the CA1 (DMSO vs. pioglitazone—0.54 ± 0.03 vs. 0.26 ± 0.02, p = 0.08), CA3 (DMSO vs. pioglitazone—2.26 ± 0.05 vs. 0.79 ± 0.05, p = 0.03), and hilus (DMSO vs. pioglitazone—0.53 ± 0.03 vs. 0.25 ± 0.02, p = 0.07), compared with the DMSO group (n = 6 in each group; Fig. 3a). The amount of extravascular IgG (indicating the degree of disruption to the BBB) was attenuated by pioglitazone administration. The integrated optical density ratio was 1.0 ± 0.2 vs. 0.6 ± 0.2 (p = 0.02) in the total hippocampus, 1.1 ± 0.2 vs. 1.0 ± 0.2 (p = 0.75) in the CA1, 1.2 ± 0.1 vs. 1.0 ± 0.3 (p = 0.09) in the CA3, and 1.6 ± 0.3 vs. 1.2 ± 0.3 (p = 0.07) in the hilus, in the DMSO versus pioglitazone groups, respectively (n = 6 in each group; Fig. 3b).

Pilocarpine-induced brain damage in the pioglitazone and DMSO groups. (a, Nissl staining) Twenty-four hours after status epilepticus was induced, the severity of neuronal loss, and the number of irregular shaped neurons and indistinct nucleoli were significantly higher in the CA3 of the DMSO group compared with the pioglitazone group (arrow). (b) The BBB damage (IgG extracted) was significantly higher in the total hippocampus (diffuse IgG immunoreactivity) in the DMSO group compared with the pioglitazone group. IOD, integrated optical density; Hippo, hippocampus; BBB, blood–brain barrier. Scale bar in the Hippo: 400 μm; in the CA1, CA3, and the hilus: 100 μm. The horizontal lines indicate means
Loss of PPAR-γ Enhanced Hippocampal Neuronal Excitability
Further analysis of the effect of PPAR-γ on neuronal excitability was done by comparing transgenic mice with PPAR-γ 50% expression (PpargC/C) and those with PPAR-γ 25% expression (PpargC/−). There was a significant increase in the voltage-gated sodium current in brain slices from the PpargC/− mice, compared with those from the PpargC/C mice (Fig. 4a). The lowest current amplitude that lead to the firing of action potentials from resting potential was significantly lower in the PpargC/− mice (43.7 ± 1.69 pA) compared with the PpargC/C mice (77.1 ± 3.61 pA) (Fig. 4b). A 130-pA injection yielded a significantly higher number of action potentials in the PpargC/− mice (9.1 ± 0.4), compared with the PpargC/C group (4.1 ± 0.7) (Fig. 4c, d).

Comparison of hippocampal CA1 neuronal excitability in transgenic mice with 25% PPAR-γ (PpargC/−) and 50% PPAR-γ (PpargC/C) expression. (a) The voltage-gated sodium current was significantly higher in the brain slices of PpargC/− mice compared with those from the PpargC/C mice. (b) It can be seen that the lowest current amplitude that led to the firing of action potentials from a resting potential was significantly smaller in the brain slices from the PpargC/− mice compared with the PpargC/C mice. (c, d) A 130-pA injection yielded a significantly higher number of action potentials in the brain slices from the PpargC/− mice compared with those from the PpargC/C mice. The horizontal lines indicate means
Pioglitazone Inhibited the Sodium Current (I Na) Activity in Hippocampal Neurons
To investigate the possible underlying mechanisms associated with the attenuation of seizure severity by pioglitazone, the patch-clamp technique was used in a whole-cell configuration, to evaluate INa activity in hippocampal neurons. The INa in response to a 50-ms depolarizing pulse from a holding potential of − 80 mV could be readily elicited. The result showed that pioglitazone can suppress the transient INa in a dose-dependent manner at potentials between − 20 and + 20 mV (Fig. 5a). It was also found that pioglitazone only affected the peak amplitude of INa with no change in the overall current–voltage relationship (Fig. 5b). In addition, the time course of activation and inactivation of INa was not altered by pioglitazone.

Inhibitory effect of pioglitazone on INa in hippocampal neurons. The cells were bathed in Ca2+-free Tyrode’s solution and the recording pipette was filled with Cs+-containing solution. (a) Superimposed INa traces obtained in the (a) absence and presence of (b) 3 μM and (c) 10 μM pioglitazone. The upper part in (a) indicates the voltage protocol applied. (b) The average current–voltage relationship of peak INa obtained in the absence (filled squares) and presence (open squares) of 10 μM pioglitazone. Notably, pioglitazone can suppress the peak amplitude of INa with no change in the overall current–voltage relationship
Discussion
In the present study, the potential role of PPAR-γ in pilocarpine-induced seizures and excitotoxicity was examined. It was found that in vivo, the PPAR-γ agonist pioglitazone significantly reduced the severity of pilocarpine-induced acute seizures and ameliorated neuronal and BBB damage. It was also shown in vitro that the hippocampal slices from mice with 25% PPAR-γ expression had higher neuronal excitability compared with those from mice with 50% PPAR-γ expression.
To the best of our knowledge, this is the first study to demonstrate that short-term pretreatment with a PPAR-γ agonist decreased seizure severity and BBB damage in pilocarpine-induced seizure modeling [23, 24]. BBB dysfunction leads to extravasation of serum albumin into the cerebral cortex microenvironment and increases leukocyte movement into the brain parenchyma. This causes local inflammation and activates glutamate receptors on endothelial cells as well as immune responses [25, 26]. Less severe seizures and status epilepticus in the pioglitazone group were also associated with reduced BBB damage. Although the mechanisms behind BBB disruption and epileptogenesis are not entirely clear, it is known that BBB damage does contribute to epileptogenesis [23, 27]. In animal studies, epileptogenesis following BBB injury seems to involve early dysfunction of astrocytes [24]. Pioglitazone has been shown to inhibit astrocyte activation in neuropathic pain and animal Alzheimer models [28, 29]. The question of whether the reduction in BBB damage observed in the current study was caused by inhibition of astrocyte activation, and whether it is capable of ameliorating epileptogenesis, warrants further investigation.
Pilocarpine-induced seizures has long been regarded as the standard animal temporal lobe seizure model. In the present study, daily PPAR-γ treatment with pioglitazone for 14 days before the administration of pilocarpine was linked to a substantial decrease in the excitotoxicity induced by pilocarpine. As evidenced in this study, PPAR-γ deficiency tends to yield higher neuronal excitability, which serves as a potential target for ameliorating epileptogenesis, and also warrants further investigation.
The anti-inflammatory effect of PPAR-γ is well known and plays an important role in many disorders, including cardiovascular and renal disease [30, 31]. Many recent studies have shown that thiazolidinediones (TZDs) are anti-inflammatory and protect against neuronal death in the CNS, such as in the animal model of Alzheimer’s disease [28, 32, 33]. In pentylenetetrazol-induced acute seizures, the kindling model and E1 mice, treatment with rosiglitazone or pioglitazone decreased microglial activation, attenuated the expression of pro-inflammatory COX-2, iNOS, and tumor necrosis factor-α [34, 35], and also reduced seizure severity. Clinical and preclinical studies have provided strong support for the idea that the processes of inflammation within the brain may constitute a common and crucial mechanism in the pathophysiology of seizures and epilepsy. In addition, increasing evidence suggests that inflammation participates in epileptogenesis and ictogenesis [36]. Therefore, modulation of inflammation may be a potential therapy for epilepsy by inhibiting epileptogenesis and ictogenesis. The results of the current study support the notion that PPAR-γ is actively involved in epileptogenesis.
In our study of the hippocampal slices, we did not administer pilocarpine, which has been associated with systemic inflammation [37]. The authors hypothesized that anti-inflammation might be one of several mechanisms involved in the attenuation of seizure severity by PPAR-γ or pioglitazone. It has been suggested that TZDs also have PPAR-γ-independent pathways. There have been studies showing that PPAR-γ is required for modulating lipid metabolism in macrophages, but that its inhibitory effects on cytokine production and inflammation may be receptor independent [38]. Furthermore, TZDs are also involved in the Janus kinase and STAT signaling pathways through PPAR-γ-independent anti-inflammatory actions [39].
The administration of pioglitazone before the induction of status epilepticus was done to evaluate the drug’s pretreatment effect on seizure prophylaxis, instead of its post-status epilepticus modulation effect. In the present study, the drug’s pretreatment effect would at least partially relate to its acute sodium channel-attenuation effect.
Epilepsies are neuronal excitability disorders, which are characterized by spontaneous and recurrent seizures. Action potentials are initiated by rapid Na+ permeability, which is regulated by the Na+ channel, that in turn causes depolarization. We hypothesize that pioglitazone might have PPAR-γ-independent effects not only on inflammation but also on neuronal excitability. Pioglitazone and troglitazone have been found to directly inhibit voltage-dependent L-type Ca2+ channels, which reduce peripheral resistance and exert hypotensive effects in vascular smooth muscle cells [40, 41]. These studies suggested that TZDs might affect ion channels directly, which could be a potential treatment for epilepsy and warrants further investigation.
In conclusion, the results of the current study indicate that PPAR-γ agonists, such as pioglitazone, have potential for the modulation of neuronal excitability and for the treatment of acute seizures. Loss of PPAR-γ exacerbates neuronal excitability and pilocarpine-induced excitotoxicity. Therefore, modulation of PPAR-γ could be a gateway to potential new anti-epileptic therapies.
References
Sillanpaa M, Schmidt D (2006) Natural history of treated childhood-onset epilepsy: prospective, long-term population-based study. Brain 129:617–624
Kersten S, Desvergne B, Wahli W (2000) Roles of PPARs in health and disease. Nature 405:421–424
Tontonoz P, Spiegelman BM (2008) Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem 77:289–312
Schlachetzki JC, Winkler J (2015) The innate immune system in Parkinson's disease: a novel target promoting endogenous neuroregeneration. Neural Regen Res 10:704–706
Kapadia R, Yi JH, Vemuganti R (2008) Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci 13:813–826
Sundararajan S, Jiang Q, Heneka M, Landreth G (2006) PPARgamma as a therapeutic target in central nervous system diseases. Neurochem Int 49:136–144
Chuang YC, Lin TK, Huang HY, Chang WN, Liou CW, Chen SD, Chang AY, Chan SH (2012) Peroxisome proliferator-activated receptors γ/mitochondrial uncoupling protein 2 signaling protects against seizure-induced neuronal cell death in the hippocampus following experimental status epilepticus. J Neuroinflammation 9:184
Wong SB, Cheng SJ, Hung WC, Lee WT, Min MY (2015) Rosiglitazone suppresses in vitro seizures in hippocampal slice by inhibiting presynaptic glutamate release in a model of temporal lobe epilepsy. PLoS One 10:e0144806
Simeone TA, Matthews SA, Samson KK, Simeone KA (2017) Regulation of brain PPARgamma2 contributes to ketogenic diet anti-seizure efficacy. Exp Neurol 287:54–64
Tsai YS, Kim HJ, Takahashi N, Kim HS, Hagaman JR, Kim JK, Maeda N (2004) Hypertension and abnormal fat distribution but not insulin resistance in mice with P465L PPARgamma. J Clin Invest 114:240–249
Tsai YS, Tsai PJ, Jiang MJ, Chou TY, Pendse A, Kim HS, Maeda N (2009) Decreased PPAR gamma expression compromises perigonadal-specific fat deposition and insulin sensitivity. Mol Endocrinol 23:1787–1798
Chen CY, Chen TM, Shyu AB (1994) Interplay of two functionally and structurally distinct domains of the c-fos AU-rich element specifies its mRNA-destabilizing function. Mol Cell Biol 14:416–426
Tsai YS, Pendse A, Moy SS, Mohri I, Perez A, Crawley JN, Suzuki K, Maeda N (2006) A de novo deafwaddler mutation of Pmca2 arising in ES cells and hitchhiking with a targeted modification of the Pparg gene. Mamm Genome 17:716–722
Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM (1999) PPAR-gamma is required for placental, cardiac, and adipose tissue development. Mol Cell 4:585–595
Huang CW, Cheng JT, Tsai JJ, Wu SN, Huang CC (2009) Diabetic hyperglycemia aggravates seizures and status epilepticus-induced hippocampal damage. Neurotox Res 15:71–81
Huang CW, Wu SN, Cheng JT, Tsai JJ, Huang CC (2010) Diazoxide reduces status epilepticus neuron damage in diabetes. Neurotox Res 17:305–316
Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, Coulter DA (2007) Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci 27:14012–14022
Ferraro TN, Golden GT, Smith GG, St Jean P, Schork NJ, Mulholland N, Ballas C, Schill J et al (1999) Mapping loci for pentylenetetrazol-induced seizure susceptibility in mice. J Neurosci 19:6733–6739
Chang YC, Huang AM, Kuo YM, Wang ST, Chang YY, Huang CC (2003) Febrile seizures impair memory and cAMP response-element binding protein activation. Ann Neurol 54:706–718
Pitkanen A, Schwartzkroin PA, Moshe SL (2006) Models of seizures and epilepsy. Elsevier Academic Press, Burlington
Huang CW, Lin KM, Hung TY, Chuang YC, Wu SN (2018) Multiple actions of rotenone, an inhibitor of mitochondrial respiratory chain, on ionic currents and miniature end-plate potential in mouse hippocampal (mHippoE-14) neurons. Cell Physiol Biochem 47:330–343
Lin CH, Hsu SP, Cheng TC, Huang CW, Chiang YC, Hsiao IH, Lee MH, Shen ML et al (2017) Effects of anti-epileptic drugs on spreading depolarization-induced epileptiform activity in mouse hippocampal slices. Sci Rep 7:11884
Oby E, Janigro D (2006) The blood–brain barrier and epilepsy. Epilepsia 47:1761–1774
Friedman A, Heinemann U (2012) Role of blood–brain barrier dysfunction in epileptogenesis. In: Noebels JL et al. (eds) Jasper’s basic mechanisms of the epilepsies. National Center for Biotechnology Information (US)
Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, Friedman A (2004) Lasting blood–brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci 24:7829–7836
Fabene PF, Navarro Mora G, Martinello M, Rossi B, Merigo F, Ottoboni L, Bach S, Angiari S et al (2008) A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med 14:1377–1383
Vezzani A, Granata T (2005) Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 46:1724–1743
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O'Banion K, Klockgether T et al (2005) Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain 128:1442–1453
Griggs RB, Donahue RR, Morgenweck J, Grace PM, Sutton A, Watkins LR, Taylor BK (2015) Pioglitazone rapidly reduces neuropathic pain through astrocyte and nongenomic PPARgamma mechanisms. Pain 156:469–482
Hasegawa H, Takano H, Komuro I (2010) Therapeutic implications of PPARgamma in cardiovascular diseases. PPAR Res. https://doi.org/10.1155/2010/876049
Kiss E, Popovic ZV, Bedke J, Adams J, Bonrouhi M, Babelova A, Schmidt C, Edenhofer F et al (2010) Peroxisome proliferator-activated receptor (PPAR)gamma can inhibit chronic renal allograft damage. Am J Pathol 176:2150–2162
Landreth G, Jiang Q, Mandrekar S, Heneka M (2008) PPARgamma agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics 5:481–489
Landreth GE, Heneka MT (2001) Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer’s disease. Neurobiol Aging 22:937–944
Mohazab RA, Javadi-Paydar M, Delfan B, Dehpour AR (2012) Possible involvement of PPAR-gamma receptor and nitric oxide pathway in the anticonvulsant effect of acute pioglitazone on pentylenetetrazole-induced seizures in mice. Epilepsy Res 101:28–35
Okada K, Yamashita U, Tsuji S (2006) Ameliorative effect of pioglitazone on seizure responses in genetically epilepsy-susceptible EL mice. Brain Res 1102:175–178
Vezzani A, French J, Bartfai T, Baram TZ (2011) The role of inflammation in epilepsy. Nat Rev Neurol 7:31–40
Marchi N, Fan Q, Ghosh C, Fazio V, Bertolini F, Betto G, Batra A, Carlton E et al (2009) Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol Dis 33:171–181
Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM (2001) PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med 7:48–52
Park EJ, Park SY, Joe E, Jou I (2003) 15d-PGJ2 and rosiglitazone suppress Janus kinase-STAT inflammatory signaling through induction of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 in glia. J Biol Chem 278:14747–14752
Asano M, Nakajima T, Iwasawa K, Morita T, Nakamura F, Imuta H, Chisaki K, Yamada N et al (1999) Troglitazone and pioglitazone attenuate agonist-dependent Ca2+ mobilization and cell proliferation in vascular smooth muscle cells. Br J Pharmacol 128:673–683
Zhang F, Sowers JR, Ram JL, Standley PR, Peuler JD (1994) Effects of pioglitazone on calcium channels in vascular smooth muscle. Hypertension 24:170–175
Acknowledgments
This work was supported in part by grants from the Taiwan National Science Council (NSC-102-2314-B-006-051-MY3), Ministry of Science and Technology, ROC, (105-2314-B-006-013, 106-2314-B-006-034, 106-2320-B-006-055, 107-2314-B-006-018, 107-2320-B-006-019), National Cheng Kung University Hospital (20180254), and Chi-Mei Medical Center (CMNCKU10303). Part of this abstract was presented at the Taiwan Neurological Society Annual Meeting, 2013.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hung, TY., Chu, FL., Wu, D.C. et al. The Protective Role of Peroxisome Proliferator-Activated Receptor-Gamma in Seizure and Neuronal Excitotoxicity. Mol Neurobiol 56, 5497–5506 (2019). https://doi.org/10.1007/s12035-018-1457-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1457-2