
Abstract
The fragile X syndrome (FXS) arises from loss of expression or function of the FMR1 gene and is one of the most common monogenic forms of intellectual disability and autism. During the past two decades of FXS research, the fragile X mental retardation protein (FMRP) has been primarily characterized as a cytoplasmic RNA binding protein that facilitates transport of select RNA substrates through neural projections and regulation of translation within synaptic compartments, with the protein products of such mRNAs then modulating cognitive functions. However, the presence of a small fraction of FMRP in the nucleus has long been recognized. Accordingly, recent studies have uncovered several mechanisms or pathways by which FMRP influences nuclear gene expression and genome function. Some of these pathways appear to be independent of the classical role for FMRP as a regulator of translation and point to novel functions, including the possibility that FMRP directly participates in the DNA damage response and in the maintenance of genome stability. In this review, we highlight these advances and discuss how these new findings could contribute to our understanding of FMRP in brain development and function, the neural pathology of fragile X syndrome, and perhaps impact of future therapeutic considerations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
As a significant monogenic basis for intellectual disability, studies of fragile X syndrome (FXS) and the FMR1 gene have been in the vanguard of efforts to devise therapeutic approaches towards neurodevelopmental disorders [1, 2]. Notable advancements in our understanding of the molecular basis for FXS pathology include the discovery of RNA-binding capacity by fragile X mental retardation protein (FMRP) and its association with ribosomes, identification of prospective mRNA substrates, and the placement of FMRP within a synaptic signaling pathway amenable to pharmacological manipulation [3,4,5,6,7,8,9,10]. Together, these findings have served as the foundation for a well-validated model where FMRP controls translation of select mRNAs in response to signaling through metabotropic glutamate receptors (mGluRs) and that loss of FMRP results in dysregulated synaptic protein synthesis, leading to aberrant synaptic connectivity that is manifested in the behavior phenotypes of FXS [11, 12].
Animal models for FXS display behavior phenotypes consistent with the above mechanisms for FMRP function at synapses, and the findings that at least some of these behavior phenotypes could be rescued by antagonists of mGluRs provoked considerable interest and excitement among basic and clinical scientists, as well as families affected by FXS [13, 14]. Unfortunately, a carefully controlled clinical trial of mGluR5 antagonists in FXS patients did not elicit the anticipated therapeutic benefits [15]. Several potential confounding factors that could contribute to this outcome have been suggested and include the age of patients enrolled, the power of the placebo effect, the effects of other medications, unknown differences between animal models and humans in regard to pharmacodynamics and pharmacokinetics of a drug, a relative lack of biomarkers to monitor in patients, the challenge of extrapolating animal behavior studies to those of humans, and perhaps a limited understanding of how FMRP works in brain development and in the circuits that control behaviors examined in the patient cohort [2, 15, 16]. These possibilities have yet to be clearly resolved. While other compounds that target synaptic dysfunction associated with Fmr1 mutation have shown promise in animal models for FXS (see [2] for a compilation), they await testing in FXS patients at the same level of experimental power used for the trials with mGluR5 antagonists.
However, another plausible explanation for the unexpected results from the mGluR5 clinical trials is that FMRP has other critical functions in neural tissue that are not amenable to rescue by targeting synaptic protein synthesis or other elements of synaptic function. Although the bulk of FMRP resides in the cytoplasm, early characterizations of FMRP revealed the presence of nuclear localization and nuclear export sequences, and that the latter, when removed by alternative splicing or mutation, led to the accumulation of FMRP in the nucleus [17,18,19]. Immunogold labeling of brain sections showed that FMRP is endogenously present within the nucleoplasm of neurons [20] and thus situated to exert regulatory effects throughout a cell.
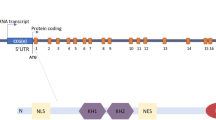
The regulatory potential of FMRP is revealed upon examination of its highly modular architecture (Fig. 1), which includes three K homology (KH) domains and one glycine-arginine (RGG) box that have demonstrated RNA and/or protein binding capacity. FMRP can self-interact via two distinct regions, most often as a dimer, and this ability increases its valency within a protein complex [21, 22]. The significance of having two dimerization regions is unknown, but it may confer distinct regulatory functions [22]. In addition to binding RNA, many interactions of FMRP with other proteins have been reported. A sizeable fraction of the known FMRP-interacting proteins function in some context of cytoplasmic mRNA transport or regulation of mRNA stability and/or translation [23], but others that include the nuclear proteins ADAR, RBP14, and NUFIP [24,25,26,27,28], and the potassium channels Slack and BK [29, 30], suggest novel FMRP functions. Of particular interest is a pair of Agenet domains comprising the first ~ 110 amino acids of FMRP (Fig. 1). Agenet domains are related to Tudor domains and mediate interactions with methylated lysine and arginine residues of proteins, which for FMRP include methylated histone H3 [31] and possibly methylated arginine residues of the FUS RGG region, as a Y96L mutation in the methyl binding pocket of the second FMRP Agenet domain disrupts interaction with FUS [32, 33]. FMRP thus has unusual capacity for pleiotropic function in neurons through its ability to serve as a scaffold with the potential to connect combinations of protein, RNA, and chromatin. As will be illustrated in the following paragraphs, such pleiotropy provides a basis for FMRP not only to modulate neuronal responses and outputs at synapses, the cell body, and within the nucleoplasm, but also to participate in cellular housekeeping functions that are essential for neural development and function.

Multi-domain structure of the FMR protein. a Linear organization of FMRP shows Agenet (AG1 and AG2) and KH (KH0, KH1, and KH2) domains, as well as an unstructured region that contains a nuclear export sequence (NES), a RGG box and the C terminal domain. FMRP can exist as a monomer (b) and as two alternative dimerization forms (c,d). e As a highly modular protein, FMRP encompasses different RNA and protein interaction sites, mediating the participation of FMRP in multiple unrelated pathways. Domains are not drawn to scale. Numbers in a represent amino acids
FMRP Modulates RNA Function as a Nuclear Protein
Processing within the nucleus (capping, splicing, 3′ end formation) and nucleocytoplasmic export are essential steps to the function of many RNA species. More recently, there has been a growing appreciation for the roles of nuclear RNA in mediating elements of chromatin function, including gene activation, silencing, and DNA repair. While a connection between the RNA binding capacity of FMRP and regulation of chromatin function has yet to be established, multiple studies have uncovered roles for FMRP in processing and editing of pre-mRNA molecules (Fig. 2).

FMRP interacts with nuclear elements involved in mRNA maturation and export pathways. a FMRP interaction with RBM14 supports growing evidence suggesting that FMRP contributes to the regulation of alternative splicing on select mRNAs. b The involvement of FMRP in RNA editing pathways has been well established and is supported by the finding that FMRP interacts with ADAR, a central RNA editing component. c Among other evidence, interactions between FMRP and the nucleocytoplasmic export factor NXF2 suggest that FMRP may be required for nucleocytoplasmic transport of specific RNAs
Alternative Splicing
About 95% of multi-exon transcripts from humans show evidence for alternative splicing, and the frequency of this transcriptome and proteome-expanding event is elevated in neural tissue, where it contributes to all elements of neural function from development to synaptic plasticity [34]. As an RNA binding protein, alternative splicing of select pre-mRNA is a potential function for nuclear FMRP (Fig. 2), and an initial insight into this possibility came with the observation that FMRP autoregulates alternative splicing of Fmr1 pre-mRNA [35]. Recent developments in RNA sequencing, which allow for direct detection of rare alternative splicing events, and a screen using cultured cells and RNAi knockdown suggest that Drosophila FMRP may impact alternative splicing of select pre-mRNA substrates [36, 37]. Recently, mouse FMRP was found to co-precipitate in an RNA-dependent manner with RNA-binding protein 14 (RBM14), a known factor in alternative splicing [27]. Subsequent analyses showed that the skipping/inclusion ratio of select exons from Protrudin and Tau transcripts was altered in both Fmr1 KO hippocampal tissue and in cultured cells where Fmr1 was knocked down. Alternative splicing events are often regulated in response to physiological and developmental signals [34, 38] and thus different populations of cells from neuronal tissues may have distinct profiles of alternatively spliced transcript variants. Detailed RNA sequencing studies with a variety of differentiated neuronal tissues or even single cells [39] will be required to more completely assess the impact of Fmr1 mutation on patterns of alternative splicing.
Nucleocytoplasmic Transport
Another candidate function for nuclear FMRP is nucleocytoplasmic transport of RNA (Fig. 2). A connection between FMRP and nucleocytoplasmic transport has been explored with the finding that FMRP interacts with NXF2, an mRNA nuclear export factor [40]. Further studies by the same research group then showed that the FMRP/NXF2 interaction destabilized the mRNA of NXF1, an essential ubiquitously expressed mRNA export factor [41]. Silencing RNAs targeted to NXF1 enhanced the nuclear accumulation of modified FMRP that had its endogenous nuclear export sequence removed, suggesting that FMRP can also exit the nucleus as part of an NXF1 mRNA export complex [42]. This same study also showed FMRP association with nascent transcripts emanating from lampbrush chromosomes of Xenopus oocytes, in addition to a prominent FMRP staining of the chromosome axis. Whether this association of FMRP with nuclear transcripts is limited to a subset of RNA substrates has yet to be resolved, but the observations from these papers suggest that loss of FMRP could alter the nuclear export of at least some mRNA substrates, as well as the overall dynamics of nucleocytoplasmic RNA trafficking.
RNA Editing
Post-transcriptional editing of mRNAs via Adenosine Deaminase Acting on RNA (ADAR) is critical for neural function. ADAR proteins have been shown to act on mRNAs coding for neurotransmitter receptor and channel subunits, along with other proteins having synaptic function [43], and thus mutations in ADAR proteins lead to neurological defects or lethality. Studies with Drosophila [24], zebrafish [25], and most recently with mice [26] show that loss of FMRP results in aberrant editing of at least some mRNA transcripts coding for synaptic receptor subunits, ion channels, and other signaling proteins that directly participate in synaptic function. Moreover, some of the altered pattern of edits in neural tissue mutant for Fmr1 elicits the synthesis of protein isoforms with previously characterized alterations in their activities, with the resulting outputs being consistent with synaptic phenotypes associated with FXS [26]. Both enhancement and inhibition of editing are observed in the Drosophila Fmr1 mutant, in what seems to be a transcript/site-specific manner [24], but only enhancement of editing is seen in the mRNAs tested from zebrafish and mouse Fmr1 mutant tissues. While certain isoforms of ADAR proteins from vertebrates are cytoplasmic, the Drosophila ADAR is nuclear, and mouse FMRP co-localizes with ADAR2 in the nucleus, where it is reported to inhibit ADAR2 activity [26]. Similar to the situation with alternative splicing, a thorough account of the impact of Fmr1 mutation on RNA editing in neurons has yet to be undertaken. Although a contribution to FXS phenotypes by alterations in alternative splicing, nucleocytoplasmic trafficking, or editing of FMRP-associated RNAs has not yet been directly demonstrated, the known contributions of some of the proteins encoded by these RNAs to synaptic plasticity makes such a possibility seem quite plausible (Fig. 2).
FMRP Regulates mRNAs That Code for Epigenetic Factors
Chromatin Structure and Gene Transcription Are Affected by Fmr1 Mutation
Several biochemical screens for RNA substrates of FMRP have been conducted [7, 8, 44, 45]. Although initial interest in candidate substrates focused on mRNAs that code for synaptic proteins, about 13% of FMRP-associated transcripts code for transcription factors or proteins that modulate properties of chromatin [44, 46]. Significantly, many of these nuclear factors have been connected with abnormalities in development of the nervous system and in autism spectrum disorder (ASD). As FMRP is associated with translation regulation, an imbalance in the expression of factors that globally alter transcriptional gene expression could present an entirely new class of cellular dysfunction that contributes to the pathology of FXS. Korb et al. [46] began their study with an examination of protein levels of several chromatin modification and transcription factors and found an increase of these proteins in cultured neurons from FMRP KO mice. Histone modifications associated with open chromatin and transcription were notably upregulated in both cultured KO neurons and cerebellar neurons from Fmr1 KO mice as judged by ChIP sequencing, and a subsequent RNA-seq analysis of ~ 16,750 genes from WT and FMRP KO neurons showed that over 1500 were upregulated, with many being connected to neural function and autism spectrum disorder. Despite FMRP KO neurons displaying a lack of change in repressive chromatin marks, nearly 1300 genes were downregulated in the FMRP KO neurons. Transcription factors are present in this group of downregulated genes, and the decrease in their transcription is proposed to result from downstream responses to global changes in chromatin. The scope of this gene misregulation in response to loss of FMRP has significant implications for understanding the cellular pathology of FXS and, as observed by Korb et al., reveals an unexpected synergy between transcriptional and translational regulation that is modulated by FMRP [46]. Moreover, it points to transcription factors and chromatin regulators as possible targets for FXS therapy.
Epigenetic Control of FXS Phenotypes
By analyzing previously reported expression profiles of genes regulated by the transcription and chromatin regulatory factors that were identified in the FMRP RNA substrate screen [44], and comparing them to the genes misregulated in the FMRP KO background, Korb et al. were able to compile numbers, percentage, and significance of overlapping gene groups [46]. The most significant overlap came from comparison of the gene expression profile of cells exposed to the BET (Bromo and Extra-Terminal domain) protein inhibitor JQ1 versus that of FMRP KO neurons. The BET protein family includes Brd4, which binds acetylated histones to promote transcription, including genes whose protein products function at synapses. Interestingly, the transcript for Brd4 was identified as a FMRP substrate in the Darnell et al. screen [44].
In addition to having an inhibitor of Brd4 in hand, Korb et al. note several other reasons why Brd4 is a compelling target for FXS therapy [46], perhaps the most significant being that it interacts with other chromatin regulators found to be misregulated as a result of FMRP mutation and may then serve as a hub that connects multiple FMRP targets. Moreover, in the light of multiple classes of histone modifications being misregulated, inhibition of a factor that generally functions downstream of histone modifications might give the best chance for effective modulation of the gene expression defects associated with FMRP mutation. With these considerations, Korb et al. then moved to test JQ1 in FMRP KO cultured neurons and Fmr1 KO mice [46]. Assays that monitored expression levels of chromatin regulators, global transcription regulation, and neural anatomy in regard to dendritic spine density all showed significant restoration of FMRP KO-associated anomalies towards the wild type. That the cumulative effect of this epigenetic and transcriptional misregulation associated with Fmr1 mutation contributes to the phenotypes of FXS is best evidenced by the rescue of mouse Fmr1 KO behavior deficits by JQ1.
FMRP Contributes to Genome Stability
Maintenance of genome stability through accurate repair of DNA damage from endogenous and exogenous sources, and suppressing the movement of mobile genetic elements, is crucial for cell function and viability. This is especially important for the nervous system, as neurons are relatively long-lived cells, and thus the accumulation of mutations could enhance the possibility of future dysfunction not only just for individual neurons, but also for the circuits they are connected to. Moreover, the need for neurogenesis arising from division of neural stem cells and neural progenitor cells during development, and perhaps adulthood, makes genome stability a paramount concern for these cell types, as all subsequent daughters will inherit the genome of the parental cell. The importance of genome stability to neural function is illustrated by disorders in the nervous system believed to arise from somatic mosaicism in the brain [47]. It is thus intriguing that several recent studies, highlighted below, indicate that FMRP has a role in repressing expression of transposable elements and that its nuclear localization is enhanced upon inducing certain types of DNA damage [31, 48,49,50,51].
FMRP, the PIWI Pathway, and Regulation of Transposition
The Argonaute family of proteins binds small RNAs to regulate gene expression at both transcriptional and post-transcriptional levels [52]. FMRP has previously been associated with the miRNA pathway, where it has interactions with Argonaute proteins (AGO) and various miRNAs [53,54,55]. PIWI proteins receive their name from the Drosophila gene P-element induced Wimpy testis (PIWI) and represent a subclass of the Argonaute family of proteins that mainly bind RNAs derived from transposable elements (TE) as a means of repressing their expression and thus suppressing the potential for deleterious transposition events [56, 57]. Because of their role repressing transposition in the germline, a phenotype common to mutations in genes of the PIWI pathway is sterility, and certain alleles of the Drosophila fragile X gene strongly impair fertility in both sexes as well. In addition to these phenotypic similarities, several studies show that dFMRP interacts in vivo and in vitro with the PIWI proteins Aubergine and Piwi [48,49,50]. The N-terminal ~ 180 amino acids of dFMRP, encompassing the Agenet domains, and much of KH0, is essential for the in vitro interaction with Piwi [50], and this is consistent with prior findings that Agenet/Tudor domains in other proteins mediate interactions with PIWI members [56]. Mutation of dFmr1 reduces levels of several species of piRNAs and increases transcripts from both germline and somatic transposons [49, 50]. In conjunction with the aforementioned interactions with AGO proteins, a picture is emerging in which dFMRP plays a myriad of roles within the small RNA pathways. Depending upon its subcellular localization, dFMRP can interact with Aub in the cytoplasm, where it may facilitate piRNA processing and degradation of TE RNA, and mediate TE transcriptional silencing in the nucleus, through interactions with Piwi (Fig. 3).

FMRP interacts with components of the PIWI-interacting RNA pathway (piRNA). The PIWI clade of Argonaute proteins interacts with piRNA to silence expression of transposable elements (TE) at the post-transcriptional level. In Drosophila, interactions between FMRP, Piwi, and Aubergine (see text for references) suggest a model that is consistent with FMRP interacting with Aubergine in the cytoplasm in a pathway that leads to RNA degradation, whereas in the nucleus Piwi interacts with FMRP to target sites of TE transcription to produce transcriptional silencing by formation of repressive chromatin
While many studies of the PIWI pathway focus on its role during germline development, piRNAs also function in somatic tissues, including the nervous system, and have been shown to modulate synaptic plasticity in Aplysia [58]. In Drosophila, an allele of aub that results in ectopic somatic expression during larval development is able to modify the overgrowth of larval neuromuscular junction synapses that are observed in a dFmr1 mutant background [49], indicating the dFMRP/Aub interaction functions in neuronal cells as well. Potentially relevant for understanding consequences of FMR1 mutation is the observation that mutations of aub result in increased expression of several transposons in the Drosophila brain [59]. A tentative possibility in light of these findings is that mutations in FMR1 may lead to activation of transposition in neurons, resulting in deleterious effects that may contribute to phenotypes of Fragile X patients.
FMRP and the DNA Damage Response
An initial observation into a connection between FMRP and genotoxic stress was first reported during efforts to explore a role for FMRP in cell cycle regulation [60]. As part of these studies, Drosophila Fmr1 mutants were exposed to genotoxic agents that included a chemical mutagen (methyl methanesulfonate), an inducer of replication stress (hydroxyurea), and gamma-irradiation, with dFmr1 mutants consistently being unable to recover from these stresses with the same efficiency displayed by wild-type controls. The authors propose that upregulation of CyclinB in dFmr1 mutants caused cells to override the G2/M checkpoint, triggering cells with DNA damage to proceed to mitosis and contributing to a diminished ability to recover from genotoxic stress. They then showed that heterozygosity for cycB could partially rescue G2/M checkpoint defects and the survival of dFmr1 mutants exposed to genotoxic agents. The partial rescue by cycB heterozygosity hinted at the possibility that FMRP had additional roles in mitigating the effects of genotoxic stress.
A significant insight into such a role came with a report from Alpatov et al. that strongly suggest that FMRP directly interacts with chromatin to modulate the DNA damage response (DDR) via its second Agenet domain [31]. While further examining a role for FMRP with the DDR, these investigators found that DNA damage induced by replication stress significantly increased levels of the phosphorylated form of the histone H2A variant H2A.X (γH2A.X) in an FMRP-dependent manner and also enhanced the ability to detect nuclear FMRP by immunofluorescence microscopy. As γH2A.X is widely recognized as a mark for double-strand breaks in DNA, the reported co-localization of FMRP with γH2A.X is significant, since it indicates that at least some of the nuclear FMRP associates with sites of DNA damage. The induction of γH2A.X during the DDR was found to be reduced in Fmr1 KO cells. Addition of wild-type Fmr1 to Fmr1 KO cells could restore the deficiency in γH2A.X induction, but Fmr1 constructs with Agenet domain mutations that impair binding to methylated histones were less efficient, suggesting that the interaction of FMRP with histones is critical and that the presence of FMRP at chromatin modulates the phosphorylation status of H2A.X.
Meiotic recombination during gamete development is an example where double-strand break (DSB) formation and repair is controlled by cellular endonucleases and components of the repair machinery, and anti-FMRP antibodies were found to decorate chromosome arms in meiotic spermatocytes. SPO11 is the conserved endonuclease that initiates DSB formation during meiotic recombination, and comparison of wild-type and Fmr1 KO spermatocytes revealed no difference in binding of anti-SPO11 antibodies to chromosome spreads, indicating that FMRP does not facilitate initiation of the double-strand break. However, extended retention of repair-associated proteins such as BRCA1 and ATR was seen with Fmr1 KO spermatocytes, suggesting that FMRP can be involved in the repair of physiologically programmed DNA breakage. Perhaps most significantly, an R138Q substitution in FMRP that was identified in an individual diagnosed with symptoms of FXS [61] also impairs binding of FMRP to methyl histones and is unable to fully complement FMRP KO mouse embryonic fibroblasts in assays that monitored DDR and γH2A.X induction. However, this mutant protein is able to rescue the excessive internalization of an AMPA receptor subunit in FMRP KO neurons, a phenotype that is associated with the exaggerated long-term depression (LTD) phenotype of FMRP KO brain slices, implying that the R138Q mutation does not impair the ability of FMRP to adequately regulate protein synthesis needed for normal LTD [31]. Although there may be pleiotropic effects associated with the FMRP R138Q substitution, as this mutant protein also fails to interact with the β4 regulatory subunits of BK channels [62], there is also a very real possibility that disruption of the DDR, and perhaps other critical outputs from the nucleus, contributes to the symptoms of FXS in the R138Q individual.
The precise role FMRP plays in the DDR pathway remains unknown. A possible model of FMRP action in the DDR that is consistent with the results from Alpatov et al. [31] is represented in Fig. 4. FMRP may bind chromatin at the site of the DNA break, facilitating recruitment of ATR and the subsequent phosphorylation of H2A.X by ATR. Interactions of FMRP with chromatin are likely mediated by methylated H3K79, a histone modification that is widespread through the genome. DNA breaks would lead to the opening of chromatin, making methylated H3K79 accessible to FMRP, which directly or indirectly, recruits ATR. A similar mechanism has been proposed to explain accessibility of 53BP1 to DNA breaks [63]. Whether the role of FMRP in DDR is RNA dependent has yet to be determined.

A model illustrating a possible role of FMRP in the DNA damage response. a, b A DNA double-strand break results in a more open chromatin structure around the DNA break, and preexisting methylated H3K79 becomes more accessible. c FMRP associates with the chromatin neighboring the DNA break through interactions between H3K79me and the Agenet domain 2. The specific mechanism that helps targeting FMRP to the break site is unknown, but one possibility is that recruiting is mediated by RNA molecules with sequence complementarity to the DNA in the region. d Finally, FMRP mediates the incorporation of active ATR (or ATM), which spreads phosphorylation of H2AX through nucleosomes in the region
Future Directions for Nuclear FMRP
FMRP is very well situated to modulate nuclear functions through the presence of methyl histone binding Agenet domains, a capacity to engage in protein-protein interactions, and an array of RNA-binding domains that bind select RNA substrates possibly destined for editing, alternative splicing, and/or nucleocytoplasmic trafficking. Cataloguing defects in splice and editing patterns and discerning the degree that loss of FMRP has on facilitating production of a splice variant or edit will be needed. To focus on identifying alterations in splice patterns/edits that elicit experimentally verified modifications to the properties of ion channels and synaptic receptors could uncover additional targets that might be amenable to pharmacological modification. Information on how FMRP exerts effects on splicing and editing during development and in response to stimulation and the signaling pathways that direct such events has yet to be elucidated.
The findings from Alpatov et al. [31] raise a plethora of questions that are potentially relevant to the pathology of FXS. What is the connection between nuclear FMRP and DNA damage? Could FMRP have a direct role in repair of DNA damage, and if so, how? Recent studies have demonstrated a role for various classes of non-coding RNAs and RNA binding proteins in the DNA damage response [64,65,66,67,68], and the capacity for FMRP to interact with histone marks associated with DNA damage, in addition to RNA binding, would make it rather well positioned to be part of such a process. Is the association of FMRP with chromatin limited to the presence of certain types of DNA damage? Alpatov et al. report nuclear accumulation of FMRP upon exposure to agents that induce replication stress [31], and similar observations were made with a Drosophila fragile X model [51]. Loss of FMRP impairs adult hippocampal neurogenesis [69], and the possibility that this deficit could render Fmr1 mutant neural stem cells and/or neural progenitor cells sensitive to replication stress is intriguing. Seemingly unexplored at this time is whether FMRP is connected with repair of other types of endogenous DNA damage not associated with dividing cells, most notably that induced by reactive oxygen species (ROS) arising from oxidative stress. Mice with a disruption of Fmr1 show an age-dependent increase in certain markers for oxidative stress [70], and metabolic imbalances that could induce oxidative stress have been observed in both mouse and insect models for FXS [71, 72]. Animals that are mutant for Fmr1 may suffer a double jeopardy of having enhanced levels of endogenous DNA damage in all neural cell types and a deficit in repair capacity for at least those that replicate DNA and divide. A deficiency in repair of DNA damage, along with the potential for enhanced mobilization of transposons, could render Fmr1 mutant animals and humans more susceptible to mosaicism. There is considerable heterogeneity in the degree of intellectual disability among FXS patients [73], and perhaps the above events could contribute to this phenomenon. The possibility that such events could also impact the response of FXS individuals to pharmacological treatments may warrant consideration.
Is FMRP binding to chromatin limited to the DDR and specialized developmental processes such as meiosis, or is it more prevalent throughout cellular physiology? Of potential interest are the observations that DNA breaks can be “planned” as an integral component of forming chromatin loops that facilitate enhancer-promoter communication required for transcription activation [74, 75]. It is then interesting to note that double-strand breaks are induced by neural activity [76] and are associated with transcription of neural immediate early genes that contribute to synaptic plasticity [77]. Whether FMRP contributes to the repair of physiologically programmed damage that arises from transcription activation pathways remains to be determined. Finally, the modifications to histones control essentially all events associated with chromatin (packaging/condensation, transcription regulation, replication, recombination and repair, and establishment/maintenance of genome architecture). There is a growing appreciation for the roles of non-coding RNAs, particularly long non-coding RNAs (lncRNA), in controlling genome function [78, 79], and a common mechanism of their function is to interact with a chromatin-bound protein and serve as a docking site for binding of other regulatory proteins (e.g., histone-modifying enzymes). The analyses of screens for FMRP RNA ligands have focused on mRNA substrates [7, 44, 45]. Mining of these data sets for potential lncRNAs may prove insightful.
Summary
The potential for misregulated gene expression arising from disruptions in RNA editing, alternative splicing, and nucleocytoplasmic trafficking of select RNAs, coupled with possible defects in maintaining genome stability, DNA repair, and perhaps other elements of chromatin regulation, could have additive effects that significantly contribute to the neural pathology and phenotypes of fragile X syndrome. An expansion of the model for FMRP function seems warranted (Figs. 2, 3, and 4), and the sum of these models suggest that a full accounting of FMRP functions will be needed to best address therapeutic possibilities for FXS.
References
Ehninger D, Li W, Fox K, Stryker MP, Silva AJ (2008) Reversing neurodevelopmental disorders in adults. Neuron 60:950–960
Scharf SH, Jaeschke G, Wettstein JG, Lindemann L (2015) Metabotropic glutamate receptor 5 as drug target for fragile X syndrome. Curr Opin Pharmacol 20:124–134
Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G (1993) The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 74:291–298
Khandjian EW, Corbin F, Woerly S, Rousseau F (1996) The fragile X mental retardation protein is associated with ribosomes. Nat Genet 12:91–93
Tamanini F, Meijer N, Verheij C, Willems PJ, Galjaard H, Oostra BA, Hoogeveen AT (1996) FMRP is associated to the ribosomes via RNA. Hum Mol Genet 5:809–813
Siomi MC, Zhang Y, Siomi H, Dreyfuss G (1996) Specific sequences in the fragile X syndrome protein FMR1 and the FXR proteins mediate their binding to 60S ribosomal subunits and the interactions among them. Mol Cell Biol 16:3825–3832
Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y et al (2001) Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107:477–487
Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB (2001) Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107:489–499
Huber KM, Gallagher SM, Warren ST, Bear MF (2002) Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A 99:7746–7750
Ashley CT Jr, Wilkinson KD, Reines D, Warren ST (1993) FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262:563–566
Bear MF, Huber KM, Warren ST (2004) The mGluR theory of fragile X mental retardation. Trends Neurosci 27:370–377
Bassell GJ, Warren ST (2008) Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60:201–214
McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK et al (2005) Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 45:753–764
Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP (2005) Suppression of two major fragile X syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49:1053–1066
Berry-Kravis E, Des Portes V, Hagerman R, Jacquemont S, Charles P, Visootsak J, Brinkman M, Rerat K et al (2016) Mavoglurant in fragile X syndrome: results of two randomized, double-blind, placebo-controlled trials. Sci Transl Med 8:321ra325
Jeste SS, Geschwind DH (2016) Clinical trials for neurodevelopmental disorders: at a therapeutic frontier. Sci Transl Med 8:321fs321
Eberhart DE, Malter HE, Feng Y, Warren ST (1996) The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum Mol Genet 5:1083–1091
Fridell RA, Benson RE, Hua J, Bogerd HP, Cullen BR (1996) A nuclear role for the fragile X mental retardation protein. EMBO J 15:5408–5414
Sittler A, Devys D, Weber C, Mandel JL (1996) Alternative splicing of exon 14 determines nuclear or cytoplasmic localisation of fmr1 protein isoforms. Hum Mol Genet 5:95–102
Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM (1997) Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci 17:1539–1547
Zhang Y, O'Connor JP, Siomi MC, Srinivasan S, Dutra A, Nussbaum RL, Dreyfuss G (1995) The fragile X mental retardation syndrome protein interacts with novel homologs FXR1 and FXR2. EMBO J 14:5358–5366
Hu Y, Chen Z, Fu Y, He Q, Jiang L, Zheng J, Gao Y, Mei P et al (2015) The amino-terminal structure of human fragile X mental retardation protein obtained using precipitant-immobilized imprinted polymers. Nat Commun 6:6634
Pasciuto E, Bagni C (2014) SnapShot: FMRP interacting proteins. Cell 159(218–218):e211
Bhogal B, Jepson JE, Savva YA, Pepper AS, Reenan RA, Jongens TA (2011) Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat Neurosci 14:1517–1524
Shamay-Ramot A, Khermesh K, Porath HT, Barak M, Pinto Y, Wachtel C, Zilberberg A, Lerer-Goldshtein T et al (2015) Fmrp interacts with Adar and regulates RNA editing, synaptic density and locomotor activity in zebrafish. PLoS Genet 11:e1005702
Filippini A, Bonini D, Lacoux C, Pacini L, Zingariello M, Sancillo L, Bosisio D, Salvi V et al (2017) Absence of the fragile X mental retardation protein results in defects of RNA editing of neuronal mRNAs in mouse. RNA Biol 14(11):1580–1591
Zhou LT, Ye SH, Yang HX, Zhou YT, Zhao QH, Sun WW, Gao MM, Yi YH et al (2017) A novel role of fragile X mental retardation protein in pre-mRNA alternative splicing through RNA-binding protein 14. Neuroscience 349:64–75
Bardoni B, Schenck A, Mandel JL (1999) A novel RNA-binding nuclear protein that interacts with the fragile X mental retardation (FMR1) protein. Hum Mol Genet 8:2557–2566
Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, Navaratnam D, Kaczmarek LK (2010) Fragile X mental retardation protein controls gating of the sodium-activated potassium channel slack. Nat Neurosci 13:819–821
Deng PY, Rotman Z, Blundon JA, Cho Y, Cui J, Cavalli V, Zakharenko SS, Klyachko VA (2013) FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77:696–711
Alpatov R, Lesch BJ, Nakamoto-Kinoshita M, Blanco A, Chen S, Stutzer A, Armache KJ, Simon MD et al (2014) A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell 157:869–881
Blokhuis AM, Koppers M, Groen EJ, van den Heuvel DM, Dini Modigliani S, Anink JJ, Fumoto K, van Diggelen F et al (2016) Comparative interactomics analysis of different ALS-associated proteins identifies converging molecular pathways. Acta Neuropathol 132:175–196
He Q, Ge W (2017) The tandem Agenet domain of fragile X mental retardation protein interacts with FUS. Sci Rep 7:962
Raj B, Blencowe BJ (2015) Alternative splicing in the mammalian nervous system: recent insights into mechanisms and functional roles. Neuron 87:14–27
Didiot MC, Tian Z, Schaeffer C, Subramanian M, Mandel JL, Moine H (2008) The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic Acids Res 36:4902–4912
Brooks AN, Duff MO, May G, Yang L, Bolisetty M, Landolin J, Wan K, Sandler J et al (2015) Regulation of alternative splicing in Drosophila by 56 RNA binding proteins. Genome Res 25:1771–1780
Stoiber MH, Olson S, May GE, Duff MO, Manent J, Obar R, Guruharsha KG, Bickel PJ et al (2015) Extensive cross-regulation of post-transcriptional regulatory networks in Drosophila. Genome Res 25:1692–1702
Tapial J, Ha KCH, Sterne-Weiler T, Gohr A, Braunschweig U, Hermoso-Pulido A, Quesnel-Vallieres M, Permanyer J et al (2017) An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res 27:1759–1768
Lein E, Borm LE, Linnarsson S (2017) The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science 358:64–69
Lai D, Sakkas D, Huang Y (2006) The fragile X mental retardation protein interacts with a distinct mRNA nuclear export factor NXF2. RNA 12:1446–1449
Zhang M, Wang Q, Huang Y (2007) Fragile X mental retardation protein FMRP and the RNA export factor NXF2 associate with and destabilize Nxf1 mRNA in neuronal cells. Proc Natl Acad Sci U S A 104:10057–10062
Kim M, Bellini M, Ceman S (2009) Fragile X mental retardation protein FMRP binds mRNAs in the nucleus. Mol Cell Biol 29:214–228
Rosenthal JJ, Seeburg PH (2012) A-to-I RNA editing: effects on proteins key to neural excitability. Neuron 74:432–439
Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C et al (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146:247–261
Ascano M Jr, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M et al (2012) FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492:382–386
Korb E, Herre M, Zucker-Scharff I, Gresack J, Allis CD, Darnell RB (2017) Excess translation of epigenetic regulators contributes to fragile X syndrome and is alleviated by Brd4 inhibition. Cell 170(1209–1223):e1220
McConnell MJ, Moran JV, Abyzov A, Akbarian S, Bae T, Cortes-Ciriano I, Erwin JA, Fasching L et al (2017) Intersection of diverse neuronal genomes and neuropsychiatric disease: the brain somatic mosaicism network. Science 356:eaal1641
Megosh HB, Cox DN, Campbell C, Lin H (2006) The role of PIWI and the miRNA machinery in Drosophila germline determination. Curr Biol 16:1884–1894
Bozzetti MP, Specchia V, Cattenoz PB, Laneve P, Geusa A, Sahin HB, Di Tommaso S, Friscini A et al (2015) The Drosophila fragile X mental retardation protein participates in the piRNA pathway. J Cell Sci 128:2070–2084
Jiang F, Lu F, Li P, Liu W, Zhao L, Wang Q, Cao X, Zhang L et al (2016) Drosophila homolog of FMRP maintains genome integrity by interacting with Piwi. J Genet Genomics 43:11–24
Zhang W, Cheng Y, Li Y, Chen Z, Jin P, Chen D (2014) A feed-forward mechanism involving Drosophila fragile X mental retardation protein triggers a replication stress-induced DNA damage response. Hum Mol Genet 23:5188–5196
Peters L, Meister G (2007) Argonaute proteins: mediators of RNA silencing. Mol Cell 26:611–623
Jin P, Zarnescu DC, Ceman S, Nakamoto M, Mowrey J, Jongens TA, Nelson DL, Moses K et al (2004) Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat Neurosci 7:113–117
Caudy AA, Myers M, Hannon GJ, Hammond SM (2002) Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev 16:2491–2496
Ishizuka A, Siomi MC, Siomi H (2002) A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev 16:2497–2508
Iwasaki YW, Siomi MC, Siomi H (2015) PIWI-interacting RNA: Its biogenesis and functions. Annu Rev Biochem 84:405–433
Sienski G, Donertas D, Brennecke J (2012) Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell 151:964–980
Rajasethupathy P, Antonov I, Sheridan R, Frey S, Sander C, Tuschl T, Kandel ER (2012) A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell 149:693–707
Perrat PN, DasGupta S, Wang J, Theurkauf W, Weng Z, Rosbash M, Waddell S (2013) Transposition-driven genomic heterogeneity in the Drosophila brain. Science 340:91–95
Liu W, Jiang F, Bi X, Zhang YQ (2012) Drosophila FMRP participates in the DNA damage response by regulating G2/M cell cycle checkpoint and apoptosis. Hum Mol Genet 21:4655–4668
Collins SC, Bray SM, Suhl JA, Cutler DJ, Coffee B, Zwick ME, Warren ST (2010) Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am J Med Genet A 152A:2512–2520
Myrick LK, Deng PY, Hashimoto H, Oh YM, Cho Y, Poidevin MJ, Suhl JA, Visootsak J et al (2015) Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc Natl Acad Sci U S A 112:949–956
Huyen Y, Zgheib O, Ditullio RA Jr, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS et al (2004) Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432:406–411
Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P et al (2012) Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 488:231–235
Wei W, Ba Z, Gao M, Wu Y, Ma Y, Amiard S, White CI, Rendtlew Danielsen JM et al (2012) A role for small RNAs in DNA double-strand break repair. Cell 149:101–112
d'Adda di Fagagna F (2014) A direct role for small non-coding RNAs in DNA damage response. Trends Cell Biol 24:171–178
Dutertre M, Lambert S, Carreira A, Amor-Gueret M, Vagner S (2014) DNA damage: RNA-binding proteins protect from near and far. Trends Biochem Sci 39:141–149
Dutertre M, Vagner S (2017) DNA-damage response RNA-binding proteins (DDRBPs): perspectives from a new class of proteins and their RNA targets. J Mol Biol 429:3139–3145
Guo W, Allan AM, Zong R, Zhang L, Johnson EB, Schaller EG, Murthy AC, Goggin SL et al (2011) Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat Med 17:559–565
el Bekay R, Romero-Zerbo Y, Decara J, Sanchez-Salido L, Del Arco-Herrera I, Rodriguez-de Fonseca F, de Diego-Otero Y (2007) Enhanced markers of oxidative stress, altered antioxidants and NADPH-oxidase activation in brains from fragile X mental retardation 1-deficient mice, a pathological model for fragile X syndrome. Eur J Neurosci 26:3169–3180
Davidovic L, Navratil V, Bonaccorso CM, Catania MV, Bardoni B, Dumas ME (2011) A metabolomic and systems biology perspective on the brain of the fragile X syndrome mouse model. Genome Res 21:2190–2202
Weisz ED, Towheed A, Monyak RE, Toth MS, Wallace DC, Jongens TA (2018) Loss of Drosophila FMRP leads to alterations in energy metabolism and mitochondrial function. Hum Mol Genet 27:95–106
Rousseau F, Heitz D, Tarleton J, MacPherson J, Malmgren H, Dahl N, Barnicoat A, Mathew C et al (1994) A multicenter study on genotype-phenotype correlations in the fragile X syndrome, using direct diagnosis with probe StB12.3: the first 2,253 cases. Am J Hum Genet 55:225–237
Fong YW, Cattoglio C, Tjian R (2013) The intertwined roles of transcription and repair proteins. Mol Cell 52:291–302
Le May N, Fradin D, Iltis I, Bougneres P, Egly JM (2012) XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol Cell 47:622–632
Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC et al (2013) Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat Neurosci 16:613–621
Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX et al (2015) Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161:1592–1605
Rutenberg-Schoenberg M, Sexton AN, Simon MD (2016) The properties of long noncoding RNAs that regulate chromatin. Annu Rev Genomics Hum Genet 17:69–94
Kopp F, Mendell JT (2018) Functional classification and experimental dissection of long noncoding RNAs. Cell 172:393–407
Acknowledgements
The authors apologize for omission of any relevant works due to space constraints.
Funding
Support from NIH MH108956 and past support from NIH and FRAXA Research Foundation are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Dockendorff, T.C., Labrador, M. The Fragile X Protein and Genome Function. Mol Neurobiol 56, 711–721 (2019). https://doi.org/10.1007/s12035-018-1122-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1122-9