
Abstract
Amyloid beta-peptide (Aβ), the neurotoxic component of senile plaques in Alzheimer’s disease (AD) brains, is known to trigger cell cycle reentry in post-mitotic neurons followed by apoptosis. However, the underlying mechanisms remain unclear. Recently, we have reported that Aβs stimulate the expression of inhibitor of differentiation-1 (Id1) to induce sonic hedgehog (SHH) (Hung et al., Mol Neurobiol 53(2):793–809, 2016), and both are mitogens capable of triggering cell cycle progression. In this work, we tested the hypothesis that Aβ-induced Id1 and SHH contribute to cell cycle reentry leading to apoptosis in neurons. We found that Aβ triggered cell cycle progression in the post-mitotic neurons, as indicated by the increased expression of two G1-phase markers including cyclin D1 and phosphorylated retinoblastoma protein (pRb), two G2-phase markers such as proliferating cell nuclear antigen (PCNA) and incorporation of 5-bromo-2′-deoxyuridine (BrdU) into newly synthesized DNA, as well as the mitotic marker histone H3 phosphorylated at Ser-10. As expected, Aβ also enhanced caspase-3 cleavage in the cortical neurons. Id1 siRNA, the neutralization antibody against SHH (SHH-Ab), and the cyclin-dependent kinase (CDK)-4/6 inhibitor PD0332991 all attenuated, in part or in full, the Aβ-induced expression of these cell cycle markers. Indeed, exogenous recombinant Id1 protein and the biologically active N-terminal fragment of SHH (SHH-N) were both sufficient to enhance the expression of cell cycle markers independent of Aβ. Taken together, our results revealed the critical roles of Id1 and SHH mediating Aβ-dependent cell cycle reentry and subsequently caspase-dependent apoptosis in the fully differentiated post-mitotic neurons, at least in vitro.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Most nonproliferating cells, including fully differentiated neurons, remain in G0 (or quiescent) state without progression into cell cycle. In the proliferating cells, cell cycle is divided into four phases: G1 (the first gap), S (DNA synthesis), G2 (the second gap), and M (mitosis); entry of each phase is under strict regulation by a highly complex signaling network involving distinct types of cyclins and cyclin-dependent kinases (CDKs). First, CDK4 and CDK6, associated with D-type cyclins (cyclin D1, D2, and D3), regulate the G0/G1 transition. Subsequently in G1, expression of cyclin E is induced in association with CDK2 that is required for G1/S transition. At the end of G1 phase, the cyclin D/CDK4/6 complex is required for the phosphorylation of retinoblastoma protein (pRb), thereby precluding its binding to the E2F transcription factors; activation of E2F thus stimulates the synthesis of cyclin E and CDK2 for S-phase entry. Cyclin E/CDK4 complex also further phosphorylates pRb, resulting in positive feedforward loops that rapidly increase both E2F and cyclin E/CDK2 as the cells approach G1/S transition. DNA synthesis in S phase requires the activity of cyclin A/CDK2 complex. At the completion of DNA replication, cyclin A forms a complex with CDK1, allowing the cells to enter G2 phase. Finally, CDK1 forms a complex with cyclin B and the cells go into mitosis (for reviews, please see [1,2,3]).
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly. Accumulation of extracellular senile plaque containing excessive aggregation of amyloid beta-peptide (Aβ) derived from amyloid precursor protein (APP) is a prominent pathological hallmark of AD [4]. One neurotoxic mechanism of Aβs is the trigger of aberrant cell cycle reentry in the differentiated post-mitotic neurons with subsequent induction of apoptosis. Several lines of evidence suggest the relationships between cell cycle reentry and neuronal loss in AD. For example, marker proteins known to be involved in cell cycle progression are expressed in post-mitotic AD neurons [2, 3, 5,6,7]. Cell cycle reentry and accumulation of cyclin D1 leading to cell death have been reported in the brains of AD patients with mutation in PS1 gene [8]. AD neurons that reenter cell cycle usually arrest at checkpoints before mitosis phase [2]. Despite these observations, exactly how Aβ may trigger cell cycle reentry in the differentiated neurons remains to be fully defined.
In our recent study, we have reported that Aβ may induce inhibitor of differentiation-1 (Id1) and then sonic hedgehog (SHH) in differentiated rat cortical neurons [9]. The Id family is composed of Id1, Id2, Id3, and Id4, each with a differential expression pattern through embryonic to adult brain [10,11,12]. Lacking the basic helix-loop-helix (bHLH) sequence that functions as the DNA-binding domain [13], Id also represents inhibitor of DNA binding that inhibits bHLH proteins via formation of a nonfunctional complex in a dominant negative fashion [13]. Id proteins have been shown to act as global regulators of lineage commitment for cell fate determination, apoptosis induction, immortalization of primary cells to function as oncoproteins, and promotion of tumor invasiveness [14]. Hedgehog was initially identified as a segmental polarity gene in Drosophila melanogaster, whose mutations affect segment number and polarity [15]. Mutations of the Hh gene are generally embryonic lethal resulting in a lawn of denticles on the ventral surface, hence the name [16]. SHH is a mammalian member of the hedgehog family known to regulate polarity of the central nervous system (CNS) [17]. The biologically active N-terminal fragment of SHH protein (SHH-N), once secreted, exerts its actions through binding to the Patched [18], which is a 12-pass transmembrane protein that in the absence of SHH-N suppresses another 7-pass transmembrane protein called Smoothened [19]. In the presence of SHH-N, Smoothened may activate its downstream transcription factors of Gli family [20]. Cyclopamine, a steroidal alkaloid that can bind to the heptahelical bundle of Smoothened receptor and therefore inhibit its biological functions [21], is commonly used to block the canonical signaling cascades of SHH. Currently known target genes affected by SHH/Gli pathways include N-Myc [22], Bmi1 [23], and cyclin D1 [24] that participate respectively in the regulation of cellular proliferation, self-renewal, and cell cycle progression.
Id1 inhibits differentiation with promotion of cell cycle progression [25, 26]. Indeed, overexpression of Id1 can induce cell cycle reentry and proliferation of Sertoli cells [27]. SHH is also known to function as a mitogen [28] capable of promoting G1 cyclin expression with sustained cell cycle progression in mammalian neural precursors [24]. Therefore, in the present study, we tested the hypothesis that induction of Id1 and SHH may contribute to Aβ-mediated cell cycle reentry in post-mitotic neurons, thereby resulting in neuronal apoptosis.
Materials and Methods
Reagents and Preparations of Aβs
The Smoothened inhibitor cyclopamine (Cat. No. GR-334; Biomol International, LP, Plymouth Meeting, PA, USA) and the CDK4/6 inhibitor PD0332991 (Cat. No. S1116; Selleckchem, Houston, TX, USA) were both dissolved in dimethyl sulfoxide (DMSO; Cat. No. JTB-9033-04; Capitol Scientific, Inc., Austin, TX, USA) as the stock solution of 10 and 5 mM, respectively. Recombinant full-length human Id1 protein (amino acids 2–155) with an 11-arginine tag on its C-terminus was purchased from Abcam (Cat. No. ab134896; Cambridge, MA, USA) and prepared as a stock solution of 1 mg/ml. To block the SHH functions, either the rabbit polyclonal (Cat. No. SC-9024) or the mouse monoclonal neutralization antibody (Cat. No. SC-365112) was applied to the cortical cultures, as specified in the figure legends. Both antibodies were from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA) and prepared as stock solutions of 200 μg/ml; the corresponding negative controls, namely the rabbit IgG (Cat. No. 12-370) and the mouse IgG (Cat. No. 12-371), were both from Merck Millipore (Billerica, MA, USA) and prepared as stock solutions of 1 mg/ml. 5-Bromo-2′-deoxyuridine (BrdU; Cat. No. 550891; BD Biosciences, San Jose, CA, USA) was prepared as the 1-mM working solution in neurobasal medium supplemented with B27 (GIBCO/Invitrogen Corporation, Carlsbad, CA, USA), which was the culture medium for primary rat cortical neurons. Cannabidiol (CBD; Cat. No. 1481; Tocris Bioscience, Bristol, UK) was prepared as a 13.5-mM stock solution and stored at − 20 °C.
Preparation of Aβs was based on our previously published paper [29]. Aβ25-35 (Cat. No. A4559, Sigma-Aldrich, St. Louis, MO, USA) was dissolved in autoclaved ddH2O to make a stock solution of 2 mM, dispensed into aliquots, and stored at − 80 °C until use. One day prior to experimentation, aliquots of Aβs were incubated at 37 °C for 24 h to allow aggregation into fibrils. Aβ1-42 (Cat. No. 20276, AnaSpec, Inc., San Jose, CA, USA) was prepared based on a previous report [30]. Briefly, Aβ1-42 was first resuspended in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP; Cat. No. 100528, Sigma-Aldrich) to make a stock solution of 1 mM and then dispensed into aliquots. Thereafter, HFIP was allowed to evaporate in the lamina flow for overnight and stored at − 80 °C until use. Prior to experimentation, Aβ1-42 was dissolved in dry DMSO (Cat. No. 1029310500, Merck, Darmstadt, Germany) to make a stock solution of 5 mM, diluted to 100 μM in 1× phosphate-buffered saline (PBS), and then incubated at 4 °C for 24 h to allow aggregation.
Primary Cortical Culture
All the procedures for animal care and preparation of fetal rat cortical cultures were performed humanely in accordance with the guidelines described in the “User Manual of Laboratory Animal Center at National Yang-Ming University.” All the experimental procedures concerning animals have been approved by the Institutional Animal Care and Use Committee (IACUC) of National Yang-Ming University with the approval number 1021234. Primary neuronal cultures were prepared from embryonic day 18 (E18) fetal Sprague-Dawley (SD) rat brains as previously described [31]. The cultures were maintained in neurobasal medium supplemented with B27 at 37 °C in a humidified incubator with 5% CO2. Cortical cells were cultured for 7–9 days in vitro (DIV) to allow growth of dendrites before experimentation.
Cell Survival Assays
MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) reduction assay was performed as previously described [32]. Hoechst staining to assess the extents of cell survival was performed as described in our previous publications [33, 34]. Cortical cells were grown on coverslips in 24-well culture dishes prior to experimental manipulations. For counting of Hoechst-stained surviving cells with normal nuclear morphology, at least 3 vision fields were randomly selected to obtain the averaged numbers of nuclei per vision field on each coverslip. The “Cell Survival (%)” was defined as the mean numbers of surviving cells per vision field in the experimental groups divided by those of the control cultures and then multiplied by 100%.
Western Blotting
Western blotting was performed as previously described [32, 35]. The rabbit antibody against Id1 (1:250; Cat. No. sc-488, Santa Cruz Biotechnology, Inc.) was diluted in signal enhancer HIKARI solution 1 (Cat. No. NT08044-71R, Nacalai Tesque, Kyoto, Japan). The mouse antibody against β-actin (1:1000; Cat. No. MAB1501; Merck Millipore, Billerica, MA, USA), rabbit monoclonal antibody against cyclin D1 (1:1000; Cat. No. ab134175, Abcam), rabbit monoclonal antibody against the retinoblastoma protein (pRb) phosphorylated at Ser-807/811 (phosphor-pRb; 1:500; Cat. No. 8516, Cell Signaling Technology, Danvers, MA, USA), rabbit monoclonal antibody against proliferating cell nuclear antigen (PCNA; 1:500; Cat. No. ab92552, Abcam), rabbit monoclonal antibody against histone H3 phosphorylated at Ser-10 (1:100; Cat. No. ab32107, Abcam), rabbit monoclonal antibody against cleaved caspase-3 (1:500; Cat. No. 9664, Cell Signaling Technology), and rabbit polyclonal antibody against caspase-3 (1:1000; Cat. No. 9662, Cell Signaling Technology) were all diluted in blocking buffer (5% nonfat dry milk in TBST buffer containing 0.05% Tween 20, 137 mM NaCl, and 20 mM Tris–HCl, pH 7.5). The horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse secondary antibodies were applied in fresh blocking buffer at 1:5000 to detect the respective primary antibodies. Immunoreactive signal was detected using ECL-Plus Western blotting detection reagents (Merck Millipore). The blots were examined under the Luminescence/Fluorescence Imaging System LAS-4000 (FUJIFILM, Tokyo, Japan). Quantification of protein expression was accomplished by using Multi Gauge analysis software (FUJIFILM). In all the Western blotting experiments, the signal intensity of β-actin served as a control for equal loading of proteins in each lane.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde at 37 °C for 15 min before hybridization with the primary antibodies except for the rabbit antibodies against PCNA, where the cells were fixed in − 20 °C methanol for 5 min. After fixation, cells were washed in PBS, blocked with 2% normal goat serum and 0.03% Triton X-100 in PBS, and then incubated with primary antibodies at 4 °C overnight according to previously published methods [35]. The same rabbit antibodies against cyclin D1 (1:100), phosphor-pRb (1:100), phosphor-histone H3 (1:100), and PCNA (1:100) for Western blotting were also used for immunocytochemistry. The rabbit monoclonal antibody against histone H3 (Cat. No. 4499, Cell Signaling Technology) was used at 1:100 dilutions. The mouse monoclonal antibodies against MAP-2 (1:100; Cat. No. MAB378, CHEMICON International, Inc., Temecula, CA, USA) or NeuN (1:100; Cat. No. ABN78, CHEMICON/Millipore Corp.) were applied to stain the mature neurons. For detection of BrdU, the samples were pretreated with 2 N HCl at room temperature for 30 min to denature the DNA; this was followed by washing twice with 0.1 M sodium borate buffer (pH 8.5) and PBS before blocking. A rat primary antibody against BrdU (1:200; Cat. No. 6326, Abcam) was used for detection of BrdU+ cells. The following secondary antibodies were used: Hilyte Fluor 594-labeled goat anti-mouse IgG (1:100; Cat. No. 61507-H594, AnaSpec), Hilyte Fluor 488-labeled goat anti-rabbit IgG (1:100; Cat. No. 61506-H488, AnaSpec), and FITC-conjugated rabbit anti-rat IgG (1:100; Cat. No. ab6730, Abcam). For confocal microscopy, the samples were observed under a laser-scanning confocal microscope (Zeiss LSM700, Oberkochen, Germany) equipped with filter sets to detect the corresponding fluorescence signals.
siRNA Transfection
Transfection of siRNA into primary cortical neurons was performed as previously described [36]. All Accell SMART pool siRNAs were purchased from Thermo Scientific Dharmacon Inc. (Lafayette, CO, USA). Cells were incubated with 1 μM scrambled siRNA (sc siRNA) or target siRNA for 72 h in Neurobasal supplemented with B27 at DIV4–7. The four target sequences for the Id1 siRNA mixture (Id: 25261) were as follows: 5′-GUUUU GUAUUGUAUAUUAC-3′, 5′-CAAACACUUUAGAUAACGU-3′, 5′-GGCUGAGAAUAUUGUUUUA-3′, and 5′-CCUCAGAACCGCAAAGUGA-3′. A nontargeting Accell siRNA pool (Cat. No. D-001950-01-05) was used as the negative control in all siRNA transfection experiments.
Statistical Analysis
Results are expressed as mean ± SEM from the sample number (N). For Western blotting, each N represents the results derived from one independent experiment using one different culture. For MTT assays, each N represents the data derived from one independent experiment using one different culture. The assays were performed in triplicates or quadruplicates to obtain the averaged reading values, which were later calculated into cell survival in percentage, for each experimental condition. Combined results from these replicated experiments are shown. For Hoechst staining, each N represents data collected from a single coverslip. In any given experiment, mean values of cell survival in percentage were derived from the combined results of 3–4 replicated experiments, each using one independent culture, comprising a total of 9–14 coverslips. Unless otherwise specified in the corresponding figure legends, two groups were compared by Student’s t test; multiple groups were first analyzed by one-way analysis of variance (ANOVA) followed by a post hoc Student-Newman-Keuls test. A P value of less than 0.05 was considered significant.
Results
Inhibition of Aβ Neurotoxicity by Id1 siRNA, Id1 Inhibitor CBD, and Cell Cycle Blocker PD0332991
In our recent study, we have demonstrated the “Aβ → Id1 → HIF-1 → SHH” signaling cascade in primary cortical neurons [9]. In this work, we attempted to further establish the signal pathway of “Aβ → Id1/SHH → cell cycle reentry → neuronal apoptosis.” As the first step, we examined whether blockade of Id1 expression/function and cell cycle progression may protect cortical neurons against Aβ toxicity. Consistent with our earlier report showing Aβ25-35-induced Id1 expression at later time points of 24–48 h, in the current study, we demonstrated that both Aβ25-35 (Fig. 1a) and Aβ1-42 (Fig. 1b) also enhanced Id1 expression at earlier time points of 2 and 24 h in primary cortical cultures. Further, Aβ25-35-induced Id1 can be co-localized to MAP-2 signal in primary cortical cultures, indicating neuronal expression of this protein (Fig. 1c).

Effects of Aβs on the expression of Id1 in primary rat cortical cultures. a, b Western blots showing the effects of Aβs on the expression of Id1 protein. Cultures were treated with 10 μM Aβ25-35 (a) or 5 μM Aβ1-42 (b) for 2 or 24 h. Control cultures were also subjected to medium exchange for the same periods of time without Aβs. Mean ± SEM from N = 4–5 in a and N = 6–7 in b. * denotes P < 0.05 compared with corresponding control cultures without Aβ treatment. c Cultures were treated with 10 μM Aβ25-35 for 24 h before confocal microscopy. Micrographs demonstrate co-localization of Id1 (green) and MAP-2 (red) immunostaining signals, as indicated by the white arrows; Hoechst staining (blue) of nuclei served as counterstaining. Scale bar = 5 μm
To establish the causative relationship between Aβ-induced Id1 expression and Aβ-mediated neurotoxicity, siRNA was applied to suppress Id1 expression in rat cortical cultures. Western blotting revealed that prior transfection of Id1 siRNA into more mature neurons at DIV4–7 completely suppressed Aβ25-35-induced Id1 protein at DIV8 (Fig. 2a). More importantly, Id1 siRNA also increased total neurite length when transfected into cortical cultures at DIV1–4 (Fig. 2b), indicating that Id1 knockdown enhanced neuronal differentiation in the immature neurons where Id1 expression was still higher. In addition to stimulating neurite outgrowth by Id1 knockdown, an increased total neurite length may also be resulted from alteration of cell numbers potentially induced by decreased Id1 expression. However, Hoechst staining excluded this possibility because total cell numbers were not significantly altered in the cultures transfected with either sc siRNA or Id1 siRNA (data not shown). These results indicated successful suppression of Id1 by siRNA in cortical neurons at the levels of both protein expression and biological function. Once the Id1 siRNA effects were confirmed, we then tested whether Id1 plays a significant role in mediating Aβ25-35/1-42 neurotoxicity. Results based on the MTT assay revealed that Id1 siRNA attenuated Aβ25-35-induced cell death at 24 h, at least in part (Fig. 2c). Id1 siRNA also partially suppressed Aβ1-42 toxicity (Fig. 2d). Similar findings were observed for Aβ25-35 (Fig. 2e) and Aβ1-42 (Fig. 2f) when Hoechst staining was conducted to examine the extents of cell survival. CBD is a pharmacological inhibitor of Id1 [37]. We found that CBD significantly rescued Aβ25-35-induced cell death by both MTT (Fig. 2g) and Hoechst staining (Fig. 2h).

Id1 plays a critical role in Aβ-induced cell death in cortical cultures. a Cortical cultures were transfected with scrambled siRNA (sc siRNA) or Id1 siRNA for 72 h followed by exposure to 10 μM Aβ25-35 for an additional 24 h before detection of Id1 by Western blotting. Mean ± SEM from N = 3. * and # denote P < 0.05. b Primary cortical cultures were transfected with scrambled siRNA (sc siRNA) or Id1 siRNA (Id1 siRNA) before immunostaining with an antibody against MAP-2 (red). Scale bar = 20 μm. The representative micrographs are shown in the upper panels; quantitative results of neurite length using ImageJ software are shown in the lower panel. Mean ± SEM from N = 3. * and # denote P < 0.05. c–f Primary cortical cultures were treated with sc siRNA or Id1 siRNA for 72 h before exposure to 10 μM Aβ25-35 (c, e) or 5 μM Aβ1-42 (d, f) for an additional 24 h before determination of cell survival by MTT assay (c, d) or Hoechst staining (e, f). Mean ± SEM from N = 8–9 in c and N = 9 in d–f. * and # denote P < 0.05. g, h Cortical cultures were treated with 10 μM Aβ25-35 with or without 1.5 μM CBD for 48 h. Cell survival was then determined by MTT assay (g) or Hoechst staining (h). Mean ± SEM from N = 3. * and # denote P < 0.05
PD0332991, a CDK4/6-specific inhibitor without other nonspecific activities against a panel of 36 additional protein kinases [38], is a cell cycle blocker that has been tested for arresting tumor growth in the intracranial xenograft model of glioblastoma multiforme [39] and in multiple myeloma [40]. Given the hypothesis that Aβ-mediated cell cycle reentry in fully differentiated neurons contributes to its neurotoxicity, it is expected that pharmacological blockade of cell cycle progression in post-mitotic neurons should mitigate Aβ toxicity. Consistent with this notion, MTT assay revealed that PD0332991 significantly inhibited cell death induced by both Aβ25-35 (Fig. 3a) and Aβ1-42 (Fig. 3c); similarly, PD0332991 also attenuated toxicity induced by Aβ25-35 (Fig. 3b) and Aβ1-42 (Fig. 3d) based on Hoechst staining. Overall, results shown in Figs. 1, 2, and 3 together indicate a detrimental effect of Id1 induced by Aβs in primary rat cortical cultures, which can be in part abolished by Id1 knockdown or Id1 inhibition; furthermore, pharmacological blockade of cell cycle progression is sufficient to suppress neurotoxicity resulting from exposure to Aβs.

PD0332991, the CDK4/6 inhibitor capable of blocking cell cycle progression, protects cortical cultures against neurotoxicity of Aβs. Cortical cultures were exposed to 10 μM Aβ25-35 (a, b) or 5 μM Aβ1-42 (c, d) with or without 1 μM PD0332991 for 24 h before MTT assay (a, c) and Hoechst staining (b, d). Mean ± SEM from N = 3 in a, c and N = 9 in b, d. * and # denote P < 0.05
Roles of Id1/SHH on Aβ25-35-Induced Reentry into G1 Phase in Post-mitotic Neurons
In the proliferating cells, entry from the quiescent G0 state into G1 phase requires association of CDK4/6 with D-type cyclins including cyclin D1. At the end of G1 phase, the cyclin D/CDK4/6 complex is also required for the phosphorylation of pRb, an active transcriptional repressor when bound to transcription factors such as members of the E2F family. Phosphorylation, and hence inactivation, of pRb by cyclin D/CDK4/6 complex in late G1 phase causes the release of E2F, allowing transcription of genes important for DNA synthesis, such as cyclin E and CDK2, for entry into S phase. We therefore selected cyclin D1 and phosphor-pRb as two G1-phase markers during cell cycle reactivation in the post-mitotic neurons. Results shown in Fig. 4a indicated that Aβ25-35 transiently induced the expression of cyclin D1 in primary cortical cultures, achieving statistical significance at 4–16 h; furthermore, pRb phosphorylation at Ser-807/811 (pRb-Pi) was also enhanced by Aβ25-35 at 16–48 h. Because our primary rat cortical culture is a neuron-enriched co-culture system that contains approximately 15% nonneuronal cells including astrocytes [31], immunocytochemistry was therefore conducted to identify the cell types with heightened expression of cyclin D1 in response to Aβ25-35. Results demonstrated an increased number of cyclin D1+ cells co-localized to the MAP-2 signal, indicative of neuronal expression of cyclin D1, in the cortical cultures treated with Aβ25-35 for 8 h (Fig. 4b); similar results were obtained for phosphor-pRb in the cortical cultures treated with Aβ25-35 for 24 h (Fig. 4c). Thus, Aβ25-35 exposure triggers activation of cell cycle programs in the differentiated post-mitotic cortical neurons. Consistent with our hypothesis, the Id1 siRNA in part suppressed the expression of cyclin D1 that was induced by Aβ25-35 (Fig. 4d). Indeed, application of recombinant Id1 proteins with an 11-arginine tag (Id1-Tag), thus capable of penetrating directly through the plasma membrane, was sufficient to induce expression of cyclin D1 independent of Aβ25-35 (Fig. 4e). Knockdown of Id1 by siRNA (Fig. 4f) and PD0332991 (Fig. 4g) both suppressed the extents of pRb phosphorylation induced by Aβ25-35. Id1 contributes to Aβ-induced SHH expression in cortical neurons [9]. We found that the neutralization antibody against SHH (SHH-Ab) fully suppressed Aβ-induced expression of cyclin D1 (Fig. 4h) and phosphorylation of pRb (Fig. 4i); moreover, application of the biologically active N-terminal fragment of SHH (SHH-N) was sufficient to dose-dependently enhance the expression of cyclin D1 (data not shown). Taken together, these results highlight the crucial roles of Id1 and SHH in mediating Aβ25-35-dependent induction of cyclin D1 and pRb phosphorylation, the markers for G1 phase, during cell cycle progression.



Aβ25-35-induced expression of cyclin D1 and phosphorylation of pRb in the post-mitotic neurons are mediated by Id1 and SHH. a Primary cortical cultures were treated with 10 μM Aβ25-35 for indicated times before detection of cyclin D1 expression and pRb phosphorylation at Ser-807/811 (pRb-Pi) by Western blotting. Mean ± SEM from N = 4 for cyclin D1 and N = 5 and pRb-Pi. * denotes P < 0.05 compared with the corresponding control cultures without Aβ treatment. b, c Rat cortical cultures were exposed to 10 μM Aβ25-35 for 8 h (b) or 24 h (c) before immunostaining with the antibodies against cyclin D1 (green; b) or pRb-Pi (green; c) along with MAP-2 (red; b, c); Hoechst staining (blue) served as counterstain. In both b and c, scale bars = 50 μm in the upper panels and 10 μm in the lower panels. Note that Aβ25-35 enhanced the expression of cyclin D1 (b) and pRb-Pi (c), both of which were localized in the MAP-2+ cells suggesting neuronal expression of these two proteins, as indicated by the white arrows. d Cortical cultures were transfected with sc siRNA or Id1 siRNA for 72 h at DIV6–9 prior to treatment with 10 μM Aβ25-35 for an additional 24 h. Expression of cyclin D1 was detected by Western blotting. Mean ± SEM from N = 5. * and # denote P < 0.05. e Cortical cultures were treated with or without 4 μg/ml recombinant Id1 protein tagged with 11 arginines (Id1-Tag) for 8 h before Western blotting to detect the expression level of cyclin D1. Mean ± SEM from N = 4. * denotes P < 0.05. f The experimental conditions were the same as described above in d except pRb-Pi was detected by Western blotting. Mean ± SEM from N = 5. * and # denote P < 0.05. g Cultures were exposed to 10 μM Aβ25-35 for 24 h with or without 1 μM PD0332991 before Western blotting to detect pRb-Pi. Mean ± SEM from N = 3. * and # denote P < 0.05. h, i Cultures were exposed to 10 μM Aβ25-35 for 8 h with the neutralization mouse monoclonal antibody against SHH (SHH-Ab; 2 μg/ml) or the same amount of mouse IgG before detection of cyclin D1 (h) or pRb-Pi (i). Mean ± SEM from N = 3 in both h and i. * and # denote P < 0.05. The “ns” denotes “not significant”
Roles of Id1/SHH on Aβ25-35-Induced Reentry into S Phase in Post-mitotic Neurons
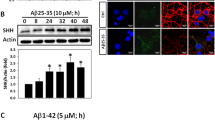
PCNA, an auxiliary protein of DNA polymerase delta, is involved in the control of DNA replication [41] and therefore commonly used as a molecular marker for labeling cells in S phase during cell cycle progression [42]. We found that Aβ25-35 time-dependently increased the expression levels of PCNA, reaching statistical significance at 24–40 h (Fig. 5a). Double immunofluorescence confocal microscopy revealed co-localization of PCNA+ cells to MAP-2+ cortical neurons upon Aβ25-35 exposure (Fig. 5b). Knockdown of Id1 suppressed the expression of both endogenous as well as Aβ25-35-induced PCNA (Fig. 5c), whereas Id1-Tag was sufficient to enhance PCNA expression in the differentiated cortical neurons independent of Aβ25-35 (Fig. 5d). Similar to its inhibitory effects on expression of cyclin D1 (Fig. 4h) and phosphorylation of pRb (Fig. 4i), SHH-Ab was capable of blocking its biological functions that completely abolished Aβ25-35-mediated PCNA induction (Fig. 5e). Interestingly, such an Aβ25-35 effect was not suppressed by cyclopamine (Fig. 5f), suggesting that the canonical SHH mediator Smoothened is not involved. Similar to its effects on cyclin D1 expression, exogenous SHH-N also dose-dependently induced PCNA expression in differentiated cortical neurons (Fig. 5g). Consistent with its neuroprotective effects against Aβ25-35/Aβ1-42 toxicity (Fig. 3), the CDK4/6 inhibitor PD0332991 significantly suppressed the expression of endogenous as well as Aβ25-35-induced PCNA in differentiated cortical neurons (Fig. 5h).


Aβ25-35-induced PCNA expression in the post-mitotic neurons is mediated by Id1 and SHH. a Primary cortical cultures were treated with 10 μM Aβ25-35 for indicated times before detection of PCNA expression by Western blotting. Mean ± SEM from N = 4. * denotes P < 0.05 compared with the control cultures without Aβ treatment. b Rat cortical cultures were treated with 10 μM Aβ25-35 for 40 h before immunostaining with antibodies against PCNA (green) and MAP-2 (red); Hoechst staining (blue) served as counterstain. Scale bars = 50 μm in the upper panels and 10 μm in the lower panels. Note that Aβ25-35 enhanced PCNA expression in the MAP-2+ cells suggesting its neuronal expression, as indicated by the white arrows. c Cortical cells were transfected with Id1 siRNA or scrambled siRNA for 72 h. This was followed by exposure to 10 μM Aβ25-35 for an additional 24 h before detection of PCNA expression. Mean ± SEM from N = 3. * and # denote P < 0.05. The “ns” denotes “not significant.” d Primary cortical cultures were treated with or without 4 μg/ml Id1-Tag for 16 h before detection of PCNA expression. Mean ± SEM from N = 3. * denotes P < 0.05. e Cortical cells were treated with 10 μM Aβ25-35 with 2 μg/ml of rabbit SHH neutralization antibody (SHH-Ab) or normal rabbit IgG for 24 h before detection of PCNA expression. Mean ± SEM from N = 3. * and # denote P < 0.05. The “ns” denotes “not significant.” f Primary cortical cultures were treated with 10 μM Aβ25-35 in the presence of 20 μM cyclopamine (CPM) or the same amount of solvent DMSO for 24 h before detection of PCNA. Mean ± SEM from N = 3. * and # denote P < 0.05. g Primary cortical cultures were treated with the biologically active N-terminal fragment of SHH (SHH-N) at indicated concentrations for 24 h before detection of PCNA expression. Mean ± SEM from N = 3. * denotes P < 0.05 compared with the control cultures without SHH-N treatment. h Cortical cells were treated with 10 μM Aβ25-35 with 1 μM PD0332991 for 24 h before detection of PCNA expression. Mean ± SEM from N = 3. * and # denote P < 0.05
In addition to PCNA, we also treated the post-mitotic cortical neurons with BrdU to label the cells undergoing DNA synthesis as an index for cell cycle reactivation. NeuN is a marker for mature neuronal nuclei [43]. Double immunofluorescence confocal microscopy confirmed that the Aβ25-35-induced BrdU signal co-localized in the NeuN+ cells, indicative of neuronal incorporation of these BrdU during Aβ25-35-induced DNA synthesis (Fig. 6a). We therefore used the numbers of NeuN+/BrdU+ cells as an S-phase index. Results shown in Fig. 6b indicated that blockade of Id1 by its siRNA completely abolished Aβ-induced increases in the numbers of NeuN+/BrdU+ cells. Exogenous Id1-Tag protein was also sufficient to increase the numbers of NeuN+/BrdU+ cells (Fig. 6c), and quantitative analyses confirmed this finding (Fig. 6d). Furthermore, SHH-Ab completely blocked Aβ-mediated increases in the numbers of NeuN+/BrdU+ cells (Fig. 6e, f), whereas exogenous SHH-N showed a dose-dependent effect in increasing the extents of NeuN/BrdU co-localization (Fig. 6g, h). Finally, the CDK4/6 inhibitor PD0332991 known to block cell cycle progression completely suppressed Aβ25-35-mediated increases in the numbers of NeuN+/BrdU+ cells (Fig. 6i). Together, results shown in Figs. 5 and 6 revealed the critical roles of Id1 and SHH in Aβ-mediated entry into S phase in the fully differentiated cortical neurons.



Aβ-induced BrdU incorporation into the newly synthesized DNA in the post-mitotic neurons is mediated by Id1 and SHH. a Cortical cultures were treated with 10 μM Aβ25-35 and 10 μM BrdU for 16 h before immunostaining with antibodies against BrdU (green) and NeuN (red); DAPI staining (blue) served as counterstain. Scale bars = 50 μm in the upper panels and 10 μm in the lower panels. Note that Aβ25-35 increased the numbers of BrdU+ cells that were localized in the NeuN+ neurons, as indicated by the white arrows. b Cortical cultures were transfected with sc siRNA or Id1 siRNA for 72 h followed by exposure to 10 μM Aβ25-35 and 10 μM BrdU for an additional 16 h before immunostaining. The numbers of NeuN+/BrdU+ cells were counted and calculated. Mean ± SEM from N = 3. * and # denote P < 0.05. c Cortical cultures were treated with 4 μg/ml Id1-Tag and 10 μM BrdU for 24 h before immunostaining with antibodies against BrdU (green) and NeuN (red); DAPI staining (blue) served as counterstain. Scale bars = 50 μm in the upper panels and 10 μm in the lower panels. Note that Id1-Tag increased the numbers of NeuN+/BrdU+ cells as indicated by the white arrows. d The numbers of NeuN+/BrdU+ cells in c were counted and calculated. Mean ± SEM from N = 5. * denotes P < 0.05. e Cortical cultures were exposed to 10 μM Aβ25-35 and 10 μM BrdU in the presence of SHH-Ab (2 μg/ml) or the same amount of mouse IgG for 24 h before immunostaining with the antibodies against BrdU (green) and NeuN (red); DAPI (blue) served as a counterstain. Scale bars = 50 μm in the upper panels and 10 μm in the lower panels. Note that Aβ25-35-induced increases in the numbers of NeuN+/BrdU+ cells were abolished by the SHH-Ab. f The numbers of NeuN+/BrdU+ cells in e were counted and calculated. Mean ± SEM from N = 3. * and # denote P < 0.05. g Cortical cultures were treated with 30 or 300 ng/ml SHH-N in the presence of 10 μM BrdU for 24 h before immunostaining with antibodies against BrdU (green) and NeuN (red); DAPI staining (blue) served as counterstain. Scale bars = 50 μm in the upper panels and 10 μm in the lower panels. Note that SHH-N at 30 and 300 ng/ml dose-dependently increased the numbers of NeuN+/BrdU+ cells, as indicated by the white arrows. h The numbers of NeuN+/BrdU+ cells in g were counted and calculated. Mean ± SEM from N = 3. * denotes P < 0.05 as compared to the control cultures without SHH-N treatment. i Cortical cultures were treated with 10 μM Aβ25-35, 1 μM PD0332991, or both in the presence of 10 μM BrdU for 16 h before immunostaining. The numbers of NeuN+/BrdU+ cells were counted and calculated. Mean ± SEM from N = 4. * and # denote P < 0.05
Roles of Id1/SHH on Aβ25-35-Induced Reentry into M Phase in Post-mitotic Neurons
Phosphorylation of histone H3 on Ser-10 in the N-terminal tail is essential for cell cycle progression [44]; indeed, the correlation between histone H3 on Ser-10 phosphorylation and chromatin condensation during mitosis has also been extensively studied [45]. We therefore used H3 phosphorylation as an index for mitosis. We found that Aβ25-35 time-dependently enhanced phosphorylation of histone H3 at Ser-10 during 16–40 h of exposure time (Fig. 7a). Double immunofluorescence confocal microscopy revealed that the phosphor-H3 histone was localized in the MAP-2+ cortical neurons (Fig. 7b). Knockdown of Id1 by siRNA significantly suppressed Aβ25-35-induced phosphorylation of histone H3 (Fig. 7c), whereas exogenous Id1-Tag protein was sufficient to enhance H3 phosphorylation (Fig. 7d). Consistently, SHH-Ab (Fig. 7e), but not cyclopamine (Fig. 7f), abolished Aβ25-35-mediated H3 phosphorylation; furthermore, exogenous SHH-N dose-dependently enhanced the extents of H3 histone phosphorylation independent of Aβ25-35 (Fig. 7g). As expected, PD0332991 also abolished both endogenous as well as the Aβ25-35-induced H3 phosphorylation at Ser-10 (Fig. 7h). These results together confirm the crucial roles of Id1/SHH in mediating Aβ-induced entry into M phase in the post-mitotic cortical neurons.


Aβ-induced phosphorylation of histone H3 at Ser-10 in the post-mitotic neurons is mediated by Id1 and SHH. a Primary cortical cultures were treated with 10 μM Aβ25-35 for indicated times before detection of phosphor-histone H3 at Ser-10 (p-H3). Mean ± SEM from N = 4. * denotes P < 0.05 compared with the control cultures without Aβ25-35 treatment. b Primary cortical cultures were treated with 10 μM Aβ25-35 for 40 h before immunostaining with antibodies against p-H3 (S10; green) and MAP-2 (red); Hoechst staining (blue) served as counterstain. In addition to phosphor-H3 at Ser-10, control cultures were also stained with the antibody against total histone H3 to demonstrate that lack of immune-positive signal for p-H3 (S10) was not due to low expression levels of histone H3. Scale bars = 50 μm in the upper panels and 10 μm in the lower panels. Note that Aβ25-35 enhances p-H3 expression in the MAP-2-positive cells, indicative of its neuronal expression as indicated by the white arrow. c Cortical cultures were transfected with sc siRNA or Id1 siRNA for 72 h, and then exposed to 10 μM Aβ25-35 for an additional 24 h before the detection of p-H3 by Western blotting. Mean ± SEM from N = 3. *, #, §, and Ψ all denote P < 0.05. d Primary cortical cultures were treated with or without 4 μg/ml Id1-Tag for 16 h before detection of p-H3. Mean ± SEM from N = 3. * denotes P < 0.05. e Cortical cells were treated with 10 μM Aβ25-35 with 2 μg/ml mouse neutralization antibody against SHH (SHH-Ab) or the same amounts of normal mouse IgG for 24 h before detection of p-H3 expression. Mean ± SEM from N = 4. * and # denote P < 0.05. The “ns” denotes “not significant.” f Cortical cells were treated with 10 μM Aβ25-35 in the presence of 20 μM cyclopamine (CPM) or the same amount of solvent DMSO for 24 h before detection of p-H3 expression. Mean ± SEM from N = 5. * denotes P < 0.05. The “ns” denotes “not significant.” g Primary cortical cultures were treated with recombinant SHH-N at indicated concentrations for 24 h before detection of p-H3. Mean ± SEM from N = 3. * denotes P < 0.05 compared with the control cultures without SHH-N. h Cortical cells were treated with 10 μM Aβ25-35 with 1 μM PD0332991 for 24 h before detection of p-H3 expression. Mean ± SEM from N = 3. * and # denote P < 0.05. The “ns” denotes “not significant”
Roles of Id1/SHH on Aβ25-35-Induced Caspase-3 Cleavage in Post-Mitotic Neurons
Apoptotic programs may be initiated following cell cycle reentry that contributes to neuronal demise. Consistent with its pro-apoptotic action, Aβ25-35 time-dependently induce cleavage of caspase-3, reaching statistical significance at 16–48 h (Fig. 8a). Aβ25-35-mediated caspase-3 cleavage was partially, but significantly, suppressed by Id1 knockdown (Fig. 8b). Given the notion that failed attempts for cell cycle reentry in the post-mitotic neurons result in apoptosis, it is predicted that cortical neurons in the S phase, hence positively labeled with BrdU, should undergo apoptosis with heightened caspase-3 cleavage. In our study, we found that Aβ25-35 increased the numbers of cells positively stained with both cleaved caspase-3 and BrdU, whereas Id1 knockdown by its siRNA significantly inhibited this Aβ25-35 effect (Fig. 7c). Similar to Id1 siRNA, the SHH-Ab also attenuated Aβ25-35-mediated caspase-3 cleavage in the differentiated cortical neurons (Fig. 7d). Consistent with its lack of effects on Aβ25-35-dependent induction of PCNA (Fig. 5f), cyclopamine again failed to block Aβ25-35-induced caspase-3 cleavage (Fig. 8e). As expected, the CDK4/6 inhibitor PD0332991 in part abrogated Aβ25-35-mediated caspase-3 cleavage (Fig. 7f). Given the notion that SHH contributes to Aβ-dependent neuronal cell cycle reentry and subsequent apoptosis, it is curious to test whether exogenous SHH-N may by itself induce caspase-3 cleavage. We found that, at the highest dosage of 300 ng/ml during a 48-h exposure, SHH-N indeed was capable of inducing caspase-3 cleavage in the cultured cortical neurons (data not shown). Together, these results indicate that Aβ25-35-induced Id1 and SHH contribute to caspase-3-dependent apoptosis in the fully differentiated primary cortical neurons.

Aβ-induced cleavage, and hence activation, of caspase-3 in the post-mitotic neurons is mediated by Id1 and SHH. a Primary cortical cultures were incubated with 10 μM Aβ25-35 for indicated times before detection of cleaved caspase-3 and pro-caspase-3 expression by Western blotting. Mean ± SEM from N = 4. * denotes P < 0.05 compared with the control cultures without Aβ25-35 treatment. b Cortical cells were transfected with Id1 siRNA or scrambled siRNA for 72 h. This was followed by exposure to 10 μM Aβ25-35 for an additional 24 h before detection of cleaved caspase-3 and pro-caspase-3. Mean ± SEM from N = 3. *, #, and § all denote P < 0.05. c Cortical cultures were transfected with sc siRNA or Id1 siRNA for 72 h followed by exposure to 10 μM Aβ25-35 and 10 μM BrdU for an additional 16 h before immunostaining with the antibodies against the cleaved caspase-3 and BrdU. The numbers of cleaved caspase-3+/BrdU+ cells were counted and calculated. Mean ± SEM from N = 3. * and # denote P < 0.05. d Cortical cells were treated with 10 μM Aβ25-35 with 2 μg/ml SHH neutralization antibody (SHH-Ab) or normal rabbit IgG for 24 h before detection of cleaved caspase-3 and pro-caspase-3. Mean ± SEM from N = 3. * and # denote P < 0.05. The “ns” denotes “not significant.” e Primary cortical cultures were treated with 10 μM Aβ25-35 in the presence of 20 μM cyclopamine (CPM) or the same amount of solvent DMSO for 24 h before detection of cleaved caspase-3 and pro-caspase-3. Mean ± SEM from N = 3. * denotes P < 0.05. The “ns” denotes “not significant.” f Primary cortical cultures were treated with 10 μM Aβ25-35 with 1 μM PD0332991 for 24 h before detection of cleaved caspase-3 and pro-caspase-3. Mean ± SEM from N = 3. *, #, and § all denote P < 0.05
Discussion
Potential pathogenic roles of cell cycle reentry in the fully differentiated post-mitotic neurons of the AD brains have long been speculated, but the detailed underlying mechanisms remain to be fully disclosed. Earlier, we have reported that Aβs, the neurotoxic peptides constituting senile plaques in the AD brains, may induce the expression of Id1, which leads to activation of HIF-1 and subsequent expression of SHH [9]. Extended from these findings, herein we provided experimental evidence supporting the crucial roles of Id1 and SHH in mediating Aβ-induced cell cycle reentry and caspase-dependent apoptosis. Using cyclin D1 and phosphorylated pRb as the G1-phase markers (Fig. 4), PCNA (Fig. 5) and BrdU incorporation into the de novo synthesized DNA (Fig. 6) as the S-phase markers, the histone H3 phosphorylated at Ser-10 as the M-phase marker (Fig. 7), as well as caspase-3 cleavage as the apoptotic marker (Fig. 8), we firmly establish the notion that Aβ-induced Id1 and SHH contribute to cell cycle reentry and subsequent caspase-dependent apoptosis in the fully differentiated post-mitotic neurons, as depicted in Fig. 9.

A diagram depicting the proposed signal transduction pathways underlying Aβ25-35-dependent neuronal cell cycle reentry and subsequent apoptosis through Id1 and SHH expression in the fully differentiated post-mitotic cortical neurons
As shown in Fig. 1c, Aβ25-35 induced the expression of Id1 in the MAP-2+ cortical neurons. Interestingly, both proteins seem to primarily co-localize at the peri-nuclear region (Fig. 1c). Since small proteins with molecular weights of less than 40 kDa can freely pass through the nuclear pore [46], the molecular weights of Id proteins ranging from 13 to 18 kDa suggest the involvement of passive diffusion in determining the subcellular distribution of these proteins. In addition, a nuclear export signal (NES) has been identified in Id1 [47]. Thus, given its nature as a bHLH protein to negatively regulate gene expression as well as the identified NES, Id1 is expected to be localized in both nucleus and cytoplasm. MAP-2, on the other hand, is a microtubule-associated protein commonly used as a cytoplasmic marker for mature neurons. Therefore, it is not surprising that both Id1 and MAP-2 can be co-localized in the cytoplasm. Furthermore, the cortical neurons treated with Aβ25-35 were less healthy; in addition to the fragmented neurites, the MAP-2 signal tended to be weaker and the MAP-2+ somata appeared to be slightly “shrunk” as compared to the control neurons, making Id1 and MAP-2 appeared to co-localize at the peri-nuclear region.
The aggregation states of Aβs may have a significant impact on interpretation of the experimental results. The Aβ25-35 used in this study was in the fibrillary form. Although Aβ1-42 was not included for the studies of cell cycle reentry, Aβ1-42 is capable of inducing Id1 (Fig. 1b) and SHH [9]. In an earlier study, we showed that the protocols for Aβ1-42 aggregation at 4 °C result in the formation of oligomers, mainly trimers and tetramers, as detected by Western blotting using the mouse monoclonal antibody 6E10 against Aβ1-16 [29]. Since oligomeric Aβ1-42 may induce Id1/SHH in primary cortical neurons, we speculated that Id1/SHH may also contribute to neuronal cell cycle reentry in cortical neurons exposed to oligomeric Aβ1-42, although further experiments are required to firmly establish this notion. Nevertheless, if this is correct, it would be intriguing to speculate that aberrant cell cycle reentry in the post-mitotic neurons may begin early in the disease stages, such as in the mild cognitive impairment (MCI) patients, prior to the accumulation of senile plaque in the AD patients at later, more advanced stages. Indeed, the reemergence of cell cycle proteins in brains as these patients progress from early stages of MCI into AD has been reported; the levels of key cell cycle proteins, such as CDK2, CDK5, and cyclin G1, have all been found to be elevated in MCI brain compared to age-matched control [48]. Detailed correlation between the pathologies and the extents of neuronal cell cycle reentry in MCI patients requires more clinical studies.
In the present work, we attempted to block the biological functions of SHH by its neutralization antibody (SHH-Ab) and the Smoothened inhibitor cyclopamine. Unexpectedly, we found that only the SHH-Ab, but not cyclopamine, effectively suppressed Aβ25-35-induced markers for cell cycle reentry, including PCNA (Fig. 5) and p-H3 (Fig. 7), as well as caspase-3 cleavage (Fig. 8) in the post-mitotic neurons. It should be noted that cyclopamine is an inhibitor of Smoothened receptor [21], thereby suppressing the canonical Smoothened/Gli signaling cascades triggered by SHH. Our findings thus appear to exclude the involvements of Smoothened receptor downstream of SHH and suggest possible contributions of noncanonical pathways. While the detailed underlying mechanisms remain to be investigated, it has been demonstrated that the large intracellular loop of human Patched1 interacts specifically with the constitutively phosphorylated cyclin B1 to block its nuclear translocation and cell cycle progression in basal cell carcinoma; furthermore, the addition of SHH disrupts this interaction and allows nuclear localization of cyclin B1 and cell cycle progression [49, 50]. Therefore, the intriguing possibility that Aβ-induced SHH binds to Patched1, thereby relieving the inhibitory binding of Patched1 to the phosphorylated cyclin B1 with subsequent cell cycle progression at M phase in the post-mitotic neurons, cannot be excluded. As the next step to approach this issue, we will knock down Patched1 in primary cortical neurons via siRNA, as has been performed in the present study, to test whether Patched1 plays a critical role contributing to SHH-mediated neuronal cell cycle reentry in the cortical cultures exposed to Aβ. Furthermore, we will also study the physical interaction between Patched1 and cyclin B1 via co-immunoprecipitation, which will be followed by knockdown of the cyclin B1 to confirm its roles in neuronal cell cycle reentry. The Patched1/cyclin B1 hypothesis, however, still cannot explain the observation that Aβ25-35-induced S-phase marker PCNA was not suppressed by cyclopamine (Fig. 5f), considering that S phase precedes M phase. Indeed, cyclopamine slightly enhanced Aβ-mediated PCNA induction with statistical significance (Fig. 5f), but not p-H3 (Fig. 7f) or caspase-3 cleavage (Fig. 8e). The biological significance of these findings remains to be further explored.
Cell cycle reentry may result in various types of cell death in the fully differentiated post-mitotic neurons. In the current study, we found that interference with Id1 (Fig. 8b) and SHH (Fig. 8d) only in part suppressed Aβ-induced caspase-3 cleavage, which may be attributed to the fact that Id1 siRNA and SHH-Ab can only partially inhibit Id1 expression and SHH activity, respectively. Therefore, it is difficult to conclude whether Id1/SHH-mediated cell cycle reentry triggers caspase-dependent apoptosis only or it also induces necrosis or other types of programmed cell death. At least in the unregulated necrosis involving passive processes like tissue trauma, we believed that Aβ-induced Id1/SHH may not play a significant role. This is because unregulated necrosis often involves cellular energy depletion, whereas in the Aβ-treated cortical neurons, energy-dependent de novo synthesis of Id1 and SHH proteins still occurs. Among various types of regulated necrosis, necroptosis, the best characterized form, is defined as necrotic cell death involving Receptor-Interacting Kinase 1 (RIP1), RIP3, and Mixed Lineage Kinase Domain-like (MLKL) [51]. Recently, necroptosis has been shown to be activated in AD [52]. Whether Id1/SHH may be connected to necroptosis is a very interesting issue that requires further investigation. Another form of neuronal cell death is “mitotic catastrophe,” which is a sequence of events that results from premature or inappropriate entry of cells into mitosis upon exposure to various stresses, including the agents influencing stability of microtubules and mitotic failure caused by defective cell cycle checkpoints [53]. It has been demonstrated that aberrant reexpression of the mitotic kinase complex CDK1/cyclin B1 in AD neurons with neurofibrillary tangles contributes to the generation of phosphorylated tau proteins [54]. The in vivo evidence also suggested that expression of cyclin B1 and phosphorylated tau in adult neurons was significantly increased with co-localization to each other after injection of okadaic acid (OA), a phosphatase inhibitor capable of inducing AD-like pathology, into rat frontal cortex; furthermore, immunohistochemical staining revealed irregular arrangement of condensed chromatin and chromosome fibers accompanied by cyclin B1 expression in neurons, suggesting that cyclin B1 may contribute to the mitotic catastrophe in differentiated neurons [55]. At present, whether Id1 and SHH are directly involved in mitotic catastrophe remains unknown. As discussed above, we speculated that binding of SHH to Patched1 may relieve its physical interaction with cyclin B1, thereby contributing to neuronal cell cycle reentry independent of Smoothened. If this contention is correct, it is likely that cyclin B1, and perhaps CDK1, may be activated to contribute to mitotic catastrophe. However, such a speculation needs further experimental evidence for confirmation.
The correlation between cell cycle reentry and AD has previously been studied [3]. In post-mortem human subjects, cell cycle markers like cyclin [56, 57] and Ki67 [57] have been detected in the hippocampal neurons of AD brains. Several earlier studies have reported that Aβ may induce cell cycle reentry in vitro. For example, Aβ increased the expression of pRb/p107 protein in primary cortical cultures from E18 rat fetal brain cortices [58]. Aβ1-42 oligomer induced co-localization of MAP-2+/BrdU+ cells in mouse primary cortical cultures from E16.5 [5]. Furthermore, in E15-16 rat primary cortical cultures, Aβ1-40 stimulated the expression of CDK4, pRb, and PCNA protein via CDK5 [59]. Despite these reports, how Aβ may reactivate cell cycle progression in the fully differentiated quiescent neurons remains to be fully investigated. In this study, we provide the first evidence supporting involvement of Id1 and SHH in Aβ-mediated cell cycle reentry and neuronal apoptosis. In addition to the effects of exogenous Aβ in stimulating the expression of cell cycle-dependent marker proteins in post-mitotic neurons indicative of cell cycle reentry, several studies have also reported that forced expression of oncogenes in post-mitotic neurons is sufficient to reactivate cell cycle progression leading to accumulation of phosphor-tau and Aβ deposition. For example, in mice conditionally expressing simian virus 40 T antigen (SV40T) in neurons, Alzheimer-like tau and amyloid pathology can be observed [60]. Furthermore, adenovirus-mediated overexpression of c-myc and ras oncogenes in post-mitotic neurons increased BrdU+ and cyclin B1+ cells, contents of phosphor-tau, and markers of early AD pathology in E18 rat primary cortical cultures [61]. These findings appear to place cell cycle events upstream of AD pathology, in direct contrast to our finding that Aβs act upstream of cell cycle reentry via induction of Id1 and subsequently SHH with resultant neuronal death. While additional studies are needed to further clarify the potential roles among AD pathology and cell cycle reactivation, we cannot exclude the possibility that both scenarios are correct resulting in the occurrence of a vicious cycle between AD pathology and cell cycle reactivation.
AD is an age-dependent neurodegenerative disorder. In the present study, primary neurons derived from fetal rat cortices were used to study the molecular mechanisms underlying Aβ-induced neuronal cell cycle reentry, which may raise the concern that whether the observed findings can actually be duplicated in clinical settings. Although this issue was not addressed in the present work, in our earlier publication, we have demonstrated the expression of SHH and Id1 in the aged (12 months old) AD transgenic mouse brains [9]. Furthermore, primary culture systems derived from fetal brains have previously been used to study Aβ-induced reactivation of cell cycle progression in both rats and mice [5, 58, 59]. In our cortical culture system, two neuronal markers, namely NeuN and MAP-2, were adopted. NeuN is expressed at both early and late post-mitotic maturation phases [62, 63], whereas MAP-2 is a marker protein for mature neurons that can be detected in dendrites and soma [64]. In the present work, we demonstrated that Aβ25-35-induced cyclin D1 (Fig. 4b), phosphor-pRb (Fig. 4c), PCNA (Fig. 5b), and p-H3 (Fig. 7b) were all localized in the MAP-2+ cortical neurons with extensive dendritic structures; furthermore, BrdU signal was also co-localized to the NeuN+ cortical neurons (Fig. 6a, c, e, g), confirming that these in vitro differentiated embryonic neurons are, at least to a certain extent, post-mitotic. Ultimately, the inherent concern of using primary embryonic neurons to addressing Aβ-induced cell cycle reactivation can only be resolved with AD transgenic mice model in vivo.
Another issue using BrdU labeling as an index for cell cycle progression is that Aβ treatment may induce DNA damage leading to incorporation of BrdU during DNA repair in addition to de novo DNA synthesis. This is, however, unlikely because, at least in the in vivo animal model, BrdU is not significantly incorporated during DNA repair and that labeling is not detected in vulnerable or dying post-mitotic neurons, even when a high dose of BrdU is directly infused into the brain [65]. In this work, Aβ25-35-induced increases in the numbers of NeuN+/BrdU+ cells were decreased by siRNA-mediated Id1 knockdown (Fig. 6b), the neutralization antibody against SHH (Fig. 6e, f), and the CDK4/6 inhibitor PD0332991 (Fig. 6i). It should be noted that Id1, SHH, and CDK4/6 are all involved in cell cycle progression rather than DNA damage/repair; furthermore, both Id1-Tag (Fig. 6c, d) and SHH-N (Fig. 6g, h) alone were sufficient to increase the numbers of NeuN+/BrdU+ cells independent of Aβ, which is unlikely to be caused by repairing DNA damage. In addition to BrdU, Aβ induction of another S-phase marker PCNA was also suppressed by Id1 siRNA (Fig. 5c) and SHH-Ab (Fig. 5e). These findings, coupled with previous reports [5, 59], suggest that the observed NeuN+/BrdU+ cells indeed represent the neurons attempting to reactivate cell cycle into S phase with DNA synthesis, rather than cells undergoing DNA damage/repair processes.
In conclusion, we demonstrated for the first time that Aβ-induced expression of Id1 and SHH, two regulators involved in cell cycle progression, may together contribute to cell cycle reentry resulting in subsequent neuronal cell death in the fully differentiated post-mitotic neurons, thus providing a novel mechanism underlying Aβ-mediated dysregulation in cell cycle progression.
References
Folch J, Junyent F, Verdaguer E, Auladell C, Pizarro JG, Beas-Zarate C, Pallas M, Camins A (2012) Role of cell cycle re-entry in neurons: a common apoptotic mechanism of neuronal cell death. Neurotox Res 22(3):195–207
Wang W, Bu B, Xie M, Zhang M, Yu Z, Tao D (2009) Neural cell cycle dysregulation and central nervous system diseases. Prog Neurobiol 89(1):1–17
Lee HG, Casadesus G, Zhu X, Castellani RJ, McShea A, Perry G, Petersen RB, Bajic V et al (2009) Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer’s disease. Neurochem Int 54(2):84–88
O’Brien RJ, Wong PC (2011) Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci 34:185–204
Varvel NH, Bhaskar K, Patil AR, Pimplikar SW, Herrup K, Lamb BT (2008) Abeta oligomers induce neuronal cell cycle events in Alzheimer’s disease. J Neurosci 28(43):10786–10793
Demir O, Singh S, Klimaschewski L, Kurnaz IA (2009) From birth till death: neurogenesis, cell cycle, and neurodegeneration. Anat Rec (Hoboken) 292(12):1953–1961
Bonda DJ, Bajic VP, Spremo-Potparevic B, Casadesus G, Zhu X, Smith MA, Lee HG (2010) Review: Cell cycle aberrations and neurodegeneration. Neuropathol Appl Neurobiol 36(2):157–163
Malik B, Currais A, Andres A, Towlson C, Pitsi D, Nunes A, Niblock M, Cooper J et al (2008) Loss of neuronal cell cycle control as a mechanism of neurodegeneration in the presenilin-1 Alzheimer's disease brain. Cell Cycle 7(5):637–646
Hung YH, Chang SH, Huang CT, Yin JH, Hwang CS, Yang LY, Yang DI (2016) Inhibitor of differentiation-1 and hypoxia-inducible factor-1 mediate sonic hedgehog induction by amyloid beta-peptide in rat cortical neurons. Mol Neurobiol 53(2):793–809
Pagliuca A, Bartoli PC, Saccone S, Della Valle G, Lania L (1995) Molecular cloning of ID4, a novel dominant negative helix-loop-helix human gene on chromosome 6p21.3-p22. Genomics 27(1):200–203
Mathew S, Chen W, Murty VV, Benezra R, Chaganti RS (1995) Chromosomal assignment of human ID1 and ID2 genes. Genomics 30(2):385–387
Deed RW, Hirose T, Mitchell EL, Santibanez-Koref MF, Norton JD (1994) Structural organisation and chromosomal mapping of the human Id-3 gene. Gene 151(1–2):309–314
Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H (1990) The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell 61(1):49–59
Norton JD (2000) ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci 113(Pt 22):3897–3905
Nusslein-Volhard C, Wieschaus E (1980) Mutations affecting segment number and polarity in Drosophila. Nature 287(5785):795–801
Mohler J (1988) Requirements for hedgehod, a segmental polarity gene, in patterning larval and adult cuticle of Drosophila. Genetics 120(4):1061–1072
Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP (1993) Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75(7):1417–1430
Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, Scott MP, Pennica D et al (1996) The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 384(6605):129–134
Alcedo J, Ayzenzon M, Von Ohlen T, Noll M, Hooper JE (1996) The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell 86(2):221–232
Riobo NA, Manning DR (2007) Pathways of signal transduction employed by vertebrate Hedgehogs. Biochem J 403(3):369–379
Chen JK, Taipale J, Cooper MK, Beachy PA (2002) Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev 16(21):2743–2748
Kenney AM, Cole MD, Rowitch DH (2003) Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 130(1):15–28
Leung C, Lingbeek M, Shakhova O, Liu J, Tanger E, Saremaslani P, Van Lohuizen M, Marino S (2004) Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 428(6980):337–341
Kenney AM, Rowitch DH (2000) Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol 20(23):9055–9067
Ling F, Kang B, Sun XH (2014) Id proteins: small molecules, mighty regulators. Curr Top Dev Biol 110:189–216
Zebedee Z, Hara E (2001) Id proteins in cell cycle control and cellular senescence. Oncogene 20(58):8317–8325
Chaudhary J, Sadler-Riggleman I, Ague JM, Skinner MK (2005) The helix-loop-helix inhibitor of differentiation (ID) proteins induce post-mitotic terminally differentiated Sertoli cells to re-enter the cell cycle and proliferate. Biol Reprod 72(5):1205–1217
Fuccillo M, Joyner AL, Fishell G (2006) Morphogen to mitogen: the multiple roles of hedgehog signalling in vertebrate neural development. Nat Rev Neurosci 7(10):772–783
Chen SD, Wu CL, Lin TK, Chuang YC, Yang DI (2012) Renin inhibitor aliskiren exerts neuroprotection against amyloid beta-peptide toxicity in rat cortical neurons. Neurochem Int 61(3):369–377
Stine WB Jr, Dahlgren KN, Krafft GA, LaDu MJ (2003) In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem 278(13):11612–11622
Ju TC, Yang YT, Yang DI (2004) Protective effects of S-nitrosoglutathione against neurotoxicity of 3-nitropropionic acid in rat. Neurosci Lett 362(3):226–231
Ju TC, Chen SD, Liu CC, Yang DI (2005) Protective effects of S-nitrosoglutathione against amyloid beta-peptide neurotoxicity. Free Radic Biol Med 38(7):938–949
Chang SH, Hwang CS, Yin JH, Chen SD, Yang DI (2015) Oncostatin M-dependent Mcl-1 induction mediated by JAK1/2-STAT1/3 and CREB contributes to bioenergetic improvements and protective effects against mitochondrial dysfunction in cortical neurons. Biochim Biophys Acta - Mol Cell Res 1853(10 Pt A):2306–2325
Wu CL, Hwang CS, Yang DI (2009) Protective effects of brain-derived neurotrophic factor against neurotoxicity of 3-nitropropionic acid in rat cortical neurons. Neurotoxicology 30(4):718–726
Wu CL, Chen SD, Yin JH, Hwang CS, Yang DI (2010) Erythropoietin and sonic hedgehog mediate the neuroprotective effects of brain-derived neurotrophic factor against mitochondrial inhibition. Neurobiol Dis 40(1):146–154
Wu CL, Yin JH, Hwang CS, Chen SD, Yang DY, Yang DI (2012) c-Jun-dependent sulfiredoxin induction mediates BDNF protection against mitochondrial inhibition in rat cortical neurons. Neurobiol Dis 46(2):450–462
McAllister SD, Christian RT, Horowitz MP, Garcia A, Desprez PY (2007) Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol Cancer Ther 6(11):2921–2927
Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X et al (2004) Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 3(11):1427–1438
Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, Ozawa T, James CD et al (2010) Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res 70(8):3228–3238
Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, Vanderkerken K, Chen-Kiang S (2008) A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res 68(14):5519–5523
Celis JE, Madsen P, Celis A, Nielsen HV, Gesser B (1987) Cyclin (PCNA, auxiliary protein of DNA polymerase delta) is a central component of the pathway(s) leading to DNA replication and cell division. FEBS Lett 220(1):1–7
van Dierendonck JH, Wijsman JH, Keijzer R, van de Velde CJ, Cornelisse CJ (1991) Cell-cycle-related staining patterns of anti-proliferating cell nuclear antigen monoclonal antibodies. Comparison with BrdUrd labeling and Ki-67 staining. Am J Pathol 138(5):1165–1172
Duan W, Zhang YP, Hou Z, Huang C, Zhu H, Zhang CQ, Yin Q (2016) Novel insights into NeuN: from neuronal marker to splicing regulator. Mol Neurobiol 53(3):1637–1647
Nowak SJ, Corces VG (2004) Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet 20(4):214–220
Prigent C, Dimitrov S (2003) Phosphorylation of serine 10 in histone H3, what for? J Cell Sci 116(Pt 18):3677–3685
Gorlich D, Kutay U (1999) Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol 15:607–660
Makita J, Kurooka H, Mori K, Akagi Y, Yokota Y (2006) Identification of the nuclear export signal in the helix-loop-helix inhibitor Id1. FEBS Lett 580(7):1812–1816
Keeney JT, Swomley AM, Harris JL, Fiorini A, Mitov MI, Perluigi M, Sultana R, Butterfield DA (2012) Cell cycle proteins in brain in mild cognitive impairment: insights into progression to Alzheimer disease. Neurotox Res 22(3):220–230
Barnes EA, Heidtman KJ, Donoghue DJ (2005) Constitutive activation of the shh-ptc1 pathway by a patched1 mutation identified in BCC. Oncogene 24(5):902–915
Barnes EA, Kong M, Ollendorff V, Donoghue DJ (2001) Patched1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J 20(9):2214–2223
Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC (2018) Neuronal cell death. Physiol Rev 98(2):813–880
Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, Readhead B, Dudley JT et al (2017) Necroptosis activation in Alzheimer’s disease. Nat Neurosci 20(9):1236–1246
Vakifahmetoglu H, Olsson M, Zhivotovsky B (2008) Death through a tragedy: mitotic catastrophe. Cell Death Differ 15(7):1153–1162
Vincent I, Jicha G, Rosado M, Dickson DW (1997) Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci 17(10):3588–3598
Chen B, Cheng M, Hong DJ, Sun FY, Zhu CQ (2006) Okadaic acid induced cyclin B1 expression and mitotic catastrophe in rat cortex. Neurosci Lett 406(3):178–182
Nagy Z, Esiri MM, Cato AM, Smith AD (1997) Cell cycle markers in the hippocampus in Alzheimer’s disease. Acta Neuropathol 94(1):6–15
Nagy Z, Esiri MM, Smith AD (1997) Expression of cell division markers in the hippocampus in Alzheimer’s disease and other neurodegenerative conditions. Acta Neuropathol 93(3):294–300
Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS (1999) Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F x DP, in B-amyloid-induced neuronal death. J Biol Chem 274(27):19011–19016
Lopes JP, Oliveira CR, Agostinho P (2009) Cdk5 acts as a mediator of neuronal cell cycle re-entry triggered by amyloid-beta and prion peptides. Cell Cycle 8(1):97–104
Park KH, Hallows JL, Chakrabarty P, Davies P, Vincent I (2007) Conditional neuronal simian virus 40 T antigen expression induces Alzheimer-like tau and amyloid pathology in mice. J Neurosci 27(11):2969–2978
McShea A, Lee HG, Petersen RB, Casadesus G, Vincent I, Linford NJ, Funk JO, Shapiro RA et al (2007) Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim Biophys Acta 1772(4):467–472
Lazarov O, Mattson MP, Peterson DA, Pimplikar SW, van Praag H (2010) When neurogenesis encounters aging and disease. Trends Neurosci 33(12):569–579
Gage F, Kempermann G, Song H (2008) Adult neurogenesis. Cold Spring Harbor Laboratory Press, New York
Kalcheva N, Weidenheim KM, Kress Y, Shafit-Zagardo B (1997) Expression of microtubule-associated protein-2a and other novel microtubule-associated protein-2 transcripts in human fetal spinal cord. J Neurochem 68(1):383–391
Bauer S, Patterson PH (2005) The cell cycle-apoptosis connection revisited in the adult brain. J Cell Biol 171(4):641–650
Funding
This study was supported by the Ministry of Science and Technology in Taiwan (MOST 103-2314-B-010-013MY3, MOST 104-2314-B-010-014-MY2, and MOST 106-2314-B-010-018MY3 to Ding-I Yang; MOST 104-2314-B-037-029 and MOST 105-2314-B-037-002 to A-Ching Chao), Kaohsiung Medical University and Hospital (KMUH 103-3T16 and KMUH 104-4R53 to A-Ching Chao), and Department of Health in Taipei City Government (10501-62-050 and 10601-62-003 to Ding-I Yang and Wei-Chao Hwang).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chao, AC., Chen, CH., Chang, SH. et al. Id1 and Sonic Hedgehog Mediate Cell Cycle Reentry and Apoptosis Induced by Amyloid Beta-Peptide in Post-mitotic Cortical Neurons. Mol Neurobiol 56, 465–489 (2019). https://doi.org/10.1007/s12035-018-1098-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1098-5