
Abstract
The amyloid precursor protein (APP), one key player in Alzheimer’s disease (AD), is extensively processed by different proteases. This leads to the generation of diverging fragments including the amyloid β (Aβ) peptide, which accumulates in brains of AD patients. Subcellular trafficking of APP is an important aspect for its proteolytic conversion, since the various secretases which cleave APP are located in different cellular compartments. As a consequence, altered subcellular targeting of APP is thought to directly affect the degree to which Aβ is generated. The mechanisms underlying intracellular APP transport are critical to understand AD pathogenesis and can serve as a target for future pharmacological interventions. In the recent years, a number of APP interacting proteins were identified which are implicated in sorting of APP, thereby influencing APP processing at different angles of the secretory or endocytic pathway. This review provides an update on the proteolytic processing of APP and the interplay of the transmembrane proteins low-density lipoprotein receptor-related protein 1, sortilin-receptor with A-type repeats, SorCS1c, sortilin, and calsyntenin. We discuss the specific interactions with APP, the capacity to modulate the intracellular itinerary and the proteolytic conversion of APP, a possible involvement in the clearance of Aβ, and the implications of these transmembrane proteins in AD and other neurodegenerative diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cognitive and memory dysfunction, accompanied by hallmark pathologies such as intraneuronal neurofibrillary tangles and extracellular amyloid plaques. The latter are composed of a heterogenous population of proteolytically cleaved amyloid β peptides (Aβ). According to the amyloid hypothesis, accumulation of Aβ in the brain is a primary cause driving AD pathogenesis and reducing Aβ would ameliorate AD symptoms [1]. Although alternative hypotheses have been formulated, mounting genetic evidence strongly suggests that alterations of sequential proteolytic processing of the amyloid precursor protein (APP) have a significant impact on AD pathology [2]. APP is proteolytically processed at many positions. The respective enzymes reside in different subcellular locations. As a consequence, altered subcellular trafficking of APP is thought to directly affect the degree to which Aβ is generated [3]. Therefore, the mechanisms underlying intracellular APP transport are critical to understand AD pathogenesis and can serve as target for future pharmacological interventions.
APP is a type-I transmembrane protein with a large extracellular/luminal moiety and a short cytoplasmic domain. Intracellular sorting, targeting, and internalization of transmembrane proteins are mediated by signals, usually short linear sequences of amino acids, in the cytoplasmic domain which are recognized by cytosolic adaptor proteins. So far, only a limited number of proteins have been identified that interact with the APP cytosolic domain and that might direct APP targeting [4]. Notably, specific intracellular targeting events can occur independently of the APP intracellular domain, such as the anterograde axonal transport of APP [5, 6]. A number of type-I transmembrane proteins have been demonstrated to modulate the intracellular itinerary of APP, its proteolytic processing and/or clearance of Aβ. Here, we review the proteolytic processing and intracellular transport of APP and summarize current evidence suggesting that transport and processing of APP is modulated by other transmembrane proteins.
Processing of APP
APP is processed by sequential proteolytic cleavages. The executing enzymes and their subcellular localization have been described, but the exact modalities and the functional meaning of the different processing steps are still not fully delineated.
The canonical processing of APP is described by the amyloidogenic and the non-amyloidogenic pathway (Fig. 1a). The latter begins with cleavage of full-length APP by α-secretase activity, which is mainly mediated by ADAM10 (A Disintegrin And Metalloprotease 10) in neurons, but also ADAM17 has been implicated in this cleavage step [7,8,9]. Cleavage by α-secretase results in the release of the ectodomain of APP—soluble APPα (sAPPα)—and the concomitant generation of a membrane retained C-terminal fragment (CTF) consisting of 83 amino acids (aa) (C83 or α-CTF; reviewed in [10]). C83 is further processed by γ-secretase, a transmembrane multiprotein complex consisting of four subunits: presenilin 1 or 2, nicastrin, APH-1, and PEN-2. y-Secretase cleaves within the transmembrane domain, a mechanism termed “regulated intramembrane proteolysis” (RIP; reviewed in [11]). APP CTFs are proteolytically processed at three positions: first at the ε-cleavage site, then the ζ-cleavage site, and finally, at the γ-cleavage site [12,13,14,15]. This leads to the release of a short peptide termed “p3” and of the APP intracellular domain (AICD) [16]. Instead of α-/γ-secretase processing, APP can also be cleaved in the amyloidogenic pathway (Fig. 1a). Here, it is first cleaved N-terminally of the Aβ sequence by the β-secretase β-site APP cleaving enzyme 1 (BACE1) [17, 18]. This results in shedding of the APP ectodomain—soluble APPβ (sAPPβ)—and the production of a 99 aa CTF (C99 or β-CTF) [19]. C99 is subsequently cleaved by γ-secretase, releasing the Aβ peptide and AICD [13, 14]. Approximately 90% of the secreted Aβ peptides are 40 aa long (Aβ40) [20]. The second most common species of Aβ, Aβ42, is more prone to aggregation than Aβ40 [21]. Aggregation of Aβ to oligomeric forms and finally so-called amyloid plaques has been proposed to be the initial step in the development of AD [22]. Thus, a shift in the ratio of Aβ40 to Aβ42 towards Aβ42 is most likely explained by a decrease of γ-secretase cleavage site specificity [23, 24], which affects the age of onset of AD [25]. ε-Cleavage occurs mainly at the carboxyl side of leucine49 of the Aβ sequence whereas cleavage at threonine48 is not as frequent [26]. Those are the starting points for two main product lines of γ-secretase cleavage. ε-Cleavage producing Aβ49, is followed by proteolytic conversion into Aβ46 (ζ-cleavage) [12] and Aβ43, finally ending in secretion of Aβ40 while ε-Cleavage generating Aβ48 is further cleaved into Aβ45 and finally Aβ42 and Aβ38.

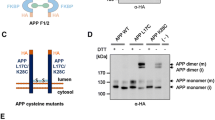
Schematic representation of different APP processing pathways. a The schematic shows the canonical APP processing pathways and the pictures (b–d) show the non-canonical pathways. a The amyloid precursor protein (APP) is first cleaved by α-secretase in the non-amyloidogenic pathway (gray) within the amyloid-β (Aβ) region (shown in red) to liberate sAPPα. The remaining α-C-terminal fragment (α-CTF) is further cleaved by γ-secretase (yellow) and releases the small 3-kDa peptide p3 and the APP intracellular domain (AICD). The amyloidogenic pathway (pink) starts with β-secretase cleavage, which liberates sAPPβ and concomitantly generates the β-C-terminal fragment (β-CTF). γ-Secretase cleavage (yellow) of the β-CTF releases the Aβ peptide, which can oligomerize and liberates the APP intracellular domain at the ε-cleavage site (AICD). b Cleavage by η-secretase gives rise to the 80–95 kDa soluble APPsη and CTFη, which is further processed by α- or β-secretase to generate Aη-α or Aη-β. c δ-Secretase generates three soluble APPsδ fragments and C-terminal fragment-δ (CTFδ), which is further cleaved by β-secretase and γ-secretase. d Meprinβ cleaves APP at positions 1, 2, or 3 of the Aβ sequence. Three additional Meprinβ cleavage sites have been identified further distal in the ectodomain of APP between Ala124/Asp125, Glu380/Thr381, and Gly383/Asp384. The indicated numbers refer to full-length APP695
Recently, three different non-canonical processing pathways were described, which start with cleavages N-terminally located to the β-secretase cleavage site (reviewed in [27]).
(I) APP can first be cleaved at the recently identified η (eta)-cleavage site 504/505 by matrix metalloproteinase 5 (MT5), also named matrix metallopeptidase 24 (MMP24) [28,29,30,31] (Fig. 1b). Subsequently, β- or α-secretase generate two peptides of 92 or 108 amino acids in length, designated as Aη-β or Aη-α peptides, respectively [28]. MT5 is a zinc-dependent metalloprotease-like ADAM10 [32] and belongs to the group of membrane type MMPs, which are mainly located at the cell surface [33] (Fig. 2). Those type-I transmembrane MMPs are activated after furin cleavage in the trans-Golgi network (TGN) and therefore presumably active at the plasma membrane [34]. MT5 is predominantly expressed in brain tissue and might play a role in remodeling extracellular matrices during development [35].

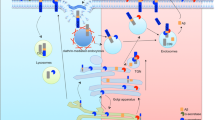
APP trafficking and processing. APP is transported in the secretory pathway (gray) from the ER to the plasma membrane via the Golgi apparatus, where it is mainly localized. APP is internalized from the plasma membrane to early endosomes. From there, it can either enter the recycling pathway (yellow) or travel back to the TGN in a retromer-mediated pathway, or can be targeted to late endosomes which fuse to lysosomes where APP is degraded (orange). APP cleavage by η-secretase or Meprinβ mainly takes place at the plasma membrane as well as non-amyloidogenic processing by α- and γ-secretase. Amyloidogenic processing of APP by β- and γ-secretase is predominantly carried out in early and late endosomes and cleavage of δ-secretase in lysosomes
(II) Three different cleavages summarized as δ-cleavage are carried out by asparagine endopeptidase (AEP), a pH-dependent soluble cysteine protease, which is active in lysosomes (Fig. 2). Cleavage of APP at Thr584 was first described in [36]; later, two additional sites were found in the ectodomain at positions N373 and N585 (APP695 numbering) (Fig. 1c) [37].
(III) APP can also be shed by Meprinβ at the cell surface, thereby competing with ADAM10 for the substrate (Fig. 2) [38, 39]. Meprinβ is a zinc-dependent metalloprotease-like [40] ADAM10 and MT5 and cleaves APP at positions 1, 2, or 3 of the Aβ sequence [41, 42]. Three additional Meprinβ cleavage sites have been identified further distal in the ectodomain of APP between Ala124/Asp125, Glu380/Thr381, and Gly383/Asp384 (Fig. 1d) [42].
Canonical and non-canonical processing of APP is mediated by enzymes with proteolytic activities at varying subcellular localizations. Depending on its subunit composition, the subcellular localization of the γ-secretase complex differs. A γ-secretase complex comprising presenilin 1 is distributed in the secretory and endocytic pathway and is concentrated at the cell surface, whereas a complex formed with presenilin 2 predominates in late endosomal and lysosomal compartments [43, 44]. While α-secretase, MT5, and Meprinβ activity is mainly localized to the cellular surface [34, 38], β-secretase activity is predominantly found in endosomal compartments and δ-cleavage occurs in lysosomes (Fig. 2) [8, 17, 37, 45, 46]. Therefore, processing of APP into amyloidogenic (Aβ) and non-amyloidogenic fragments is highly dependent on its intracellular itinerary. Consequently, altered subcellular targeting of APP directly affects the degree of Aβ generation. Thus, mistargeting of APP has been realized to cause amyloidogenic processing and a number of factors that are involved in APP subcellular targeting have been identified. Mutations in APP or in components of the γ-secretase complex lead to an increase in Aβ42 production and cause the rare early onset familial form of AD (EOAD). These genetic defects are usually not seen in the late onset form of AD (LOAD) accounting for 95% of all cases [47]. Mutations in factors that partake in APP subcellular targeting may underlie the complex pathology of LOAD. Accordingly, some of these factors discussed in this review have been genetically linked to LOAD.
Trafficking and Endocytosis of APP
The intracellular itinerary of APP has been extensively studied in undifferentiated cells [48]. After cleavage of its signal peptide in the ER, APP enters the secretory pathway and is translocated through the Golgi apparatus, where it is predominantly localized [49,50,51], to the plasma membrane (Fig. 2). Here, it can be processed or internalized. Following endocytosis, APP is targeted to early endosomes and then sorted to three different paths: (I) a subset of APP molecules undergoes recycling to the cell surface [48, 52], (II) a different fraction of APP is transported retrogradely from endosomes back to the TGN in a retromer-mediated pathway [53], and (III) some APP molecules are targeted to late endosomes which fuse with lysosomes where APP is degraded [54, 55]. APP is mainly endocytosed via clathrin-coated vesicles into early endosomes [4] and internalization of APP also depends on cholesterol [56, 57], suggesting an overlap of clathrin- and cholesterol-dependent endocytosis [57]. Thus, APP might be first clustered in cholesterol-rich-coated pits and then internalized into a specialized clathrin-dependent endocytic pathway.
Although, it has been shown that the highly conserved YENPTY motif in the C-terminus and possibly also the basolateral sorting signal of APP are essential for this process [58], the molecular machinery involved in clustering and targeting of APP to early endosomes is still unknown.
Anterograde Axonal Transport of APP
APP trafficking has also been studied in differentiated cells with a focus on neurons and axonal transport. APP moves along axons by fast anterograde transport as shown by nerve ligation [59] and pulse-chase labeling experiments [60, 61].
Live cell imaging studies documented fast axonal transport of vesicular APP distinct in morphology and transport kinetics from synaptophysin-containing vesicles [62]. Consistently, synaptophysin was not found in APP kinesin co-immunoisolations [6]. Gene-silencing experiments indicated that conventional kinesin is the main molecular motor involved in the anterograde transport of APP [59, 63, 64]. Conventional kinesin is a multimeric complex composed of two kinesin heavy chain (KHCs, kinesin-1s) and two kinesin light chain (KLCs) subunits [65]. In mammalian brain, three KHCs (kinesin-1a, b, and c) and two KLC (KLC1 and KLC2) isoforms are expressed, which exist in different combinations of kinesin-1 and KLC homodimers [66]. Kinesin-1C has been identified as the main KHC isoform associated with APP-containing transport vesicles [6]. The mode of interaction between conventional kinesin and APP-containing transport vesicles is still unclear. In general, the alternatively spliced carboxy-terminus of KLCs is assumed to mediate selective binding of conventional kinesin to different membrane-bound organelles [67,68,69], whereas the tandem repeat containing N-terminus can interact in vitro via hydrophobic patches non-specifically with different target proteins, including APP [70, 71]. So, it was shown that recombinant KLCs produced in bacteria interact non-specifically with proteins as diverse as GFP, Fe65, and PAT1a and the intracellular domain of APP, APLP1, and APLP2 in pull-down assays [71]. Also, indirect associations of APP to kinesin-1 via adaptor proteins, such as JIP1b were reported [72,73,74]. Current data showed that knockdown of JIP1b either specifically impairs anterograde transport [74], anterograde, and retrograde transport [75] or affects neither anterograde nor retrograde transport of APP [76]. Further, JIP1b APP binding studies revealed at least two interaction sites in KLC1 and/or KHC. Also, deletion mutants of JIP1b argue for multiple interaction sites for KLC1 [74]. In light of these inconsistencies and the increasing list of candidate protein-binding partners for both KIF5s and KLCs [70], the in vivo significance of JIP1b as an adaptor partner has not yet been rigorously established. Instead, indirect influences of JIP1b on activation of kinases that in turn might modulate kinesin-dependent transport should be taken into account.
Moreover, in contradiction to the model of direct or indirect interaction of the APP C-terminus with KLC1, APP is transported independently of its carboxy-terminus along the axon towards the presynaptic terminal [5, 6, 36, 77]. Thus, APP does not fulfill the criteria expected for a conventional kinesin receptor. Instead, APP most likely represents a cargo protein sorted into transport vesicles associated by a so far barely understood Rab3-dependent mechanism to kinesin-1c [6].
Modulators of Intracellular APP Transport
Intracellular transport and processing of APP can be modulated by other transmembrane proteins, such as low-density lipoprotein receptors (LDLRs), Vps10p-Doamin (Vps10p-D) receptors, and calsyntenins. LDLRs are mainly known as endocytic receptors for a wide variety of ligands, including lipid-carrying lipoproteins and proteases or protease inhibitors, but some family members also play a role in signal transduction and modulation [78,79,80]. Ligands endocytosed by LDLR family members are directed to endosomes and recycling or lysosomal compartments, while the receptors themselves are transported back to the plasma membrane [81, 82].
In mammals, there are seven core members of the LDLR family which vary in size and structure [80, 83]. All family members are type-I transmembrane proteins with large extracellular moieties and a short cytoplasmic domain. The extracellular domains consist of ligand-binding repeats (also named complement type repeats) and epidermal growth factor (EGF) homology domains (Fig. 3). The intracellular domain harbors at least one NPxY motif. Some family members contain additional structural elements that separate them from the core members. Sortilin-receptor with A-type repeats (SorLA) for example contains in addition a domain homologous to a sorting receptor for yeast vacuolar hydrolases that cycles between the TGN and endocytic compartments (Vps10p) [84]. SorLA is a unique mosaic receptor [85, 86] which combines structural features of the LDLR family by harboring EGF-type and ligand-binding repeats but presents as well the hallmark of the Vps10p-D receptor family, an N-terminal Vps10p-D [87] (Fig. 3). Additionally, SorLA contains a fibronectin type III domain, the exact function of which is still elusive but might be involved in protein-protein interactions [88]. Vps10p-D receptors are also type-I transmembrane proteins. Their large extracellular/luminal moieties contain a Vps10p-D and their short cytoplasmic domains harbor canonical motifs for intracellular sorting. The Vps10p-D makes up the entire extracellular/luminal part of sortilin and is combined with a so-called leucine-rich domain in SorCS1–3 (Fig. 3). Structural features are thought to be shared among all Vps10p-D receptors. The N-terminal part of the sortilin Vps10p-D comprises a ten-bladed β-propeller creating a large tunnel with multiple ligand-binding sites which is followed by the so-called ten conserved cysteines (10CC) domain [87, 89]. This C-terminal segment of 10CC forms five disulfide bonds [89, 90], but the amino acid identity among all Vps10p-Ds is only modest, but high for SorCS1–3 and separates the SorCS subgroup from sortilin and SorLA [87].

Schematic representation and domain structure of LRP1, members of the Vps10p-D receptor family and calsyntenin-1–3. Low-density lipoprotein receptor-related protein 1 (LRP1) is a type-I transmembrane protein. The extracellular domain consists of four ligand-binding repeats (also named complement type repeats) and epidermal growth factor (EGF) homology domains and β-propeller domains, which are important for pH-dependent release of bound ligands in endosomes. SorLA, sortilin, and SorCS1–3 are members of the vacuolar protein sorting 10 protein (Vps10p) domain receptor family. SorLA contains two additional types of domains not present in LRP1: the fibronectin type III domain and the Vps10p domain. The extracellular domain of sortilin is comprised solely of the Vps10p domain while SorCS1–3 contain an additional leucine-rich domain in the juxtamembrane region. The amino acid identity among all Vps10p-Ds is only modest, but high for SorCS1–3 and separates the SorCS subgroup from sortilin and SorLA. Calsyntenin 1–3 contain two cadherin-like domains in the ectodomain and one laminin, nectin, sex hormone (LNS) binding globulin) domain. Calsyntenin-1 and -2 contain an acidic region, which is able to bind calcium in their cytoplasmic tail and two KLC1 binding motifs. The acidic region and one KLC1 binding motif are lacking in Calsyntenin-3
The third group of type-I transmembrane proteins modulating APP transport and discussed in this review are calsyntenins (calsyntenin 1–3, Clstn 1–3). These were initially isolated as postsynaptic Ca2+-binding proteins [91] and have been also named Alcadeinα, β, and γ. They belong to the Cadherin-related family comprised of atypical Cadherins [92]. Their extracellular moiety is characterized by a repeat of two Cadherin-like domains [91, 93], an LNS domain (laminin, nectin, sex hormone-binding globulin) [94] and kinesin light chain-binding motifs in their cytoplasmic domains [95, 96]. Calsyntenin-1 and -2 contain two KLC1-binding motifs consisting of a WDDS sequence, while calsyntenin-3 is lacking one KLC1-binding motif [93]. Calsyntenin-1 and -2 additionally harbor a c-terminal calcium-binding site via an acidic amino acid stretch, which is shorter in calsyntenin-2 than in calsyntenin-1 [91, 93, 97].
Low-Density Lipoprotein Receptor-Related Protein 1
Multiple members of the LDLR family have been shown to influence APP physiology with the focus resting on low-density lipoprotein receptor-related protein 1 (LRP1) [83]. LRP1 is one of the largest gene family members with a size of approximately 600 kDa and four extracellular ligand-binding domains [98]. Like APP, LRP1 is transported along the constitutive secretory pathway. Native LRP1 is cleaved in the trans-Golgi by furin into a 515 kDa α-subunit and an 85 kDa β-subunit, which stay attached in a non-covalent manner [99, 100]. LRP1 has been shown to bind numerous different ligands to its extracellular domain [101], including APP, apolipoprotein E (apoE), and α2-macroglobulin (α2M), which are all proteins associated with AD [102,103,104]. Furthermore, LRP1 itself has been identified as a risk factor for AD [105].
LRP1 associates with APP through the N-terminal Kunitz-protease inhibitor (KPI) domain of APP [106, 107] and the APP C-terminal cytoplasmic domain [108]. As neurons express mainly APP695 lacking the KPI domain [109], the C-terminal interaction with APP appears more relevant in respect to brain function. The cytosolic interaction between APP and LRP1 is assumed to be mediated by the scaffolding protein Fe65, which binds with its PTB1 domain to the NPxY motif of LRP1 and with the PTB2 domain to the NPxY motif in the C-terminus of APP [108, 110, 111].
The LRP1 cytoplasmic tail contains two NPxY motifs, of which the distal motif is thought to be involved in its very fast endocytosis, with a half-life time of less than 30 s at the cell surface [112, 113]. LRP1 increases APP endocytosis and causes consistently an increased secretion of Aβ and sAPPβ, while a lack of LRP1 increases sAPPα (Fig. 4) [106, 111, 114,115,116]. Notably, this holds true for APP in its monomeric and dimeric form [117], although LRP1 binding to APP was significantly decreased after inducing dimerization of APP [118], which presumably affects APP trafficking.

LRP1 modulates APP trafficking. APP interacts with LRP1 presumably early in the secretory pathway before LRP1 is cleaved by furin in the TGN. APP and LRP1 are trafficking in different transport vesicles in anterograde direction to the cell surface, APP with faster transport kinetics. Internalization of APP is facilitated by LRP1, which associates with APP on the cell surface, thereby increasing sAPPβ and Aβ production
Interestingly, the influence of LRP1B on APP processing is contrary to what is shown for LRP1 [119]. Both, LRP1 and LRP1B, are highly expressed in the brain [120,121,122] and LRP1 and LRP1B only differ from each other in one additional ligand binding type repeat in ligand binding domain IV of LRP1B and a larger distance between the NPxY motifs [123]. Even though these differences are subtle, LRP1B has much slower internalization kinetics than LRP1 [123]. Assuming, that LRP1B competes with LRP1 for binding to the Fe65-APP complex, APP would be endocytosed at a lower rate, explaining the increase of sAPPα and the decrease in Aβ (Fig. 2) [119]. However, other LDLR family members, such as VLDL could act similarly [124].
Accumulating evidence suggests that LRP1 affects APP transport also in the secretory pathway (Fig. 4). APP interacts with LRP1 before it is cleaved by furin, implying an interaction of APP with LRP1 early in the secretory pathway [111]. Accordingly, APP still binds LRP1 when a dilysine ER-retention motif (KKAA) is introduced to the LRP1 cytoplasmic domain [100]. Moreover, co-localization and co-transport of LRP1 and APP along the secretory pathway has been reported recently [117]. Live cell imaging analyses revealed that LRP1 and APP are trafficked in distinct anterograde transport vesicles, whereby LRP1 is transported with velocities ≤ 1 μm/s and APP with velocities ≥ 1 μm/s. However, elevated expression levels of LRP1 altered APP transport kinetics to those observed for LRP1 before. In contrast, lowered levels of LRP1 lead to a significant decrease in APP stationary vesicles and to a significant increased mean velocity [117]. This strongly suggests that LRP1 affects APP transport by recruitment of APP into common transport vesicles, thereby regulating its cell surface localization and in turn, its processing by proteases located at the plasma membrane, including ADAM10, MT5-MMP, and Meprinβ.
LRP1 can be proteolytically processed and might therefore affect APP physiology and pathogenicity not only by modulating its transport kinetics but also through competition with APP for the secretase BACE1 [125, 126] and γ-secretase [127, 128].
Further, multiple studies showed that Aβ is taken up by LRP1 either through direct binding to the LRP1 N-terminal domain [129, 130] or bound to ApoE or α2M [131]. Here, LRP1 was demonstrated to function as one of the major Aβ clearance receptors from brain to blood through transcytotic transport over the blood-brain-barrier [132,133,134]. Also, LRP1 was shown to bind Aβ in the blood and to target it for degradation [135] and to play an important role in cellular uptake of Aβ in microglia [136].
Taken together, the actual data strongly argue that besides its role in endocytosis, LRP1 functions as a sorting receptor for APP, recruiting APP to specific transport vesicles. Here, LRP1 interestingly affects both, the secretory as well as the endocytic pathway, which might be pivotal for sorting in bipolar cells.
Sortilin-Related Receptor Containing LDLR A Repeats
SorLA (also termed “LR11” or “SORL1”) is a 250-kDa type-I transmembrane protein [85, 86] (Fig. 3) that shares several ligands with the LDL receptor family, including apolipoprotein E (ApoE), apolipoprotein A-V, LDLR-associated protein (RAP), lipoprotein lipase, platelet-derived growth factor-BB (PDGF-BB), and components of the plasminogen-activating system, such as urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA) [137,138,139,140]. These ligands probably all bind to the cluster of 11 complement-type repeats (CR) of SorLA (Fig. 3) [140, 141]. The Vps10p-D of SorLA functions as an additional ligand-binding domain. It specifically binds a variety of growth factors and neuropeptides including neurotensin, glial cell-derived neurotrophic factor (GDNF), ciliary neurotrophic factor (CNTF), and interleukin-6 [90, 141,142,143]. SorLA binds APP directly and sequesters APP in intracellular compartments which results in reduced processing into Aβ [144]. The interaction has been mapped to the carbohydrate (E2) domain of APP and the CR domains 5–8 of SorLA [144, 145]. Deletion of the CR cluster abolishes the ability of SorLA to protect APP from processing [145]. Moreover, a second interaction site involving the cytoplasmic domains of APP and SorLA has been proposed [146]. As outlined above, the Vps10p-D of SorLA functions as an independent ligand-binding site. It binds monomeric Aβ in a pH-dependent manner and is thought to facilitate its subcellular targeting to lysosomes [147, 148]. This interaction seems specific for the SorLA Vps10p-D as the Vps10p-D of sortilin has been incapable to bind Aβ [147].
SorLA is highly expressed in the brain where it shows predominant localization in neurons of the cerebral cortex, hippocampus, and cerebellum, but it is expressed as well in a variety of other organs [149]. Genetic analysis revealed that SorLA modulates the risk for late onset as well as early onset AD (reviewed in [150]). SorLA expression levels are reduced in brains of AD patients as compared to healthy non-demented subjects [151]. Mouse models supported the significance of SorLA as a negative regulator of APP processing in vivo because knockout mice exhibit higher Aβ-levels in the brain as compared with wild-type mice [152]. Moreover, overexpression of SorLA in neurons decreased Aβ levels [152]. Taken together, the current data support the notion that SorLA is a sorting receptor for APP and Aβ. Thus, understanding the determinants of SorLA’s subcellular itinerary is key to understand APP processing and degradation of monomeric Aβ.
On a subcellular level, SorLA is mainly localized to Golgi compartments and endosomes and only a minor fraction is present on the cellular surface [141, 153, 154]. This localization has been observed in undifferentiated cells [141] and recent studies demonstrated a predominant somatic-dendritic localization of SorLA in neurons and targeting of SorLA to the basolateral membrane and to sorting endosomes in polarized MDCK cells [155].
SorLA is synthesized as a precursor and converted in the trans-Golgi network to mature receptor by proprotein convertase-mediated cleavage and subsequent dissociation of its N-terminal propeptide which is a prerequisite for binding of exogenous ligands to its Vps10p-D [141]. Ectodomain shedding followed by regulated intramembrane proteolysis as described for APP has been also observed for SorLA. It serves as a substrate for ADAM17, whose active site is located in the aqueous environment of the extracellular domain and can function as α-secretase which can be stimulated by phorbol esters or by some ligands. PDGF-BB, for example, stimulates shedding of SorLA, whereas other ligands have no effect [156]. The primary cleavage elicits subsequent gamma-secretase-mediated proteolysis within the transmembrane segment. This cleavage releases the cytoplasmic domain, which is rapidly degraded [156, 157]. Interestingly, a nuclear localization of the SorLA cytoplasmic domain fused to green fluorescent protein was demonstrated and a transcriptional activity of the SorLA cytoplasmic domain suggested [157].
SorLA conveys rapid internalization of ligands, but as compared to LRP1, SorLA is endocytosed more slowly [140]. SorLA contains canonical cytoplasmic interaction motifs and a number of cytosolic adaptors have been identified. It has been demonstrated that internalization of SorLA depends on an acidic cluster and on adaptor protein-2 (AP-2) interaction [158]. SorLA binds additional cytosolic adaptors, including AP-1; Golgi-localized, gamma adaptin ear-containing, ARF-binding (GGAs); phosphofurin acidic cluster sorting protein 1 (PACS1), and retromer, which govern the itinerary between TGN and endosomal compartments. The acidic cluster of the SorLA cytoplasmic domain is a functional interaction motif for AP-1 and PACS1 [158, 159]. Both adaptors are engaged in retrograde endosome to TGN sorting and deletion of one of the adaptors results in an altered subcellular localization of SorLA [158, 159]. The interaction of AP-1 with the acidic cluster and additional amino acids in SorLA’s cytoplasmic domain underlie the polarized distribution of SorLA in neurons and other polarized cells [155]. Anterograde Golgi to endosome sorting of SorLA depends on the acidic cluster combined with a dileucine motif and on the GGA-binding motif DXXM [158]. GGA1 and GGA2 interact with SorLA and have been suggested to guide this type of sorting [158,159,160,161]. However, disruption of the GGA interaction motif in mice in vivo had no impact on APP processing but increased Aβ levels in the brain [162]. Thus, the disrupted motif might rather modulate SorLA-mediated lysosomal targeting of Aβ. The SorLA cytoplasmic domain interacts via its FANSHY motif with retromer, an adaptor complex engaged in endosome to Golgi retrieval [163]. In mammals, retromer is composed of a trimer comprised of VPS26, VPS29, and VPS35 and a dimer of two sorting nexins (SNX) [164]. Retromer deficiency in mice and flies increases production of Aβ and the retromer-dependent endosomal-trans-Golgi sorting pathway has been suggested to be implicated in late onset AD [165, 166]. In agreement, VPS35 and VPS26 expressions are reduced in vulnerable regions of AD brains and knockdown of retromer elements in cell culture led to increased Aβ production, while overexpression of retromer elements decreased Aβ levels [167]. So far, there is no evidence that retromer interacts directly with APP. Knockdown of VPS35 or SNX1 results in reduced SorLA expression [158], VPS26 interacts directly with SorLA and mutations in the SorLA cytoplasmic domain that affect the retromer binding site result in APP missorting and enhanced processing [163] and in agreement, disruption of part of the FANSHY motif in mice in vivo resulted in accumulation of SorLA in endosomes and increased APP processing [162]. This is in line with very recent time lapse imaging results in primary cortical neurons showing that after co-expression of SorLA, APP stationary vesicles and anterograde vesicles were significantly decreased while there is clear increase in the number of retrograde vesicles, suggesting that SorLA is mainly involved in retrograde transport of APP [118] (Fig. 5). Interestingly, the percentage of vesicles co-transporting APP and SorLA is only about 10% [118] and the co-transport rate of APP and SorLA was significantly reduced after inducing dimerization of APP [118], which is in line with results from Willnow and colleagues showing an impact of SorLA on the extent of APP dimerization [168].

SorLA modulates APP trafficking. SorLA is mainly localized to Golgi compartments and endosomes, where it strongly co-localizes with APP. Only a minor fraction of SorLA is present on the cellular surface and can be shed by α- and γ-secretase. After endocytosis, SorLA binds cytosolic adaptors which shuttle SorLA between endosomes and the TGN (black arrow). SorLA likely connects retromer and APP. Binding of SorLA to GGAs or AP1 in the TGN shuttles SorLA back to endosomes. Co-expression of SorLA leads to a significant decrease in APP stationary vesicles and anterograde vesicles with a concomitant increase in the number of APP containing retrograde vesicles, indicating that SorLA is mainly involved in retrograde transport of APP
Thus, SorLA likely connects retromer and APP. In addition, an interaction of SorLA and SNX27 was proposed [169]. SNX27 contains a PDZ domain and has been shown to serve as a cargo selector for the retromer complex. The binding of other transmembrane proteins occurs through the SNX27 PDZ domain and respective binding motifs in the cytoplasmic domain of the cargo [170]. In contrast, the binding of SorLA appears independent of the SNX27 PDZ domain [169]. However, SNX27 mediates sorting from endosomes to the plasma membrane [171] and in agreement with this, SNX27 enhances SorLA and APP surface levels and promotes non-amyloidogenic APP processing [169].
In conclusion, current data suggest a dual role for SorLA in modulating APP processing and Aβ accumulation. One function is retrograde sorting of APP from endosomal compartments to the trans-Golgi and thereby reducing amyloidogenic processing of APP. The other function is targeting of already processed monomeric Aβ for lysosomal degradation.
Sortilin-Related Receptor CNS Expressed 1
Another member of the Vps10p-D receptor family, sortilin-related receptor CNS expressed 1 (SorCS1), has been genetically linked to late onset AD [172,173,174,175,176]. SorCS1 is a 130 kDa type-I transmembrane protein with an N-terminal Vps10p-D followed by a leucine-rich domain, a transmembrane domain, and a short cytoplasmic tail (Fig. 3) [177]. SorCS1 binds PDGF-BB and presumably other growth factors through its Vps10p-D [156]. In addition, the SorCS1 Vps10p-D interacts directly with neurexin1 (Nrxn1) [178, 179]. Proteomic analysis of the synaptic SorCS1 interactome revealed that SorCS1 forms a complex with Nrxn1 and Nrxn2, neuroligin 1 (Nlgn1) and Nlgn3, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) subunits, Ntrk2 (also known as brain-derived neurotrophic factor receptor, TrkB) and APP [178]. Accordingly, other studies had demonstrated the interaction of SorCS1 and APP by co-immunoprecipitations before [174, 180, 181].
SorCS1 is predominantly expressed in the developing and adult brain and neuronal activity can induce its expression in the hippocampus [182,183,184]. Genetic variations in SORCS1 are associated with memory performance [185]. In addition, expression analysis revealed lower SorCS1 levels in the amygdala from AD brains as compared to unaffected brains [174]. Cellular analyses demonstrate that high SorCS1 expression levels result in a modest decrease of Aβ and a reduction of SorCS1 expression leads to an increase of Aβ levels [174, 180]. Moreover, studies in hypomorphic SorCS1 mice support the impact of SorCS1 on APP processing [180]. These observations are reminiscent of SorLA modulating APP processing. Like SorLA, SorCS1 is subject to ectodomain shedding by ADAM17 which can be stimulated by phorbol esters or PDGF-BB and is followed by γ-secretase-mediated proteolysis within the transmembrane segment [156].
SorCS1 is expressed in different alternative splice variants with identical extracellular and transmembrane moieties, but different cytoplasmic domains conveying varying trafficking properties [87, 181, 186, 187]. Whereas some cytoplasmic domains mediate rapid cellular uptake of ligands, others direct the receptor to the cellular surface and do not mediate rapid internalization. It has been demonstrated that the endocytic isoforms of SorCS1 are internalized in an AP-2-dependent manner and are capable of targeting internalized cargo to lysosomes [187]. In accordance, affinity-purified SorCS1 complexes revealed the presence of AP-2 [178] and a direct interaction of SorCS1a and AP-2 has been shown [187]. SorCS1 has been linked to SorLA and the retromer subunit Vps35 by co-immunoprecipitation [180]. However, a direct binding of SorCS1 to the retromer complex or a retromer-mediated transport of SorCS1 awaits demonstration. All analyzed SorCS1 cytoplasmic domains failed to convey Golgi to late-endosomal transport in an assay in which the SorLA cytoplasmic domain was mediating this type of intracellular transport (Fig. 6) [158, 187]. Moreover, none of the SorCS1 cytoplasmic domains interacts with GGAs [186, 188]. Therefore, it was concluded that SorCS1 isoforms are not engaged in mediating Golgi to late-endosomal transport [187]. This is an important difference to SorLA, which conveys this type of sorting [158]. Thus, it is likely that SorCS1 has a function different from SorLA in APP trafficking. In neurons SorCS1 presents mainly a somato-dendritic localization [178, 181] and depending on the splice variant, SorCS1 is localized to endosomes or to the plasma membrane [181, 186]. SorCS1 is translocated to postsynaptic sites where it regulates Nrxn and AMPAR surface trafficking and in agreement SorCS1 deficiency leads to reduced glutamatergic synaptic transmission [178]. Uptake of APP appears to be independent of SorCS1 [181], but APP and the endocytic splice variant SorCS1c share a common postendocytic pathway. Both proteins share vesicular transport characteristics and overexpression of SorCS1c, but not of SorCS1b, reduces neuronal anterograde transport of APP and increases the fraction of APP localized to stationary vesicles [181].

SoCS1c modulates APP trafficking. SorCS1c is an endocytic receptor, but internalization of APP appears to be independent of SorCS1, although SorCS1c and APP share a common postendocytic pathway. SorCS1c was shown to reduce the anterograde transport rate of APP in neurons. Further involvement of SorCS1c in APP trafficking from sorting endosomes to the Golgi needs to be determined
Taken together, current data suggest that SorCS1 is engaged in the regulation of sorting and anterograde targeting of APP. Notably, SorCS1 is genetically associated as well with type-I and -II diabetes [189,190,191,192]. Diabetes is a known risk factor for AD. Therefore, SorCS1 might link diabetes and AD pathology.
Sortilin
Sortilin (SORT1), also known as neurotensin receptor3 [193] or gp95 (glycoprotein of 95 kDa) [138] is expressed in neurons of the CNS and PNS [138, 194, 195] as well as in non-neuronal tissues like heart, lung, skeletal muscle, and testis [138].
Sortilin is synthesized as an inactive precursor which is converted in the TGN to the mature receptor by furin-mediated cleavage of a 44-residue N-terminal propeptide [196]. The propeptide of sortilin precludes binding of ligands by sterical hindrance [196]. Therefore, the unprocessed receptor is unable to bind ligands. This characteristic has been described as well for the SorLA Vps10p-D whereas receptors of the SorCS subgroup seem to bind ligands independently of their N-terminal processing [141, 186, 197]. Remarkably, sortilin binds the propeptide of SorCS1 which itself does not bind its propeptide [198]. However, the functional relevance of this interaction is not completely understood, but it has a pronounced effect on sortilin’s ability to mediate specific cellular functions [198].
In addition to its own propeptide, the extracellular/luminal domain of sortilin interacts with neuropeptides, such as neurotensin, and neurotrophic factors, such as the proforms of nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) and ciliary neurotrophic factor (CNTF) [193, 199,200,201], but as well with ligands typical for the LDLR family, such as the LDLR-associated protein (RAP), lipoprotein lipase (LpL), lipoprotein AV, and apolipoprotein E (APOE) 2, 3, 4 [196, 202,203,204]. Sortilin appears promiscuous as it binds various ligands and is unique as it is the only known non-LDLR family member that binds RAP. In addition, it interacts with other ligands of the LDLR family, whereas the Vps10p-Ds of SorCS1, -2, and -3 do not bind RAP and other typical LDLR ligands [90, 186].
Sortilin enters the secretory pathway and might transport ligands anterogradely [154]. However, once internalized from the plasma membrane, the receptor is capable to direct cargo to late endosomal compartments and to lysosomes for degradation [202]. Sortilin is internalized through clathrin-coated pits and this depends on canonical AP-2 binding motifs. Like SorLA, sortilin interacts with GGAs, AP-1, and retromer complex and these interactions convey the cycling of sortilin between the TGN and late endosomes (Fig. 7) [202, 205,206,207].

Sortilin modulates APP trafficking. Sortilin is located predominantly in the TGN and cycles between endosomes and TGN similarly as SorLA. In contrast, sortilin might shuttle APP mainly in anterograde direction and then to the cell surface. Sortilin is an endocytic receptor, which internalizes various ligands by receptor-mediated endocytosis and delivers them to lysosomes, but its possible role on APP endocytosis needs to be determined
Sortilin functions as an APOE receptor [204], a known risk factor in sporadic AD [208]. Sortilin mediates cellular uptake of Aβ bound to APOE and sortilin KO mice crossed to two different AD mouse models show increased Aβ plaque burden and significantly increased Aβ40 levels, but no changes on C-terminal fragments of APP generated by α- or β-secretase [204]. Therefore, sortilin might play a role in catabolizing Aβ peptides.
Sortilin has also been shown to co-localize and to interact with APP in neurons [154, 209]. FRET analysis and co-immunoprecipitation identified the ectodomain as well as the intracellular region as interaction domains, whereby binding via the C-terminus seemed to be more prominent [209]. The binding site in APP was mapped to amino acids 1–141 [209] within the E1 domain (aa 31–191) [210], more specifically mainly to the growth factor-like domain (GFLD) (aa 28–123). In agreement, Gustafsen et al. detected the interaction of sortilin and APP by co-immunoprecipitation, demonstrated that the extracellular domain of APP is internalized by sortilin and sorted differentially by SorLA and sortilin [154]. However, Gustafsen et al. mapped the APP-sortilin interaction via surface plasmon resonance analysis to a different region (6A) within the APP ectodomain, further distal to the E1 domain and binding was inhibited by the sortilin propeptide [154]. Although more than one binding site may exist, these potentially conflicting results need clarification. Yang et al. further identified within the APP C-terminus, the NPTYKFFE sequence (residues 759–766) as a sortilin interaction site [209]. This contains the internalization motif for clathrin-dependent endocytosis NPTY [211] and the KFFE motif, which had been shown to be important for APP transport mediated via the μ4 subunit of AP4 [212].
Microarray expression analysis of sortilin in brain tissue of the occipital lobe and cerebellum showed no significant difference between AD and control patients [213]. A different study reported unchanged sortilin protein levels in frontal or temporal cortical tissue and no association between sortilin levels and antemortem cognitive test scores [214]. However, there was a positive association between temporal cortex sortilin levels and severity of neuropathology by Braak and NIA-Reagan diagnoses [214]. Moreover, sortilin fragments were identified in senile plaques [215]. In contrast to the microarray data, protein levels of sortilin were found to be significantly increased in postmortem temporal cortex of AD patients, which may relate to the finding that sortilin overexpression leads to increased BACE1 cleavage of APP and thereby also to elevated Aβ production [216,217,218]. Remarkably, sortilin also interacts with BACE1 and influences its retrograde transport to the TGN [216]. Increased levels of sAPPβ and Aβ after sortilin overexpression in cultured cells were reported while RNAi-mediated suppression of sortilin resulted in decreased BACE1 mediated cleavage of APP [216]. Thus, sortilin seems to influence the proteolytic conversion of APP. Albeit, a different study suggests that overexpression of sortilin leads to increased sAPPα and decreased sAPPβ levels [154] while overexpressed SORLA leads to decreased sAPPα/sAPPβ products. This supports a different impact on APP trafficking of these two APP sorting molecules.
Like APP, SorLA, and SorCS1, sortilin is cleaved by α- and γ-secretases [156, 219]. The metalloproteinases ADAM10 and ADAM17 are thought to act mainly as α-secretases. ADAM10 and ADAM17 can be activated experimentally by two different reagents. Phorbol 12-myristate 13-acetate (PMA), an activator of metalloproteinases via the protein kinase C (PKC) pathway specifically activates ADAM17, whereas ionomycin, a calcium ionophore strongly activates ADAM10 in a calmodulin-dependent manner [220]. PMA has been demonstrated to activate shedding of SorLA and SorCS1 [156]. In parallel experiments, shedding of sortilin induced by PMA was only minor. Using ionomycin to induce cleavage by ADAM10 significantly increased the level of shed sortilin [221]. Analysis of mouse embryonic fibroblasts lacking either ADAM10 or ADAM17 revealed that in these cells, shedding of sortilin was mainly carried out by ADAM10 [221]. Therefore, the proteolytic conversion of sortilin in the juxtamembrane stalk region is thought to be mainly mediated by ADAM10. However, shedding of sortilin has been analyzed in different cell types [156, 221, 222] and has been reported to be activated by PKC in a tumor cell line [222]. This may indicate that sortilin could be cleaved by ADAM17 in a certain cellular environment.
Different studies indicate that sortilin plays a role in other human diseases like cardiovascular diseases (CVD) or frontotemporal lobar degeneration (FTLD). Remarkably, single nucleotide polymorphisms (SNPs) within and in the vicinity of the SORT1 gene, encoding sortilin, have been associated with CVD [223,224,225,226,227,228] and CVD is considered as a risk factor for AD [229]. A number of studies analyzed SNPs in different populations regarding AD, but in contrast to SorLA and SorCS1, no higher risk has been directly associated with AD so far in this kind of surveys [213, 230,231,232,233,234,235,236].
Although no strong genetic association has been reported for sortilin in AD so far, there are several indications that sortilin seems to be involved in another neurodegenerative disease, in FTLD [237, 238], which is the second most prevalent form of early onset dementia after AD (between age 45 and 65) [239]. One important player in FTLD is progranulin, since pathogenic mutations in progranulin (GRN) were identified throughout the gene, and all cause the disease via haploinsufficiency resulting in reduced progranulin levels [238]. Sortilin was the first identified receptor of progranulin [240, 241]. Sortilin binds progranulin with high affinity, mediates its rapid endocytosis, and subjects it to lysosomal degradation [240, 242]. Sortilin has been additionally linked to FTLD via the TAR DNA-binding protein 43 (TDP-43) which has been described as a major risk protein in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with ubiquitin inclusions (FTLD-U) [243]. TDP-43 is involved in splicing events of sortilin and progranulin [244] and dysregulation of sortilin splicing via TDP-43 was shown to lead to an altered progranulin metabolism [245].
In conclusion, sortilin is an additional member of the Vps10p family interacting with APP, shown to modulate APP processing. There is no strong genetic association for sortilin in AD so far, but sortilin might play a role in FTLD.
Calsyntenin
Calsyntenin-1 was first identified in a screen for proteins released from synapse forming spinal cord neurons [91]. A different study used the binding partner of the APP cytoplasmic domain, X11L/X11β/mint2 [246], as a bait in a yeast two-hybrid screen to identify the molecular machinery which regulates APP trafficking [97] (Fig. 8). Thereby, Alcadein (Alzheimer-related cadherin-like protein) (Alc) was found, which is identical to calsyntenin-1 [97]. The protein belongs to a family of three homologs (Fig. 3 ) in humans and in mice, alcadeinα, β, and γ or calsyntenin 1–3, which are all predominantly expressed in neurons [91, 93, 97]. In addition, there exist two splice forms of alcadeinα termed “alcadeinα1” and “alcadeinα2” [97]. For simplification, we will use the term “calsyntenin.”

Calsyntenin-1 modulates APP trafficking. Calsyntenin-1 and APP are mainly co-localized in the TGN and interact indirectly via X11L. Knockdown of calsyntenin-1 inhibits anterograde movement of APP leading to an accumulation in the TGN. Additionally, calsyntenin-1 and APP are co-localized in Rab5 positive early endosomes, but not in Rab11 positive recycling endosomes. The synaptogenic calsyntenin-3 is thought to act differently and shows a more abundant localization at the cell surface than calsyntenin-1 or -2
In situ hybridizations of different developmental murine stages (E14–P48) revealed calsyntenin-1 mRNA expression throughout the body and in all brain regions [91, 247]. In contrast, calsyntenin-2 and -3 mRNAs are restricted to the CNS. Calsyntenin-2 is mainly present in the hippocampus and the olfactory bulb, at later stages also in cortex and cerebellum while calsyntenin-3 is present in various brain areas [247]. Moreover, calsyntenin-2 and -3 are expressed in interneurons of the hippocampus and the neocortex [93, 94] and calsyntenin-1 was localized to excitatory and inhibitory postsynapses [91].
The 150 kDa full-length calsyntenin-1, -2, and -3 can be proteolytically cleaved into one secreted 115-kDa fragment and one C-terminal 34-kDa fragment (CTF) by ADAM10 as well as ADAM17 [91, 248, 249]. Cleavage of calsyntenin-1 mainly takes place in the secretory pathway [250]. Moreover, it has been shown that calsyntenin-3 is extensively shed since in mouse brain less than half of calsyntenin-3 is present as full-length protein and more than half of it as the shed ectodomain [94]. The 34-kDa CTF fragment can be further processed by γ-secretase which results in release of the intracellular domain (ICD) and a secreted Aβ-like fragment which was also termed p3 [248, 249, 251]. The calsyntenin ICD can suppress gene transactivation of the APP ICD possibly by titrating away Fe65 binding partners of APP [248].
Calsyntenins have been suggested as biomarkers in AD because altered levels were observed in AD. Thus, in presenilin 1 familial Alzheimer’s disease (FAD) mutants, which lead to an altered Aβ40/42 ratio for APP concomitantly altered C-termini for the calsyntenin-1 p335/38 fragments were observed [249]. Furthermore, elevated levels of calsyntenin-1 p338 fragments were detected in the CSF of patients of various clinical populations including sporadic AD [251]. Additionally, calsyntenin-1 full-length protein levels were decreased in the brains of AD patients [252] and an involvement of calsyntenin-3 in AD has been implicated in a recent proteomics study showing decreased calsyntenin-3 levels in CSF of FAD patients [253]. A potential role of calsyntenins in AD is in line with a survey indicating that calsyntenin-3 accumulates in dystrophic neurites surrounding amyloid-β (Aβ) plaques [254].
So far, calsyntenins have not been genetically linked to AD, but there is accumulating evidence that calsyntenin-1 is involved in APP transport. Several studies demonstrated partial co-localization of APP and calsyntenin-1, which are both present in the soma, in dendrites, and axons of neurons [252, 255]. In the soma, both proteins are mainly localized to the trans-Golgi network [252, 255, 256]. About 29–41% of vesicles in axons of the sciatic nerve, hippocampal, or cortical neurons contained both APP and calsyntenin-1 [95, 252, 257]. Additionally, immunostainings revealed that APP and calsyntenin-1 co-localize to about 48% in the central domain of growth cones in hippocampal neurons, but only to about 12% in the peripheral domain of growth cones [96, 257]. Live cell imaging analyses of cells transfected with fluorophore-tagged calsyntenin-1 or APP have been performed in different cellular systems. Velocities of antero- and retrograde moving vesicles containing one of the proteins varied in the different surveys [6, 62, 95, 96, 252, 258, 259]. However, co-transfection in neurons demonstrated co-transport of both proteins [252]. Those results suggest that APP and calsyntenin-1 co-localize and are partially co-transported (Fig. 8). However, attempts to prove a direct interaction of calsyntenin-1 and APP failed. The association of both proteins is rather bridged by the cytoplasmic interaction partner X11L [97]. X11L expression leads to a decrease in Aβ40 and sAPP production [260,261,262]. Expression of the tripartite complex X11L/calsyntenin-1/APP leads to an even more severe reduction of Aβ levels, but not calsyntenin-1/APP expression alone. X11L expression enhances the half-life time of APP and this effect is even more pronounced in the presence of calsyntenin-1 [97].
As exemplified above, a large body of evidence suggests that APP represents one of several transmembrane proteins that undergo fast axonal transport by means of conventional kinesin but does not interact directly with kinesin (reviewed in [263]). A number of studies strongly indicate that calsyntenin-1 acts as an organelle adaptor that links kinesin-1 light chain to transport vesicles because it directly binds KLC’s via its C-terminal domain [95, 96, 256]. Immunostainings showed that calsyntenin-1-positive organelles are aligned along microtubules in axons of neurons and co-localize partly with kinesin-1 [96]. Calsyntenin-1 can induce vesicle association with KLC1, as shown via live cell imaging [95]. Taken together, current results suggest that calsyntenin-1 links certain types of vesicles to kinesin.
Immunoisolates of calsyntenin-1 from mouse brains identified two different types of calsyntenin-containing transport organelles [257]. One was characterized by early endosomal markers like Rab5 and contained APP [257]. In contrast, the other calsyntenin-containing transport organelle which was characterized by the recycling vesicle marker Rab11 was lacking APP [257]. The presence of APP in early endosomal vesicles is consistent with previous studies, showing co-localization of APP with Rab5-positive endosomes [264, 265]. Rab5 immunoisolates contained mainly the calsyntenin CTF while APP immunoisolates contained calsyntenin mainly in the full-length form [257]. These data together with time lapse imaging analyses lead to the hypothesis that calsyntenin-1 docks kinesin to different endosomal carriers transporting APP and other cargo anterogradely along the axon to the growth cone [255, 257].
In line with this notion are calsyntenin-1 knockdown experiments which resulted in a significant increase in APP levels in the TGN [252], corroborating the idea that calsyntenin-1 mediates kinesin-1 transport of cargoes on post-Golgi carriers [255, 256]. Knockdown of calsyntenin in neurons leads to suppressed anterograde (64.3%) and retrograde transport (46.6%) of APP [252], but not to a complete inhibition of APP movement. This supports the assumption that calsyntenin function underlies only one of several transport mechanisms of APP.
Remarkably, knockdown or overexpression of calsyntenin seems to influence APP processing, presumably by altering APP transport. So far, the corresponding data are conflicting. A significant increase in α- and β-CTF production after knockdown of calsyntenin-1 was shown [255]. On the contrary, coexpression of calsyntenin-1 or of the ICD of calsyntenin with APP suppressed APP anterograde transport and facilitated Aβ40 and Aβ42 production [95]. In contrast, a study of Vagnoni et al. showed an increase in Aβ40 and Aβ42 levels and β-CTF and sAPPβ after knockdown of calsyntenin-1 while sAPPα and α-CTFs were reduced. Moreover, calsyntenin-1 levels are reduced in AD brains which correlates inversely with Aβ levels [252]. Therefore, further studies are required to understand the impact of calsyntenin-1 on APP processing.
Calsyntenin-1 and -2 seem to play similar cellular roles, whereas a different function for calsyntenin-3 is likely, because this protein lacks the C-terminal KLC1 binding motifs as well as the calcium-binding region in its C-terminus [93] and shows a more prominent surface localization than calsyntenin-1 and -2 [94]. This is consistent with the fact that, so far, no role for calsyntenin-3 has been demonstrated in organelle transport [96]. Albeit, the impact of calsyntenin-3 on APP transport has not been analyzed in detail. In accordance with a different function, it has been recently reported that calsyntenin-3, but not calsyntenin-1 or -2, is able to induce presynaptic differentiation [94, 247]. It shares this capacity with APP and other well characterized synaptic adhesion molecules (SAMs) [266,267,268,269,270]. One of the best described SAM’s are neuroligins, which are located at the postsynapse interacting in trans with presynaptic neurexins [271]. Neurexin genes each encode a longer α form and a shorter β form [272]. Conflicting results described an interaction of postsynaptic calsyntenin-3 with neurexin1α in a direct fashion [94, 273], but not with neurexin1β [94]. However, a different study failed to reproduce the direct interaction between calsyntenin-3 and neurexin1 in similar cell surface binding assays, raising the question whether calsyntenin-3 binds neurexin1 directly [247]. Moreover, calsyntenin-1 and -2 were reported not to interact with neurexin [94]. The physiological relevance of these findings still needs to be resolved but calsyntein-3 might affect synapse formation and or stabilization, which are important determinants in AD [274].
In conclusion, current knowledge implies that calsyntenins are not genetically linked to AD, although altered calsyntenin fragment levels have been observed in AD brains. Calsyntenin-1 and most likely calsyntenin-2 modulate APP transport and presumably its processing.
Conclusions
Intracellular transport of APP determines its processing by different proteolytic enzymes and altered subcellular trafficking of APP is thought to directly affect the degree to which Aβ is generated. The type-I transmembrane proteins LRP1, SorLA, SorCS1, sortilin, and calsyntenin-1 have been demonstrated to modulate APPs intracellular targeting and processing. Genetic linkage analyses underscore the role of SorLA in the development of EOAD and LOAD, whereas LRP1 and SorCS1 were identified in some studies as LOAD risk factors. On the other hand, a genetic association of sortilin and calsyntenin-1 with AD has not been demonstrated so far.
APP interacts directly with LRP1, SorLA, SorCS1, and sortilin and these interactions are thought to link APP to specific intracellular sorting machineries. Thus, SorLA seems to link APP to the retromer complex which has been suggested as an AD risk factor, but retromer does not interact directly with APP. In contrast, LRP1 also interacts indirectly with APP via Fe65 and calsyntenin-1 interacts indirectly with APP and this interaction is bridged by the cytosolic adaptor protein X11L. This indirect interaction could link APP through calsyntenin-1 to kinesins and enable microtubule-dependent axonal transport.
In addition to full-length APP, LRP1, SorLA, and sortilin interact directly, or in the case of sortilin indirectly, with Aβ and are expected to partake in its clearance. It is under debate which cells convey Aβ clearance. However, recent human genome-wide association studies and systems-biology approaches have identified an unexpectedly dominant role of the microglial innate immune response in increasing the risk of developing AD [275]. SorLA, LRP1, and calsyntenin-1 are expressed in microglia [276] and LRP1 associates with the lipid transporter ABCA7 on the cell membrane [277]. ABCA7 and other transmembrane proteins such as Trem2 and CD33 have been recognized as LOAD risk factors, identified in microglia, related to the immune response and Aβ clearance [275, 278, 279]. Future studies will have to prove if SorLA in addition to LRP1 functions as a microglial scavenger receptor for Aβ.
In conclusion, our understanding which proteins are modulating APPs intracellular transport has improved, but we still lack detailed information on the specific sorting steps determining APPs subcellular targeting and which miss targeting events underlie disease development.
References
Huang Y, Mucke L (2012) Alzheimer mechanisms and therapeutic strategies. Cell 148(6):1204–1222
Tanzi RE, Bertram L (2005) Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120(4):545–555
Small SA, Gandy S (2006) Sorting through the cell biology of Alzheimer’s disease: intracellular pathways to pathogenesis. Neuron 52(1):15–31
King GD, Scott Turner R (2004) Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer’s disease risk? Exp Neurol 185(2):208–219
Back S et al (2007) Beta-amyloid precursor protein can be transported independent of any sorting signal to the axonal and dendritic compartment. J Neurosci Res 85(12):2580–2590
Szodorai A et al (2009) APP anterograde transport requires Rab3A GTPase activity for assembly of the transport vesicle. J Neurosci: Off J Soc Neurosci 29(46):14534–14544
Kuhn PH et al (2010) ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J 29(17):3020–3032
Lammich S et al (1999) Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A 96(7):3922–3927
Slack BE, Ma LK, Seah CC (2001) Constitutive shedding of the amyloid precursor protein ectodomain is up-regulated by tumour necrosis factor-alpha converting enzyme. Biochem J 357(Pt 3):787–794
Lichtenthaler SF (2011) Alpha-secretase in Alzheimer’s disease: molecular identity, regulation and therapeutic potential. J Neurochem 116(1):10–21
Lichtenthaler SF, Haass C, Steiner H (2011) Regulated intramembrane proteolysis—lessons from amyloid precursor protein processing. J Neurochem 117(5):779–796
Zhao G et al (2004) Identification of a new presenilin-dependent zeta-cleavage site within the transmembrane domain of amyloid precursor protein. J Biol Chem 279(49):50647–50650
Weidemann A et al (2002) A novel epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with notch processing. Biochemistry 41(8):2825–2835
Sastre M et al (2001) Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of notch. EMBO Rep 2(9):835–841
Gu Y et al (2001) Distinct intramembrane cleavage of the beta-amyloid precursor protein family resembling gamma-secretase-like cleavage of notch. J Biol Chem 276(38):35235–35238
Haass C et al (1993) Beta-amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J Biol Chem 268(5):3021–3024
Vassar R et al (1999) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286(5440):735–741
Vassar R et al (2009) The beta-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J Neurosci 29(41):12787–12794
Haass C et al (1992) Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature 359(6393):322–325
Qi-Takahara Y et al (2005) Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J Neurosci Off J Soc Neurosci 25(2):436–445
Xia W et al (1997) Enhanced production and oligomerization of the 42-residue amyloid beta-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem 272(12):7977–7982
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580):353–356
Selkoe DJ, Wolfe MS (2007) Presenilin: running with scissors in the membrane. Cell 131(2):215–221
Weggen S, Beher D (2012) Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer’s disease. Alzheimers Res Ther 4(2):9
Duering M et al (2005) Mean age of onset in familial Alzheimer’s disease is determined by amyloid beta 42. Neurobiol Aging 26(6):785–788
Takami M et al (2009) Gamma-secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci: Off J Soc Neurosci 29(41):13042–13052
Andrew RJ et al (2016) A Greek tragedy: the growing complexity of Alzheimer amyloid precursor protein proteolysis. J Biol Chem 291(37):19235–19244
Willem M et al (2015) Eta-secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 526(7573):443–447
Tyan SH, Koo EH (2015) New tricks from an old dog: another synaptotoxic fragment from APP. Cell Res 25(11):1185–1186
Ahmad M et al (2006) Cleavage of amyloid-beta precursor protein (APP) by membrane-type matrix metalloproteinases. J Biochem 139(3):517–526
Baranger K et al (2016) MT5-MMP promotes Alzheimer’s pathogenesis in the frontal cortex of 5xFAD mice and APP trafficking in vitro. Front Mol Neurosci 9:163
Yong VW et al (2001) Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci 2(7):502–511
Visse R, Nagase H (2003) Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res 92(8):827–839
Wang X, Pei D (2001) Shedding of membrane type matrix metalloproteinase 5 by a furin-type convertase: a potential mechanism for down-regulation. J Biol Chem 276(38):35953–35960
Sekine-Aizawa Y et al (2001) Matrix metalloproteinase (MMP) system in brain: Identification and characterization of brain-specific MMP highly expressed in cerebellum. Eur J Neurosci 13(5):935–948
Tienari PJ et al (1996) Neuronal sorting and processing of amyloid precursor protein: implications for Alzheimer’s disease. Cold Spring Harb Symp Quant Biol 61:575–585
Zhang Z et al (2015) Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat Commun 6:8762
Schonherr C et al (2016) Generation of aggregation prone N-terminally truncated amyloid beta peptides by meprin beta depends on the sequence specificity at the cleavage site. Mol Neurodegener 11:19
Jackle F et al (2015) Metalloprotease meprin beta is activated by transmembrane serine protease matriptase-2 at the cell surface thereby enhancing APP shedding. Biochem J 470(1):91–103
Arolas JL et al (2012) Structural basis for the sheddase function of human meprin beta metalloproteinase at the plasma membrane. Proc Natl Acad Sci U S A 109(40):16131–16136
Bien J et al (2012) The metalloprotease meprin beta generates amino terminal-truncated amyloid beta peptide species. J Biol Chem 287(40):33304–33313
Jefferson T et al (2011) Metalloprotease meprin beta generates nontoxic N-terminal amyloid precursor protein fragments in vivo. J Biol Chem 286(31):27741–27750
Sannerud R et al (2016) Restricted location of PSEN2/gamma-secretase determines substrate specificity and generates an intracellular Abeta pool. Cell 166(1):193–208
Meckler X, Checler F (2016) Presenilin 1 and presenilin 2 target gamma-secretase complexes to distinct cellular compartments. J Biol Chem 291(24):12821–12837
Kaether C et al (2006) Amyloid precursor protein and notch intracellular domains are generated after transport of their precursors to the cell surface. Traffic 7(4):408–415
Skovronsky DM et al (2000) Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J Biol Chem 275(4):2568–2575
Rocchi A et al (2003) Causative and susceptibility genes for Alzheimer’s disease: a review. Brain Res Bull 61(1):1–24
Haass C et al (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2(5):a006270
Caporaso GL et al (1994) Morphologic and biochemical analysis of the intracellular trafficking of the Alzheimer beta/A4 amyloid precursor protein. J Neurosci: Off J Soc Neurosci 14(5 Pt 2):3122–3138
Palacios G et al (1992) Beta-amyloid precursor protein localization in the Golgi apparatus in neurons and oligodendrocytes. An immunocytochemical structural and ultrastructural study in normal and axotomized neurons. Brain Res Mol Brain Res 15(3–4):195–206
Guo Q et al (2012) Amyloid precursor protein revisited: neuron-specific expression and highly stable nature of soluble derivatives. J Biol Chem 287(4):2437–2445
Das U et al (2013) Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron 79(3):447–460
Willnow TE, Andersen OM (2013) Sorting receptor SORLA—a trafficking path to avoid Alzheimer disease. J Cell Sci 126(Pt 13):2751–2760
Haass C et al (1992) Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature 357(6378):500–503
Cole GM et al (1992) An endosomal-lysosomal pathway for degradation of amyloid precursor protein. Ann N Y Acad Sci 674:103–117
Ehehalt R et al (2003) Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol 160(1):113–123
Schneider A et al (2008) Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J Neurosci 28(11):2874–2882
Lai A, Sisodia SS, Trowbridge IS (1995) Characterization of sorting signals in the beta-amyloid precursor protein cytoplasmic domain. J Biol Chem 270(8):3565–3573
Koo EH et al (1990) Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A 87(4):1561–1565
Amaratunga A, Fine RE (1995) Generation of amyloidogenic C-terminal fragments during rapid axonal transport in vivo of beta-amyloid precursor protein in the optic nerve. J Biol Chem 270(29):17268–17272
Morin PJ et al (1993) Amyloid precursor protein is synthesized by retinal ganglion cells, rapidly transported to the optic nerve plasma membrane and nerve terminals, and metabolized. J Neurochem 61(2):464–473
Kaether C, Skehel P, Dotti CG (2000) Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol Biol Cell 11(4):1213–1224
Yamazaki T, Selkoe DJ, Koo EH (1995) Trafficking of cell surface beta-amyloid precursor protein: retrograde and transcytotic transport in cultured neurons. J Cell Biol 129(2):431–442
Ferreira A, Caceres A, Kosik KS (1993) Intraneuronal compartments of the amyloid precursor protein. J Neurosci 13(7):3112–3123
Bloom GS et al (1988) Native structure and physical properties of bovine brain kinesin and identification of the ATP-binding subunit polypeptide. Biochemistry 27(9):3409–3416
DeBoer SR et al (2008) Conventional kinesin holoenzymes are composed of heavy and light chain homodimers. Biochemistry 47(15):4535–4543
Gyoeva FK, Bybikova EM, Minin AA (2000) An isoform of kinesin light chain specific for the Golgi complex. J Cell Sci 113(Pt 11):2047–2054
Cyr JL et al (1991) Molecular genetics of kinesin light chains: generation of isoforms by alternative splicing. Proc Natl Acad Sci U S A 88(22):10114–10118
Khodjakov A et al (1998) A specific light chain of kinesin associates with mitochondria in cultured cells. Mol Biol Cell 9(2):333–343
Morfini G et al (2016) Conventional kinesin: biochemical heterogeneity and functional implications in health and disease. Brain Res Bull 126(Pt 3):347–353
Lazarov O et al (2005) Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci 25(9):2386–2395
Matsuda S, Matsuda Y, D'Adamio L (2003) Amyloid beta protein precursor (AbetaPP), but not AbetaPP-like protein 2, is bridged to the kinesin light chain by the scaffold protein JNK-interacting protein 1. J Biol Chem 278(40):38601–38606
Inomata H et al (2003) A scaffold protein JIP-1b enhances amyloid precursor protein phosphorylation by JNK and its association with kinesin light chain 1. J Biol Chem 278(25):22946–22955
Chiba K et al (2014) Quantitative analysis of APP axonal transport in neurons: role of JIP1 in enhanced APP anterograde transport. Mol Biol Cell 25(22):3569–3580
Fu MM, Holzbaur EL (2013) JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J Cell Biol 202(3):495–508
Vagnoni A et al (2013) Loss of c-Jun N-terminal kinase-interacting protein-1 does not affect axonal transport of the amyloid precursor protein or abeta production. Hum Mol Genet 22(22):4646–4652
Rusu P et al (2007) Axonal accumulation of synaptic markers in APP transgenic drosophila depends on the NPTY motif and is paralleled by defects in synaptic plasticity. Eur J Neurosci 25(4):1079–1086
Dieckmann M, Dietrich MF, Herz J (2010) Lipoprotein receptors—an evolutionarily ancient multifunctional receptor family. Biol Chem 391(11):1341–1363
Nykjaer A, Willnow TE (2002) The low-density lipoprotein receptor gene family: a cellular Swiss army knife? Trends Cell Biol 12(6):273–280
Pohlkamp T, Wasser CR, Herz J (2017) Functional roles of the interaction of APP and lipoprotein receptors. Front Mol Neurosci 10:54
Brown MS, Goldstein JL (1986) A receptor-mediated pathway for cholesterol homeostasis. Science 232(4746):34–47
May P, Bock HH, Herz J (2003) Integration of endocytosis and signal transduction by lipoprotein receptors. Sci STKE 2003(176):PE12
Wagner T, Pietrzik CU (2012) The role of lipoprotein receptors on the physiological function of APP. Exp Brain Res 217(3–4):377–387
Marcusson EG et al (1994) The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell 77(4):579–586
Jacobsen L et al (1996) Molecular characterization of a novel human hybrid-type receptor that binds the alpha2-macroglobulin receptor-associated protein. J Biol Chem 271(49):31379–31383
Yamazaki H et al (1996) Elements of neural adhesion molecules and a yeast vacuolar protein sorting receptor are present in a novel mammalian low density lipoprotein receptor family member. J Biol Chem 271(40):24761–24768
Hermey G (2009) The Vps10p-domain receptor family. Cell Mol Life Sci: CMLS 66(16):2677–2689
Campbell ID, Spitzfaden C (1994) Building proteins with fibronectin type III modules. Structure 2(5):333–337
Quistgaard EM et al (2009) Ligands bind to sortilin in the tunnel of a ten-bladed beta-propeller domain. Nat Struct Mol Biol 16(1):96–98
Westergaard UB et al (2004) Functional organization of the sortilin Vps10p domain. J Biol Chem 279(48):50221–50229
Vogt L et al (2001) Calsyntenin-1, a proteolytically processed postsynaptic membrane protein with a cytoplasmic calcium-binding domain. Mol Cell Neurosci 17(1):151–166
Gul IS et al (2017) Evolution and diversity of cadherins and catenins. Exp Cell Res 358(1):3–9
Hintsch G et al (2002) The calsyntenins—a family of postsynaptic membrane proteins with distinct neuronal expression patterns. Mol Cell Neurosci 21(3):393–409
Pettem KL et al (2013) The specific alpha-neurexin interactor calsyntenin-3 promotes excitatory and inhibitory synapse development. Neuron 80(1):113–128
Araki Y et al (2007) The novel cargo Alcadein induces vesicle association of kinesin-1 motor components and activates axonal transport. EMBO J 26(6):1475–1486
Konecna A et al (2006) Calsyntenin-1 docks vesicular cargo to kinesin-1. Mol Biol Cell 17(8):3651–3663
Araki Y et al (2003) Novel cadherin-related membrane proteins, Alcadeins, enhance the X11-like protein-mediated stabilization of amyloid beta-protein precursor metabolism. J Biol Chem 278(49):49448–49458
Herz J et al (1988) Surface location and high affinity for calcium of a 500-kd liver membrane protein closely related to the LDL-receptor suggest a physiological role as lipoprotein receptor. EMBO J 7(13):4119–4127
Willnow TE et al (1996) The low-density-lipoprotein receptor-related protein (LRP) is processed by furin in vivo and in vitro. Biochem J 313(Pt 1):71–76
Waldron E et al (2008) LRP1 modulates APP trafficking along early compartments of the secretory pathway. Neurobiol Dis 31(2):188–197
Jaeger S, Pietrzik CU (2008) Functional role of lipoprotein receptors in Alzheimer’s disease. Curr Alzheimer Res 5(1):15–25
Bu G (2009) Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10(5):333–344
Blacker D et al (1998) Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat Genet 19(4):357–360
Roses AD (1996) Apolipoprotein E and Alzheimer’s disease. A rapidly expanding field with medical and epidemiological consequences. Ann N Y Acad Sci 802:50–57
Kang DE et al (2000) Modulation of amyloid beta-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor-related protein pathway. J Clin Invest 106(9):1159–1166
Ulery PG et al (2000) Modulation of beta-amyloid precursor protein processing by the low density lipoprotein receptor-related protein (LRP). Evidence that LRP contributes to the pathogenesis of Alzheimer’s disease. J Biol Chem 275(10):7410–7415
Kounnas MZ et al (1995) LDL receptor-related protein, a multifunctional ApoE receptor, binds secreted beta-amyloid precursor protein and mediates its degradation. Cell 82(2):331–340
Trommsdorff M et al (1998) Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem 273(50):33556–33560
Sandbrink R, Masters CL, Beyreuther K et al (1996) Ann N Y Acad Sci 777:281–287
Klug W et al (2011) Phosphorylation of LRP1 regulates the interaction with Fe65. FEBS Lett 585(20):3229–3235
Pietrzik CU et al (2004) FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J Neurosci: Off J Soc Neurosci 24(17):4259–4265
Li Y et al (2000) The YXXL motif, but not the two NPXY motifs, serves as the dominant endocytosis signal for low density lipoprotein receptor-related protein. J Biol Chem 275(22):17187–17194
Reekmans SM et al (2010) Inactivation of the proximal NPXY motif impairs early steps in LRP1 biosynthesis. Cell Mol Life Sci 67(1):135–145
Cam JA et al (2005) Rapid endocytosis of the low density lipoprotein receptor-related protein modulates cell surface distribution and processing of the beta-amyloid precursor protein. J Biol Chem 280(15):15464–15470
Pietrzik CU et al (2002) The cytoplasmic domain of the LDL receptor-related protein regulates multiple steps in APP processing. EMBO J 21(21):5691–5700
Koo EH, Squazzo SL (1994) Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem 269(26):17386–17389
Herr UM et al (2017) LRP1 modulates APP intraneuronal transport and processing in its monomeric and dimeric state. Front Mol Neurosci 10:118
Eggert S et al (2017) Dimerization leads to changes in APP (amyloid precursor protein) trafficking mediated by LRP1 and SorLA. Cell Mol Life Sci. https://doi.org/10.1007/s00018-017-2625-7
Cam JA et al (2004) The low density lipoprotein receptor-related protein 1B retains beta-amyloid precursor protein at the cell surface and reduces amyloid-beta peptide production. J Biol Chem 279(28):29639–29646
Haas J et al (2011) LRP1b shows restricted expression in human tissues and binds to several extracellular ligands, including fibrinogen and apoE-carrying lipoproteins. Atherosclerosis 216(2):342–347
Strickland DK et al (1990) Sequence identity between the alpha 2-macroglobulin receptor and low density lipoprotein receptor-related protein suggests that this molecule is a multifunctional receptor. J Biol Chem 265(29):17401–17404
Kristensen T et al (1990) Evidence that the newly cloned low-density-lipoprotein receptor related protein (LRP) is the alpha 2-macroglobulin receptor. FEBS Lett 276(1–2):151–155
Liu CX et al (2001) The putative tumor suppressor LRP1B, a novel member of the low density lipoprotein (LDL) receptor family, exhibits both overlapping and distinct properties with the LDL receptor-related protein. J Biol Chem 276(31):28889–28896
Lane-Donovan C, Philips GT, Herz J (2014) More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron 83(4):771–787
von Arnim CA et al (2005) The low density lipoprotein receptor-related protein (LRP) is a novel beta-secretase (BACE1) substrate. J Biol Chem 280(18):17777–17785
von Einem B et al (2010) The role of low-density receptor-related protein 1 (LRP1) as a competitive substrate of the amyloid precursor protein (APP) for BACE1. Exp Neurol 225(1):85–93
May P, Reddy YK, Herz J (2002) Proteolytic processing of low density lipoprotein receptor-related protein mediates regulated release of its intracellular domain. J Biol Chem 277(21):18736–18743
Lleo A et al (2005) Low density lipoprotein receptor-related protein (LRP) interacts with presenilin 1 and is a competitive substrate of the amyloid precursor protein (APP) for gamma-secretase. J Biol Chem 280(29):27303–27309
Deane R et al (2004) LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43(3):333–344
Deane R et al (2008) apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest 118(12):4002–4013
Rebeck GW et al (1993) Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron 11(4):575–580
Pflanzner T et al (2011) LRP1 mediates bidirectional transcytosis of amyloid-beta across the blood-brain barrier. Neurobiol Aging 32(12):2323 e1–2323 11
Storck SE et al (2016) Endothelial LRP1 transports amyloid-beta(1-42) across the blood-brain barrier. J Clin Invest 126(1):123–136
Shibata M et al (2000) Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 106(12):1489–1499
Sagare A et al (2007) Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med 13(9):1029–1031
Kanekiyo T, Bu G (2014) The low-density lipoprotein receptor-related protein 1 and amyloid-beta clearance in Alzheimer's disease. Front Aging Neurosci 6:93
Taira K et al (2001) LR11, a mosaic LDL receptor family member, mediates the uptake of ApoE-rich lipoproteins in vitro. Arterioscler Thromb Vasc Biol 21(9):1501–1506
Petersen CM et al (1997) Molecular identification of a novel candidate sorting receptor purified from human brain by receptor-associated protein affinity chromatography. J Biol Chem 272(6):3599–3605
Klinger SC et al (2011) SorLA regulates the activity of lipoprotein lipase by intracellular trafficking. J Cell Sci 124(Pt 7):1095–1105
Gliemann J et al (2004) The mosaic receptor sorLA/LR11 binds components of the plasminogen-activating system and platelet-derived growth factor-BB similarly to LRP1 (low-density lipoprotein receptor-related protein), but mediates slow internalization of bound ligand. Biochem J 381(Pt 1):203–212
Jacobsen L et al (2001) Activation and functional characterization of the mosaic receptor SorLA/LR11. J Biol Chem 276(25):22788–22796
Larsen JV et al (2016) Cytokine-like factor 1, an essential facilitator of cardiotrophin-like cytokine:ciliary neurotrophic factor receptor alpha signaling and sorLA-mediated turnover. Mol Cell Biol 36(8):1272–1286
Larsen JV, Petersen CM (2017) SorLA and CLC:CLF-1-dependent downregulation of CNTFRalpha as demonstrated by western blotting, inhibition of lysosomal enzymes, and immunocytochemistry. J Vis Exp. https://doi.org/10.3791/55019
Andersen OM et al (2006) Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry 45(8):2618–2628
Mehmedbasic A et al (2015) SorLA complement-type repeat domains protect the amyloid precursor protein against processing. J Biol Chem 290(6):3359–3376
Spoelgen R et al (2006) Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and beta-secretase beta-site APP-cleaving enzyme. J Neurosci: Off J Soc Neurosci 26(2):418–428
Caglayan S et al (2014) Lysosomal sorting of amyloid-beta by the SORLA receptor is impaired by a familial Alzheimer’s disease mutation. Science translational medicine 6(223):223ra20
Kitago Y et al (2015) Structural basis for amyloidogenic peptide recognition by sorLA. Nat Struct Mol Biol 22(3):199–206
Hermans-Borgmeyer I et al (1998) Unique expression pattern of a novel mosaic receptor in the developing cerebral cortex. Mech Dev 70(1–2):65–76
Andersen OM, Rudolph IM, Willnow TE (2016) Risk factor SORL1: from genetic association to functional validation in Alzheimer's disease. Acta Neuropathol 132(5):653–665
Scherzer CR et al (2004) Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch Neurol 61(8):1200–1205
Andersen OM et al (2005) Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A 102(38):13461–13466
Burgert T et al (2013) SORLA-dependent and -independent functions for PACS1 in control of amyloidogenic processes. Mol Cell Biol 33(21):4308–4320
Gustafsen C et al (2013) Sortilin and SorLA display distinct roles in processing and trafficking of amyloid precursor protein. J Neurosci: Off J Soc Neurosci 33(1):64–71
Klinger SC et al (2016) Polarized trafficking of the sorting receptor SorLA in neurons and MDCK cells. FEBS J 283(13):2476–2493
Hermey G et al (2006) Tumour necrosis factor alpha-converting enzyme mediates ectodomain shedding of Vps10p-domain receptor family members. Biochem J 395(2):285–293
Bohm C et al (2006) SorLA signaling by regulated intramembrane proteolysis. J Biol Chem 281(21):14547–14553
Nielsen MS et al (2007) Sorting by the cytoplasmic domain of the amyloid precursor protein binding receptor SorLA. Mol Cell Biol 27(19):6842–6851
Schmidt V et al (2007) SorLA/LR11 regulates processing of amyloid precursor protein via interaction with adaptors GGA and PACS-1. J Biol Chem 282(45):32956–32964
Herskowitz JH et al (2012) GGA1-mediated endocytic traffic of LR11/SorLA alters APP intracellular distribution and amyloid-beta production. Mol Biol Cell 23(14):2645–2657
Jacobsen L et al (2002) The sorLA cytoplasmic domain interacts with GGA1 and -2 and defines minimum requirements for GGA binding. FEBS Lett 511(1–3):155–158
Dumanis SB et al (2015) Distinct functions for anterograde and retrograde sorting of SORLA in amyloidogenic processes in the brain. J Neurosci: Off J Soc Neurosci 35(37):12703–12713
Fjorback AW et al (2012) Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J Neurosci: Off J Soc Neurosci 32(4):1467–1480
Cullen PJ, Korswagen HC (2011) Sorting nexins provide diversity for retromer-dependent trafficking events. Nat Cell Biol 14(1):29–37
Muhammad A et al (2008) Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc Natl Acad Sci U S A 105(20):7327–7332
Wen L et al (2011) VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J Cell Biol 195(5):765–779
Holtzman DM, Herz J, Bu G (2012) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2(3):a006312
Schmidt V et al (2012) Quantitative modelling of amyloidogenic processing and its influence by SORLA in Alzheimer’s disease. EMBO J 31(1):187–200
Huang TY et al (2016) SNX27 and SORLA interact to reduce amyloidogenic subcellular distribution and processing of amyloid precursor protein. J Neurosci: Off J Soc Neurosci 36(30):7996–8011
Clairfeuille T et al (2016) A molecular code for endosomal recycling of phosphorylated cargos by the SNX27-retromer complex. Nat Struct Mol Biol 23(10):921–932
Lauffer BE et al (2010) SNX27 mediates PDZ-directed sorting from endosomes to the plasma membrane. J Cell Biol 190(4):565–574
Grupe A et al (2006) A scan of chromosome 10 identifies a novel locus showing strong association with late-onset Alzheimer disease. Am J Hum Genet 78(1):78–88
Liang X et al (2009) Genomic convergence to identify candidate genes for Alzheimer disease on chromosome 10. Hum Mutat 30(3):463–471
Reitz C et al (2011) SORCS1 alters amyloid precursor protein processing and variants may increase Alzheimer’s disease risk. Ann Neurol 69(1):47–64
Wang HF et al (2012) SORCS1 and APOE polymorphisms interact to confer risk for late-onset Alzheimer’s disease in a northern Han Chinese population. Brain Res 1448:111–116
Xu W et al (2013) The genetic variation of SORCS1 is associated with late-onset Alzheimer’s disease in Chinese Han population. PLoS One 8(5):e63621
Hermey G et al (1999) Identification and characterization of SorCS, a third member of a novel receptor family. Biochem Biophys Res Commun 266(2):347–351
Savas JN et al (2015) The sorting receptor SorCS1 regulates trafficking of neurexin and AMPA receptors. Neuron 87(4):764–780
Traunmuller L et al (2016) Control of neuronal synapse specification by a highly dedicated alternative splicing program. Science 352(6288):982–986
Lane RF et al (2010) Diabetes-associated SorCS1 regulates Alzheimer’s amyloid-beta metabolism: evidence for involvement of SorL1 and the retromer complex. J Neurosci: Off J Soc Neurosci 30(39):13110–13115
Hermey G et al (2015) SorCS1 variants and amyloid precursor protein (APP) are co-transported in neurons but only SorCS1c modulates anterograde APP transport. J Neurochem 135(1):60–75
Hermey G et al (2004) The three sorCS genes are differentially expressed and regulated by synaptic activity. J Neurochem 88(6):1470–1476
Hermey G, Schaller HC, Hermans-Borgmeyer I (2001) Transient expression of SorCS in developing telencephalic and mesencephalic structures of the mouse. Neuroreport 12(1):29–32
Oetjen S et al (2014) Spatiotemporal expression analysis of the growth factor receptor SorCS3. J Comp Neurol 522(15):3386–3402
Reitz C et al (2011) Impact of genetic variation in SORCS1 on memory retention. PLoS One 6(10):e24588
Hermey G et al (2003) Characterization of sorCS1, an alternatively spliced receptor with completely different cytoplasmic domains that mediate different trafficking in cells. J Biol Chem 278(9):7390–7396
Nielsen MS et al (2008) Different motifs regulate trafficking of SorCS1 isoforms. Traffic 9(6):980–994
Doray B et al (2012) Do GGA adaptors bind internal DXXLL motifs? Traffic 13(10):1315–1325
Clee SM et al (2006) Positional cloning of Sorcs1, a type 2 diabetes quantitative trait locus. Nat Genet 38(6):688–693
Goodarzi MO et al (2007) SORCS1: a novel human type 2 diabetes susceptibility gene suggested by the mouse. Diabetes 56(7):1922–1929
Granhall C et al (2006) High-resolution quantitative trait locus analysis reveals multiple diabetes susceptibility loci mapped to intervals <800 kb in the species-conserved Niddm1i of the GK rat. Genetics 174(3):1565–1572
Paterson AD et al (2010) A genome-wide association study identifies a novel major locus for glycemic control in type 1 diabetes, as measured by both A1C and glucose. Diabetes 59(2):539–549
Mazella J et al (1998) The 100-kDa neurotensin receptor is gp95/sortilin, a non-G-protein-coupled receptor. J Biol Chem 273(41):26273–26276
Sarret P et al (2003) Distribution of NTS3 receptor/sortilin mRNA and protein in the rat central nervous system. J Comp Neurol 461(4):483–505
Hermans-Borgmeyer I et al (1999) Expression of the 100-kDa neurotensin receptor sortilin during mouse embryonal development. Brain Res Mol Brain Res 65(2):216–219
Munck Petersen C et al (1999) Propeptide cleavage conditions sortilin/neurotensin receptor-3 for ligand binding. EMBO J 18(3):595–604
Westergaard UB et al (2005) SorCS3 does not require propeptide cleavage to bind nerve growth factor. FEBS Lett 579(5):1172–1176
Larsen JV et al (2014) Human sorCS1 binds sortilin and hampers its cellular functions. Biochem J 457(2):277–288
Nykjaer A et al (2004) Sortilin is essential for proNGF-induced neuronal cell death. Nature 427(6977):843–848
Teng HK et al (2005) ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci: Off J Soc Neurosci 25(22):5455–5463
Larsen JV et al (2010) Sortilin facilitates signaling of ciliary neurotrophic factor and related helical type 1 cytokines targeting the gp130/leukemia inhibitory factor receptor beta heterodimer. Mol Cell Biol 30(17):4175–4187
Nielsen MS et al (1999) Sortilin/neurotensin receptor-3 binds and mediates degradation of lipoprotein lipase. J Biol Chem 274(13):8832–8836
Nilsson SK et al (2008) Endocytosis of apolipoprotein A-V by members of the low density lipoprotein receptor and the VPS10p domain receptor families. J Biol Chem 283(38):25920–25927
Carlo AS et al (2013) The pro-neurotrophin receptor sortilin is a major neuronal apolipoprotein E receptor for catabolism of amyloid-beta peptide in the brain. J Neurosci: Off J Soc Neurosci 33(1):358–370
Seaman MN (2007) Identification of a novel conserved sorting motif required for retromer-mediated endosome-to-TGN retrieval. J Cell Sci 120(Pt 14):2378–2389
Mari M et al (2008) SNX1 defines an early endosomal recycling exit for sortilin and mannose 6-phosphate receptors. Traffic 9(3):380–393
Canuel M et al (2008) Sortilin mediates the lysosomal targeting of cathepsins D and H. Biochem Biophys Res Commun 373(2):292–297
Corder EH et al (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261(5123):921–923
Yang M et al (2013) The intracellular domain of sortilin interacts with amyloid precursor protein and regulates its lysosomal and lipid raft trafficking. PLoS One 8(5):e63049
Soba P et al (2005) Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J 24(20):3624–3634
Koo EH et al (1996) Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. J Cell Sci 109(Pt 5):991–998
Burgos PV et al (2010) Sorting of the Alzheimer’s disease amyloid precursor protein mediated by the AP-4 complex. Dev Cell 18(3):425–436
Reitz C et al (2013) Independent and epistatic effects of variants in VPS10-d receptors on Alzheimer disease risk and processing of the amyloid precursor protein (APP). Transl Psychiatry 3:e256
Mufson EJ et al (2010) Preservation of cortical sortilin protein levels in MCI and Alzheimer’s disease. Neurosci Lett 471(3):129–133
Hu X et al (2017) Sortilin fragments deposit at senile plaques in human cerebrum. Front Neuroanat 11:45
Finan GM, Okada H, Kim TW (2011) BACE1 retrograde trafficking is uniquely regulated by the cytoplasmic domain of sortilin. J Biol Chem 286(14):12602–12616
Saadipour K et al (2013) Amyloid beta(1)(−)(4)(2) (Abeta(4)(2)) up-regulates the expression of sortilin via the p75(NTR)/RhoA signaling pathway. J Neurochem 127(2):152–162
Tan J, Evin G (2012) Beta-site APP-cleaving enzyme 1 trafficking and Alzheimer’s disease pathogenesis. J Neurochem 120(6):869–880
Nyborg AC et al (2006) Sortilin, SorCS1b, and SorLA Vps10p sorting receptors, are novel gamma-secretase substrates. Mol Neurodegener 1:3
Horiuchi K et al (2007) Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell 18(1):176–188
Evans SF et al (2011) Neuronal brain-derived neurotrophic factor is synthesized in excess, with levels regulated by sortilin-mediated trafficking and lysosomal degradation. J Biol Chem 286(34):29556–29567
Navarro V, Vincent JP, Mazella J (2002) Shedding of the luminal domain of the neurotensin receptor-3/sortilin in the HT29 cell line. Biochem Biophys Res Commun 298(5):760–764
Qi L et al (2011) Genetic risk score and risk of myocardial infarction in Hispanics. Circulation 123(4):374–380
Takeuchi F et al (2012) Association of genetic variants influencing lipid levels with coronary artery disease in Japanese individuals. PLoS One 7(9):e46385
Arvind P et al (2014) CELSR2-PSRC1-SORT1 gene expression and association with coronary artery disease and plasma lipid levels in an Asian Indian cohort. J Cardiol 64(5):339–346
Jones GT et al (2013) A sequence variant associated with sortilin-1 (SORT1) on 1p13.3 is independently associated with abdominal aortic aneurysm. Hum Mol Genet 22(14):2941–2947
Lee JY et al (2013) A genome-wide association study of a coronary artery disease risk variant. J Hum Genet 58(3):120–126
Angelakopoulou A et al (2012) Comparative analysis of genome-wide association studies signals for lipids, diabetes, and coronary heart disease: cardiovascular biomarker genetics collaboration. Eur Heart J 33(3):393–407
Stampfer MJ (2006) Cardiovascular disease and Alzheimer’s disease: common links. J Intern Med 260(3):211–223
Andersson CH et al (2016) A genetic variant of the sortilin 1 gene is associated with reduced risk of Alzheimer’s disease. J Alzheimers Dis: JAD 53(4):1353–1363
Zeng F et al (2013) No association of SORT1 gene polymorphism with sporadic Alzheimer’s disease in the Chinese Han population. Neuroreport 24(9):464–468
Lambert JC et al (2013) Genome-wide haplotype association study identifies the FRMD4A gene as a risk locus for Alzheimer’s disease. Mol Psychiatry 18(4):461–470
Naj AC et al (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43(5):436–441
Beecham GW et al (2014) Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet 10(9):e1004606
Reynolds CA et al (2010) Analysis of lipid pathway genes indicates association of sequence variation near SREBF1/TOM1L2/ATPAF2 with dementia risk. Hum Mol Genet 19(10):2068–2078
Rogaeva E et al (2007) The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 39(2):168–177
Ward ME, Miller BL (2011) Potential mechanisms of progranulin-deficient FTLD. J Mol Neurosci: MN 45(3):574–582
Rademakers R, Neumann M, Mackenzie IR (2012) Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 8(8):423–434
Bird T et al (2003) Epidemiology and genetics of frontotemporal dementia/Pick’s disease. Ann Neurol 54(Suppl 5):S29–S31
Hu F et al (2010) Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68(4):654–667
Carrasquillo MM et al (2010) Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am J Hum Genet 87(6):890–897
Zheng Y et al (2011) C-terminus of progranulin interacts with the beta-propeller region of sortilin to regulate progranulin trafficking. PLoS One 6(6):e21023
Gendron TF, Rademakers R, Petrucelli L (2013) TARDBP mutation analysis in TDP-43 proteinopathies and deciphering the toxicity of mutant TDP-43. J Alzheimers Dis 33(Suppl 1):S35–S45
Polymenidou M et al (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14(4):459–468
Prudencio M et al (2012) Misregulation of human sortilin splicing leads to the generation of a nonfunctional progranulin receptor. Proc Natl Acad Sci U S A 109(52):21510–21515
Rogelj B et al (2006) The X11/mint family of adaptor proteins. Brain Res Rev 52(2):305–315
Um JW et al (2014) Calsyntenins function as synaptogenic adhesion molecules in concert with neurexins. Cell Rep 6(6):1096–1109
Araki Y et al (2004) Coordinated metabolism of Alcadein and amyloid beta-protein precursor regulates FE65-dependent gene transactivation. J Biol Chem 279(23):24343–24354
Hata S et al (2009) Alcadein cleavages by amyloid beta-precursor protein (APP) alpha- and gamma-secretases generate small peptides, p3-Alcs, indicating Alzheimer disease-related gamma-secretase dysfunction. J Biol Chem 284(52):36024–36033
Maruta C et al (2012) Constitutive cleavage of the single-pass transmembrane protein alcadeinalpha prevents aberrant peripheral retention of Kinesin-1. PLoS One 7(8):e43058
Hata S et al (2011) Alternative processing of gamma-secretase substrates in common forms of mild cognitive impairment and Alzheimer’s disease: evidence for gamma-secretase dysfunction. Ann Neurol 69(6):1026–1031
Vagnoni A et al (2012) Calsyntenin-1 mediates axonal transport of the amyloid precursor protein and regulates Abeta production. Hum Mol Genet 21(13):2845–2854
Ringman JM et al (2012) Proteomic changes in cerebrospinal fluid of presymptomatic and affected persons carrying familial Alzheimer disease mutations. Arch Neurol 69(1):96–104
Uchida Y et al (2013) Calsyntenin-3 C-terminal fragment accumulates in dystrophic neurites surrounding abeta plaques in tg2576 mouse and Alzheimer disease brains: Its neurotoxic role in mediating dystrophic neurite formation. Am J Pathol 182(5):1718–1726
Ludwig A et al (2009) Calsyntenins mediate TGN exit of APP in a kinesin-1-dependent manner. Traffic 10(5):572–589
Vagnoni A et al (2011) Phosphorylation of kinesin light chain 1 at serine 460 modulates binding and trafficking of calsyntenin-1. J Cell Sci 124(Pt 7):1032–1042
Steuble M et al (2010) Molecular characterization of a trafficking organelle: dissecting the axonal paths of calsyntenin-1 transport vesicles. Proteomics 10(21):3775–3788
Goldsbury C et al (2006) Inhibition of APP trafficking by tau protein does not increase the generation of amyloid-beta peptides. Traffic 7(7):873–888
Stamer K et al (2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 156(6):1051–1063
Borg JP et al (1998) The X11alpha protein slows cellular amyloid precursor protein processing and reduces Abeta40 and Abeta42 secretion. J Biol Chem 273(24):14761–14766
Tomita S et al (1999) Interaction of a neuron-specific protein containing PDZ domains with Alzheimer’s amyloid precursor protein. J Biol Chem 274(4):2243–2254
Shrivastava-Ranjan P et al (2008) Mint3/X11gamma is an ADP-ribosylation factor-dependent adaptor that regulates the traffic of the Alzheimer’s precursor protein from the trans-Golgi network. Mol Biol Cell 19(1):51–64
Brunholz S et al (2011) Axonal transport of APP and the spatial regulation of APP cleavage and function in neuronal cells. Exp Brain Res. 217(3–4):353–364. https://doi.org/10.1007/s00221-011-2870-1
Grbovic OM et al (2003) Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J Biol Chem 278(33):31261–31268
Ikin AF et al (1996) Alzheimer amyloid protein precursor is localized in nerve terminal preparations to Rab5-containing vesicular organelles distinct from those implicated in the synaptic vesicle pathway. J Biol Chem 271(50):31783–31786
Siddiqui TJ, Craig AM (2011) Synaptic organizing complexes. Curr Opin Neurobiol 21(1):132–143
Schilling S et al (2017) APLP1 is a synaptic cell adhesion molecule, supporting maintenance of dendritic spines and basal synaptic transmission. J Neurosci 37:5345–5365
Stahl R et al (2014) Shedding of APP limits its synaptogenic activity and cell adhesion properties. Front Cell Neurosci 8:410
Baumkotter F et al (2014) Amyloid precursor protein dimerization and synaptogenic function depend on copper binding to the growth factor-like domain. J Neurosci: Off J Soc Neurosci 34(33):11159–11172
Wang Z et al (2009) Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J Neurosci: Off J Soc Neurosci 29(35):10788–10801
Dean C, Dresbach T (2006) Neuroligins and neurexins: linking cell adhesion, synapse formation and cognitive function. Trends Neurosci 29(1):21–29
Dalva MB, McClelland AC, Kayser MS (2007) Cell adhesion molecules: signalling functions at the synapse. Nat Rev Neurosci 8(3):206–220
Lu Z et al (2014) Calsyntenin-3 molecular architecture and interaction with neurexin 1alpha. J Biol Chem 289(50):34530–34542
Scheff SW, Price DA (2006) Alzheimer’s disease-related alterations in synaptic density: neocortex and hippocampus. J Alzheimers Dis: JAD 9(3 Suppl):101–115
Mhatre SD et al (2015) Microglial malfunction: the third rail in the development of Alzheimer’s disease. Trends Neurosci 38(10):621–636
Thomas DM, Francescutti-Verbeem DM, Kuhn DM (2006) Gene expression profile of activated microglia under conditions associated with dopamine neuronal damage. FASEB J 20(3):515–517
Jehle AW et al (2006) ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol 174(4):547–556
Villegas-Llerena C et al (2016) Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol 36:74–81
Southam KA, Vincent AJ, Small DH (2016) Do microglia default on network maintenance in Alzheimer’s disease? J Alzheimers Dis 51(3):657–669
Acknowledgments
We thank Prof. Claus Pietrzik for critically reading the manuscript.
Funding
Funding for SE was from the TU Nachwuchsring (University of Kaiserslautern) and SK and GH were supported by Alzheimer Forschung Initiative (AFI).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Eggert, S., Thomas, C., Kins, S. et al. Trafficking in Alzheimer’s Disease: Modulation of APP Transport and Processing by the Transmembrane Proteins LRP1, SorLA, SorCS1c, Sortilin, and Calsyntenin. Mol Neurobiol 55, 5809–5829 (2018). https://doi.org/10.1007/s12035-017-0806-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0806-x