
Abstract
Epileptic seizures are generally associated with pathological changes in the hippocampus such as astrogliosis, mossy fiber sprouting, and neuronal damage. However, more than 30% of temporal lobe epilepsy in humans shows neither neuronal damage nor mossy fiber sprouting despite chronic epileptic seizures. A similar situation exists in certain commonly used strains of mice, specifically C57BL/6 and BALB/c, which exhibit epileptic seizures, but no neuronal damage upon kainic acid administration. This suggests that intrinsic factors may influence the pathological manifestations of epilepsy. Mechanisms which are behind the resistance of hippocampal cells to KA-induced neuronal death are unknown. Autophagy seems to be involved in the pathogenesis of many brain insults and to have a dual nature in neuroprotection and cell death. This study addresses the role of autophagy upon status epilepticus (SE) that has been induced by kainic acid (KA) in the C57BL/6 strain which is classified as seizure resistant. We analyzed the dynamics in the expression of autophagic and cell death markers in the hippocampus upon SE. Immunofluorescence data show that KA did not induce neuronal death in the hippocampal CA1–CA3 subfields; however, it leads to an exclusive activation of caspase-3 in the mossy fibers. We also found alterations in the expression of core proteins of the autophagic machinery. Levels of MAP1LC3, phospho-mTOR/mTOR, and Beclin 1 were significantly increased after induction of seizures. However, levels of Atg3, Atg14, Atg5-Atg12, Atg7, BAG3, Hsp70, and LAMP1 showed no significant alterations compared to controls. Although KA did not induce neuronal death, this study provides morphological and biochemical evidence that status epilepticus induced by KA activates caspase-3 in mossy fibers and induces autophagy in the C57BL/6 hippocampus. These data indicate that autophagic factors may modulate the sensitivity of pyramidal cells to KA and that autophagy may constitute a part of an endogenous neuroprotective arsenal which might be behind the resistance of C57BL/6-hippocampal cells to KA-induced neuronal death.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The induction of status epilepticus (SE) by kainic acid (KA) in rodents leads to a series of neuropathological alterations and the subsequent appearance of spontaneous recurrent seizures. Many features of mice models, such a hippocampal sclerosis and mossy fibers sprouting, resemble human temporal lobe epilepsy (TLE). SE in humans still caries approx. 30% mortality and leads generally to widespread neuronal damage [1]. The mechanisms underlying these alterations are unknown, but may in part be excitotoxic. The treatment with KA is thought to cause a cascade of molecular events that trigger axon sprouting, formation of new functional synaptic circuits, and excitability changes in several, brain areas [2]. However, it is not known whether seizures precipitate cell death in the hippocampus or whether hippocampal cell loss results in the development and maintenance of epilepsy. Inbred mouse strains differ significantly in their behavior sensitivity to a variety of convulsant agents [3]. Some inbred strains of mice do not display the characteristic pattern of hippocampal neurodegeneration observed in rats following seizures induced by systemic administration of KA [4]. This difference in hippocampal sensitivity did not appear to be due to differences in KA pharmacokinetics between strains, or differences of neuronal activity and metabolism upon KA or differences in the density and distribution of glutamate receptors [5]. Recent investigations have provided insight into the potential mechanisms of genetic protection from cell damage upon KA seizures in different mouse strains. For instance, recent works have indicated that C57BL/6 mice and hybrids of this strain were resistant to kainate-induced TLE, regardless of the severity of seizure activity during SE [6]. Since many transgenic and knockout phenotypes are available on the C57BL/6 strain background [6], investigating post-SE alterations after KA on this strain will be important for studying the molecular mechanisms that contribute to epileptogenesis.
This study addresses the role of autophagy upon SE that has been induced by KA in the C57BL/6 strain which is classified as seizure resistant. We analyzed the dynamics in the expression of cell death markers and of autophagic markers in the hippocampus of C57BL/6 mice upon SE induced by systemic application of KA. In our study, the activation of autophagy was analyzed morphologically and biochemically in strict compliance with the “Guidelines of the use and interpretations of assays for monitoring autophagy in higher eucaryotes” [7].
Material and Methods
Animals
Male C57BL/6 mice aged 9–10 weeks were purchased from Janvier Labs (France). All animals were housed in cages with littermates on 12-h light-dark schedules. Water and food were available ad libitum.
Induction of SE
KA (Sigma, USA) was dissolved in isotonic saline (pH 7.3–7.4). Mice were administered KA (30 mg/kg) intraperitoneally (i.p.). Control mice were given an equal volume of saline subcutaneously (s.c.). Behavioral seizures were scored using a modified Racine scale. Only animals reaching SE (defined as near-continuous seizure activity lasting over 10 min) were included in the study. Some animals progressed to intermittent (stage 4) followed by continuous (stage 5) rearing with forelimb clonus and falling behavior. Some mice then progressed to generalized tonic–clonic whole body convulsions (stage 6). Some mice did not progress orderly through seizure stages but rather went from immobility with rigidity to rapid jumping “popcorn” behavior, with a hyperactive running period sometimes occurring in between. The “popcorn” bouncing activity, which was also graded as stage 6 seizure activity, was fatal if it persisted (18% mortality).
Immunohistochemistry
Mice were anesthetized with an overdose of pentobarbital (100 mg/kg i.p.) at 6 h (n = 6), 24 h (n = 6), 48 h (n = 6), 7 days (n = 3), and 14 days (n = 3) after induction of SE. Then, mice underwent transcardiac perfusion with 4% phosphate-buffered paraformaldehyde. Control animals (n = 6) were subjected to the same procedure. Brains were post-fixed by immersion in 4% paraformaldehyde/PBS for 1 h and placed overnight in 30% sucrose/PBS. Coronal sections (12-μm thick) were cut on a cryostat and stored at −70 °C until use. For immunofluorescence, slides were pre-incubated for 1 h at room temperature in PBS, 5% NGS (Sigma), and primary antibodies were applied at 4 °C for 24 h in PBS plus 5% normal goat serum. Antibodies raised against caspase-3 (p17, Cell Signaling, Germany), LC3 (Cell Signaling, Germany), and LAMP1 (Cell Signaling, Germany) were used. After several washes in 0.1 M PBS, sections were incubated with Alexa 488- or 568-conjugated anti-mouse IgG (1:200, 2 h, room temperature, in 0.1 M PBS; Molecular Probes, Eugene), washed again, and incubated with Alexa 568- or 488-conjugated anti-rabbit IgG (1:200, 2 h, in 0.1 M PB), respectively. After rinsing the sections with 0.1 M PBS, they were mounted in Dako fluorescent mounting medium. Fluorescent images were acquired using an Axio-Cam digital camera mounted on a Zeiss microscope (Carl Zeiss, Jena). For the assessment of non-specific immunostaining, alternating sections from each experimental group were incubated without the primary antibody.
Evaluation of Neuronal Viability
For the evaluation of neuronal viability, coronal brain cryosections (12 μm) were stained with cresyl violet. Neuronal viability was evaluated as the percentage of viable neurons in CA1–CA3 hippocampal subfields in a section 6-mm rostral to the tentorial incision. Neuronal viability was expressed as the percentage of viable neurons.
TUNEL Staining
In Situ Cell Death Detection Kit (Roche, Germany) was employed for the detection of DNA fragmentation and apoptotic bodies in hippocampal cells. Normal nuclei did not stain with the TUNEL method.
Preparation of Tissue and Western Blot
Mice were anesthetized with an overdose of pentobarbital (100 mg/kg i.p.) at 6 (n = 6), 24 (n = 6), and 48 h (n = 6) after induction of SE. Aliquots were stored at −80 °C, and 30 μg of total protein was used per lane. Samples were resuspended to contain 30 μg of total protein in loading buffer, heated for 5 min at 95 °C, and separated using a XCellSureLockNovex Mini-Cell electrophoresis system from Invitrogen (Carlsbad, USA) on NuPAGENovex 4–12% Bis-Tris Gels (Invitrogen). Gels were ran at 200 V for 55 min. The matched gel was soaked in transfer buffer (25 mM Tris, 190 mM glycine, 20% methanol) and then transferred to a PVDF membrane for 1 h at 25 V using a Bio-Rad Semi-Dry Blotting apparatus. Blots were blocked in 20 ml Roti-Block solution (Carl Roth, Germany) in order to reduce unspecific binding of antibodies. Antibodies raised against caspase-3, cleaved caspase-3, μ-calpain, calpastatin, phospho-mTOR(Ser2448) mTOR, MAP1LC3, Beclin 1, Atg3, Atg5, Atg7, Atg5-Atg12, Atg14 LAMP1 (Cell Signaling, USA), BAG3 (Santa Cruz, USA), and Hsp70 (StressGen, Germany) were used at 1:1000 dilution. β-Actin (Sigma-Aldrich, USA) (dilution 1:40,000) was used as control protein; anti-rabbit IgG (Santa Cruz, USA) (1:30,000) and anti-mouse IgG (P0447 Dako, Germany) (1:30,000) were used as secondary antibodies. Protein expression levels were quantified using gel analysis software: ImageJ (v1.44p for Windows, National Institute of Health, Bethesda, USA). Data were reported as means ± SEM of n > or = 3 experiments. Significant differences between means were statistically assessed by ANOVA. Significant differences between animals at each time point were assessed by Dunnett’s test, and *p < 0.05, **p < 0.01, and ***p < 0.001 were considered statistically significant.
Results
Morphology and Viability of Hippocampal Cells upon SE
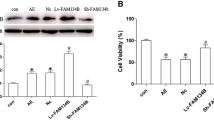
To examine the neuronal morphology in the hippocampus after KA-induced seizures, cresyl violet was used for staining. Neurons in the CA1-CA3 regions did not show any spectacular changes upon SE stress. Some scattered neurons exhibited slight swelling without necrotic changes (Fig. 1a). Moreover, the in Situ Cell Death Detection Kit (Roche, Germany) was employed for the detection of DNA fragmentation and apoptotic bodies in hippocampal cells. Here again, we did not detect any TUNEL-positive cell in the CA1–CA3 regions in SE animals (Fig. 1b (a, b)). As a positive control, we used brain sections from animals treated with pilocarpine with a selective neurodegeneration in the CA1 region (Fig. 1b (c)).


(a) Neuronal morphology in the hippocampus was examined with cresyl violet staining. Neurons in the CA1–CA3 regions did not show any spectacular changes upon SE stress. Some scattered neurons exhibited slight swelling without necrotic changes. (b, a, b) By using the in Situ Cell Death Detection Kit employed for the detection of DNA fragmentation and apoptotic bodies in hippocampal cells, we did not detect any TUNEL-positive cell in the CA1–CA3 regions in SE animals. (b, c) Positive controls for TUNEL from pilocarpine-treated animals. (c) Western blot analysis showed only a slight increase in expression levels of the active form of caspase-3 in the hippocampus of epileptic animals (*p < 0.05 different from controls). (d) μ-Calpain and calpastatin levels exhibit no significant changes in the hippocampus of epileptic animals at all examined time points
For Western blot analysis, we have used two antibodies: one directed against the caspase-3 (Fig. 1c) which detects endogenous levels of full-length caspase-3 (35 kDa) and the large fragment of caspase-3 resulting from cleavage (17–19 kDa). The second one (Fig. 1c) directed against the cleaved caspase-3 (Asp175) detects only the endogenous levels of the large fragment (17/19 kDa) of activated caspase-3 resulting from cleavage adjacent to Asp175. This antibody does not recognize full-length caspase-3 or other cleaved caspases. Both antibodies show a slight increase in the active form of caspase-3 in the hippocampus of epileptic animals at all examined time points (Fig. 1c). μ-Calpain and calpastatin levels did not reveal any alterations in the hippocampus of KA-treated animals (Fig. 1d). Although the calpastatin levels exhibit a slight decrease, this was statistically not different from controls and was not accompanied by the appearance of cleavage products.
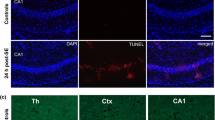
However and surprisingly, on morphological levels, the immunofluorescence analysis showed an accumulation of the cleaved caspase-3 exclusively in the mossy fibers, i.e., axons from the granule cells of the dentate gyrus, especially those located in the hilus and in the stratum lucidum of the hippocampal CA3 sector (Fig. 2). This strong immunostaining of the active form of caspase-3 in mossy fibers occurs without any localization in the cytosol of granule cells. Active caspase-3 was absent in neuronal bodies in the dentate gyrus and in pyramidal cells of the CA1–CA3 subfields (Fig. 2b, c).

The immunofluorescence analysis of the active form of caspase-3 showed a dramatic accumulation of the cleaved caspase-3 exclusively in the mossy fibers, i.e., axons from the granule cells of the dentate gyrus b 24 h post-SE and c 48 h post-SE. This strong immunostaining of the active form of caspase-3 in mossy fibers occurs without any detection in the cytosol of granule cells. Active caspase-3 was absent in neuronal bodies in the dentate gyrus and CA1-CA3-subfields (a). DG dentate gyrus, c-Cp3 cleaved caspase
Effects of KA on Autophagic Markers
We were able to detect considerable alterations in the levels of central proteins of the autophagic machinery. Protein levels of MAP1LC3, in its activated form LC3-II indispensable protein of the autophagosomal membrane, were significantly increased as early as 6 h after SE (Fig. 3a). To semi-quantify the amount of autophagosome formation after SE relative to naive controls, we examined hippocampi from mice with SE for evidence of an LC3 shift from an apparent LC3 I (18 kD) to LC3 II (16 kD) position. By Western blot analysis, the time-course study of LC3 demonstrated that the ratio of LC3 II to LC3 I was significantly increased, peaking at 6 h after induction of epilepsy (Fig. 3a, b). To confirm that SE induces autophagosome formation, we examined the immunoreactivity of LC3 in the hippocampus of controls and epileptic animals. LC3, the microtubule-associated protein 1A light-chain 3, is normally located throughout the cytoplasm, but becomes concentrated in autophagosomes during autophagy. Indeed, LC3-staining appeared as aggregates/punctated dots in the hippocampus of epileptic mice (Fig. 3b).

a Immunoblot and statistical analysis of MAP1LC3 levels after kainic acid-induced SE. A dramatic, almost twofold upregulation in the ratio of LC3-II to LC3-I can be observed as early as 6 h after SE. Looking at the single forms of LC3 contributing to the ratio mentioned above, it becomes obvious that the reported increase is due to an upregulation of LC3-II rather due to a decrease in the amount of inactive LC3-I. b Immunoreactivity of LC3 in the hippocampus of controls and epileptic animals. LC3, the microtubule-associated protein 1A light-chain 3, is normally located throughout the cytoplasm in controls but becomes concentrated in autophagosomes during autophagy. Indeed, LC3 staining appeared as aggregates/punctated dots in the hippocampus of epileptic mice
Likewise, Beclin 1, one of the most important inductors of autophagy, showed significantly elevated protein levels at the same time-point (Fig. 4a). The ratio p-mTOR/mTOR was also significantly increased after SE (Fig. 4b). Furthermore, we were able to show a significant upregulation in Atg3 levels at 6-h post SE. There were no significant alterations in protein levels of Atg5 and Atg7 compared to controls, whereas Atg14 showed dramatic upregulation 6 and 48 h post SE, the latter being significant (Fig. 4c–f). Co-chaperone BAG3 and chaperone protein Hsp70 by tendency showed upregulated protein levels after SE; however, there were no significant alterations compared to controls (Fig. 4g, h).


Immunoblot and statistical analysis of several members of the Atg family after kainic acid-induced SE. a Beclin 1, one of the most important inductors of autophagy, showed significantly elevated protein levels at 6 h post-SE. b The ratio of phosphorylated to unphosphorylated mTOR shows a significant upregulation at time point 48 h after SE. Looking at the single forms of mTOR contributing to this ratio, there are a significant upregulation of mTOR levels at 24 h after SE and a significant upregulation of phospho-mTOR levels at 48 h after SE to be mentioned. c Note the significant upregulation of Atg3 at time point 6 h after SE compared to unchanged levels at 24 and 48 h after SE. d No significant alterations of Atg5-Atg12 levels at any point in time after SE. e No significant alterations of Atg7 levels at any point in time after SE. f Note significantly elevated levels of Atg14 at time point 48 h after SE compared to, not significantly, altered levels at 6 and 24 h after SE. g Although elevation in BAG3 levels at 6 h after SE to be noted, there was no significant alterations of BAG3 levels at any point in time after SE. h A slight no significant upregulation of Hsp70 levels is observed after SE
Lysosomal Activity Is Increased Following SE
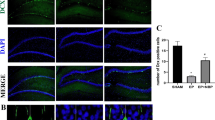
Lysosomal marker LAMP-1 protein levels appeared to be increased at 6 and 24 h after SE, without showing significant alterations compared to controls (Fig. 5a). To evaluate whether the increased number of autophagosomes was due to enhanced autophagic flux or to failed lysosomal degradation, lysosomal activity was investigated by immunohistochemistry using antibodies directed against LAMP-1. The numbers of LAMP-1-positive dots were quantified. An increased number of lysosomes would reflect an increase in autophagic flux. In hippocampal neurons, the number of LAMP1-positive vesicles was strongly increased by SE (Fig. 5b).

LAMP-1 protein levels appeared to be increased at 6 and 24 h after SE, without showing significant alterations compared to controls (a). Lysosomal activity was also investigated by immunohistochemistry. In hippocampal neurons, the density of LAMP1-positive vesicles was strongly increased by SE (b)
Discussion
Some mice strains sensitive to the acute effects of KA such as C3H, C57BL/6, and BALB/c, developed no pyramidal cell damage or mossy fiber sprouting, while other strains developed the greatest pyramidal cell loss and mossy fiber sprouting despite moderated seizure severity. It has been found that the genetic background of mice is an important factor of their response to many kinds of experimental insults such as epilepsy or cerebral ischemia [8]. However, it is not excluded that other endogenous/intrinsic factors might confer resistance against hippocampal damage upon SE. We know that prolonged high-grade seizure activity does not result in cell death or synaptic reorganization in genetically protected mouse strains. This is very interesting when taking into consideration the variability of hippocampal pathology in human temporal lobe epilepsy. However, the mechanisms underlying this protection are still unknown.
C57BL/6 mice were initially reported to be resistant to excitotoxic damage caused by kainic acid [4]. Our study corroborated previous studies and confirmed the resistance of these mice to KA. However, by evaluating apoptosis and autophagy markers, we found in this resistant strain an exclusive accumulation of the active form of caspase-3 in the mossy fibers and elevated protein levels of autophagy markers in the whole hippocampus. This underlines, therefore, an autophagic response to KA administration and an unexpected activation of caspase-3 in the mossy fibers.
We here report an activation of caspase-3 in the mossy fibers and the presence of enhanced autophagy in living neurons after SE in mice. Our results show that many of the neurons expressing enhanced autophagy were morphologically intact. Moreover, in our model, the dynamics of autophagy are region dependent, because strong autophagy is activated only in the hippocampal CA1-CA3 neurons. The best-known function of autophagy is protective in order to maintain cell survival under conditions such as starvation or pathogen infection [9,10,11,12,13,14,15]. Of all things, the potentially vulnerable CA1–CA3 neurons exhibited a triggered transient autophagic activity upon SE. Thus, from the current analysis, the picture is that pyramidal cells may possess autonomous features that regulate the KA-induced phenotype of cell death. The enhanced autophagy may perhaps confer a potential “protective” mechanism against excitotoxicity induced by KA in C57BL/6 mice.
What it is the function of the neuronal autophagy in the present study? There are three possibilities: (1) the autophagy response is just an epiphenomenon, (2) or it can be an active player in the cell death machinery, (3) or a protective mechanism. It cannot be an active player in the cell death machinery, because there is no cell damage in the hippocampus upon SE at the examined time points. The post-epileptic alterations of autophagy dynamics in cells could be considered as a part of an endogenous neuroprotective arsenal or a stress response which may serve to regulate the excitotoxicity effects of KA. In this context, we have recently reported that pharmacological inhibition of autophagy using 3-MA or bafilomycin A1 enhances susceptibility to proapoptotic signals induced by serum deprivation in hippocampal HT22 cells [13,14,15,16]. A strong evidence for a death-inhibiting role of autophagy has been shown in several models of cerebral ischemia [17]. Pharmacological inactivation of autophagy or specific knockdown of autophagy genes have been shown to be deleterious for cell viability. Such experiments support the function of autophagy as protective mechanism to maintain cell survival [13, 17, 18].
A very striking finding in this study is the exclusive expression of the active form of caspase-3 in hippocampal mossy fibers, i.e., axons from the granule cells of the dentate gyrus, especially those located in the hilus and in the stratum lucidum of the hippocampal CA3 sector. This strong immunostaining of the active form of caspase-3 in mossy fibers occurs without any activation in the cytosol of granule cells. Active caspase-3 was absent in neuronal bodies in the dentate gyrus and CA1-CA3-subfields. While the expectation was to find granule cells also expressing active caspase-3, our results showed no activation in this cell type. We can assume that granule cells may die of apoptosis at late stages post-SE. However, apoptotic cell bodies were not observed at later stages (14–21 days post-SE) by using the TUNEL method. In order to check if the accumulation of cleaved caspase-3 in the mossy fibers might be associated with any caspase activity, we have analyzed potential substrates of caspase-3. For instance, it is known that the endogenous calpain inhibitor, calpastatin, was degraded not by calpain but by caspases during apoptosis. Calpastatin fragmentation might have yet unidentified functions to facilitate the cross talk between caspases and calpains during apoptosis. Thus, it is possible that caspase-3 might indirectly activate calpain via calpastatin degradation. However, in our setting, we did not observe any significant alterations neither of calpastatin nor of calpain levels after SE induction. Although the calpastatin levels exhibit a slight decrease, this was not accompanied by the appearance of cleavage products.
Given the cytoarchitecture of neurons, it is possible that the activation of the apoptotic machinery might have differential effects that depend on spatial subcellular localization of the activated caspase-3. Specifically, neurons in which caspase-3 is initially activated in the proximity of the nucleus will be more likely to exhibit oligonucleosomal DNA degradation. However, neurons in which caspase-3 is initially activated in a distant process will be more likely to exhibit dendritic and axonal cytoskeletal proteolysis. It is quite complicated to distinguish between such specific spatial mechanisms if the apoptotic signal spreads rapidly through the cell. An early morphological change that accompanies cell death is a retraction of the neuritic network. The molecular mechanisms involved in the retraction and collapse of the axonal network during neuronal apoptosis are still unknown. It is not clear whether axonal damage is an outcome of the death process that occurs within the cell body or whether damage to the axonal network per se serves to trigger apoptosis of the whole neuron.
Although caspase-3 is a key protein in apoptosis execution, evidence also indicates a possible non-apoptotic role in the regulation of synaptic plasticity [19,20,21,22,23]. As matter of fact, many proteins that are involved in neuronal plasticity are caspase-3 substrates. In this context, Mattson et al. [24, 25] have shown that apoptosis can occur locally in synapses and dendrites of cultured neurons independently of the cell body. The presence of active caspase-3 in the mossy fibers may mediate a kind of “axonopathy” involving deregulation of axonal transport or proteolysis of cytoskeletal components. The relevance of this finding is unclear, but it may prime or mark axons for further structural alterations at later stages.
In conclusion, we have shown for the first time that upon SE activation of caspase-3 in hippocampal mossy fibers occurred, and autophagy in hippocampal cells is enhanced in a region-specific manner. We hypothesize that triggered autophagy could be involved in the resistance of C57BL/6 hippocampal cell mice to KA-induced excitotoxicity. Much more work is needed in order to find out the requirement under which activation of autophagy reflects a protective or a harmful response to neuronal stress.
References
DeLorenzo RJ, Pellock JM, Towne AR, Boggs JG (1995) Epidemiology of status epilepticus. J Clin Neurophysiol 12:316–325
Smith BN, Dudek FE (2001) Short- and long-term changes in CA1 network excitability after kainate treatment in rats. J Neurophysiol 85:1–9
Schauwecker PE (2002) Complications associated with genetic background effects in models of experimental epilepsy. Prog Brain Res 135:139–148
Schauwecker PE, Steward O (1997) Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci U S A 94:4103–4410
Kurschner VC, Petruzzi RL, Golden GT, Berrettini WH, Ferraro TN (1998) Kainate and AMPA receptor binding in seizure-prone and seizure-resistant inbred mouse strains. Brain Res 780:1–8
Steward O, Schauwecker PE, Guth L, Zhang Z, Fujiki M, Inman D et al (1999) Genetic approaches to neurotrauma research: opportunities and potential pitfalls of murine models. Exp Neurol 157:19–42
Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463–477
Liu L, Hamre KM, Goldowitz D (2012) Kainic acid-induced neuronal degeneration in hippocampal pyramidal neurons is driven by both intrinsic and extrinsic factors: analysis of FVB/N↔C57BL/6 chimeras. J Neurosci 32:12093–12101
Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T (2001) Beclinphosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2:330–335
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T (2004) The role of autophagy during the early neonatal starvation period. Nature 432:1032–1036
Rami A, Kögel D (2008) Apoptosis meets autophagy-like cell death in the ischemic penumbra: two sides of the same coin? Autophagy 4:422–426
Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N (2013) Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 1833:3448–3459
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889
Steiger-Barraissoul S, Rami A (2009) Serum deprivation induced autophagy and predominantly an AIF-dependent apoptosis in hippocampal HT22 neurons. Apoptosis 14:1274–1288
Rami A (2009) Review: autophagy in neurodegeneration: firefighter and/or incendiarist? Neuropathol Appl Neurobiol 35:449–461
Fekadu J, Rami A (2016) Beclin-1 deficiency alters autophagosome formation, lysosome biogenesis and enhances neuronal vulnerability of HT22 hippocampal cells. Mol Neurobiol 53:5500–5509
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B et al (2008) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118:2190–2199
D'Amelio M, Sheng M, Cecconi F (2012) Caspase-3 in the central nervous system: beyond apoptosis. Trends Neurosci 35:700–709
Snigdha S, Smith ED, Prieto GA, Cotman CW (2012) Caspase-3 activation as a bifurcation point between plasticity and cell death. Neurosci Bull 28:14–24
Wang JYY, Luo ZG (2014) Non-apoptotic role of caspase-3 in synapse refinement. Neurosci Bul 30:667–670
Li Z, Sheng M (2012) Caspases in synaptic plasticity. Mol Brain 5:15
Dash PK, Blum S, Moore AN (2000) Caspase activity plays an essential role in long-term memory. Neuroreport 11:2811–2816
Mattson MP, Keller JN, Begley JG (1998) Evidence for synaptic apoptosis. Exp Neurol 153:35–48
Chan SL, Mattson MP (1999) Caspase and calpain substrates: roles in synaptic plasticity and cell death. J Neurosci Res 58:167–190
Acknowledgements
This work was supported by the Adolf-Messer-Stiftung (grant to A. Rami—“Molecular mechanisms of autophagy”). We thank Alena Konoplew for the technical assistance. A.P. Benz was supported partly by the AMS and the FPF (Frankfurter Promotionsförderung). We thank Professor J. Stehle for support of our work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rami, A., Benz, A. Exclusive Activation of Caspase-3 in Mossy Fibers and Altered Dynamics of Autophagy Markers in the Mice Hippocampus upon Status Epilepticus Induced by Kainic Acid. Mol Neurobiol 55, 4492–4503 (2018). https://doi.org/10.1007/s12035-017-0665-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0665-5