
Abstract
Neuronal apoptotic cell death plays an important role in many neurological disorders, including Alzheimer’s disease, Parkinson’s disease, and ischemic stroke. Spatholobi Caulis (SC) has been widely used in traditional herbal medicine for the treatment of cancer, inflammation, viral infection, and anemia. However, the protective effects of SC extract (SCE) against apoptotic cell death in the brain have not been reported. We investigated the protective effects of SCE against neuronal injury etoposide-induced neurotoxicity and in rats subjected to focal transient ischemic stroke middle cerebral artery occlusion (MCAO) for 45 min, followed by 7 days of reperfusion. The in vitro study demonstrated that SCE protected cells against etoposide-induced cell viability loss in SH-SY5Y cells. Apoptotic phenotypes, such as cleaved PARP and caspase-3, and oxidative stress in etoposide-treated cells were ameliorated by SCE treatment. In MCAO-reperfusion injury, SCE promoted neuronal survival and level of brain-derived neurotrophic factor (BDNF) by reducing glial activation, oxidative stress, and apoptosis in the ipsilateral cortex. These results indicated that SCE exerted protective effects under etoposide treatment and in a MCAO-reperfusion model by reducing JNK and p38 MAPK activation. This study presents the first evidence that SCE has therapeutic potential for the treatment of ischemic stroke or neurological disorder-related cell death.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke is one of the most common neurological diseases and is characterized by deprived blood circulation to the brain, which is caused by occlusion of the cerebral artery. The pathophysiology of ischemic stroke is complicated and is mediated by various processes, such as inflammatory, oxidative, excitotoxicity, and apoptotic mechanisms [1, 2]. Ischemic stroke causes brain damage and neuronal cell death due to the activation of ischemic processes, which result in the depletion of oxygen and glucose [3, 4]. Moreover, ischemic stroke exhibits the reduction and stop of cerebral blood flow by platelet or fibrin clots within the blood vessel, which is an important mediator of cerebral ischemic stroke injury [5, 6]. Loss of blood supply affects neuronal damage in the ischemic area. During the reperfusion after middle cerebral artery occlusion (MCAO), the brain is reintroduced oxygenation and glucose. These events cause the elevation of oxidative stress due to free radical and reactive oxygen species (ROS) generation and apoptotic cell death in the ischemic region of the brain [7, 8]. Previous research involving animal models and clinical evidence has shown that herbal-based interventions can be an important part of the prevention or treatment of ischemic stroke [9,10,11].
Spatholobi Caulis (SC) is a traditional herbal medicine that is produced from the vine stem of Spatholobus suberectus Dunn (also called Ji Xue Teng) and has been shown to have anti-anemic, anticancer, anti-inflammatory, antiviral, and anti-osteoarthritic effects [12,13,14,15,16]. Previous research has shown that SC ethanolic extract (SCE) inhibits H2O2-induced oxidative stress and increases cell growth in the PC12 cell line, which was derived from a transplantable rat pheochromocytoma [17]. However, the protective effects of SCE on brain injury and its molecular mechanism have not been reported. The present study revealed that SCE has protective effects under etoposide-induced cytotoxic conditions in SH-SY5Y cells and under focal transient ischemic stroke in a MCAO-reperfusion injury model. Furthermore, our study clarified the underlying molecular mechanisms of SCE against etoposide- and MCAO-reperfusion-induced neuronal damage.
Materials and Methods
Reagents and Antibodies
Donepezil hydrochloride (Aricept) was purchased from Eisai Korea Inc. (Seoul, Korea); 4% paraformaldehyde (PFA) solution and 2′-7′-dichlorofluorescin diacetate (DCFDA) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA) and Invitrogen Molecular Probes (Eugene, OR, USA), respectively. JC-1 (chloride salt) was obtained from Biotium (Hayward, CA, USA). Triton X-100 and 3% goat serum were purchased from Biobasic (Biobasic Inc., Ontario, Canada) and Gibco (Gibco BRL, Grand Island, NY, USA), respectively. TdT-mediated dUTP-X nick end labeling (TUNEL) staining kit was purchased from Roche Diagnostics (Mannheim, Germany). Anti-poly (ADP-ribose) polymerase (PARP), cysteinyl aspartate specific proteinase-3 (caspase-3), phosphorylated (p) p53, p-JNK, total (t) JNK, p-p38, and t-p38 were purchased from Cell Signaling Technology. Anti-β-actin was obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Secondary antibodies for immunostaining were purchased from Vector Laboratories (Vector Laboratories Inc., CA, USA) or Molecular Probes (Eugene, OR, USA). Secondary antibodies for Western blotting were obtained from Thermo Scientific (Thermo Scientific, MA, USA). Acetonitrile, methanol (Fisher Scientific, UK), and trifluoroacetic acid (Sigma-Aldrich Co.) were high-performance liquid chromatography (HPLC) grade, and ultrapure water from Milli-Q system (Millipore) was used for the mobile phase preparation.
Preparation of Ethanolic Extract of Spatholobi Caulis
Dried vine stem of SC was purchased from a local vendor at Hyundai Herbal Market (Yeongcheon, Korea) and deposited in the herbal bank of the KM-Application Center, Korea Institute of Oriental Medicine (KIOM; Daegu, Korea) after verification by Professor Ki Hwan Bae of the College of Pharmacy, Chungnam National University (Daejeon, Korea). Ethanolic extract of SC was extracted in 70% ethanol (30 g/390 ml) at 40 °C in a shaking incubator for 24 h. After extraction, the solution was filtered through filter paper (Whatman filter paper #1, Whatman Ltd., Maidstone, UK), and the filtrate was then lyophilized (yield 10.3297%). The freeze-dried SCE powder (50 mg) was then dissolved in 1 ml 50% DMSO (v/v) and filtered through a 0.22-μm syringe filter. Each standard component for HPLC analysis in this study was prepared by dissolving 1 mg in 1 ml of methanol. The respective five standard mixtures were diluted to 20 μg/ml in methanol. The SCE was dissolved in methanol at 10 mg/ml, which was sonicated at room temperature for 30 min and filtered using a 0.2-mm syringe membrane filter (Whatman Ltd.).
Cell Culture
SH-SY5Y cells (kindly provided by Prof. Jaewon Lee, Pusan National University, Korea) are human neuroblastoma-derived cell line and had neuron-like characteristic. These cells can differentiate into neurons by induction of retinoic acid (RA). SH-SY5Y cells were cultured in a humidified 95% air (including 20% O2) and 5% CO2 incubator at 37 °C with RPMI 1640 media (Lonza, Walkersville, MD, USA) supplemented with heat-inactivated 10% fetal bovine serum (HyClone Laboratories, UT, USA), 2 mM glutamine, and 1% penicillin/streptomycin antibiotic mixture (Corning Incorporated, NY, USA).
Cell Viability Analysis
Cell viability was evaluated by MTT assay. Cells (5 × 103 cells/ml) were seeded in 96-well plates. For MTT assay, 100 μl of 0.25 mg/ml MTT solution in phosphate-buffered saline (PBS) was added to each well. After incubation at 37 °C for 2 h, MTT solution was removed, and cells were lysed by solubilization solution (1:1 DMSO:ethanol). Color development was measured at 560 nm using a microplate reader (SpectraMax i3, Molecular Devices, CA, USA). To identify the molecule critical for the protective effects of SCE, cells were treated with each inhibitor 1 h prior to treatment of SCE and etoposide.
Western Blot Analysis
Protein concentrations were determined using the bicinchoninic acid (BCA) assay kit (Thermo Scientific, MA, USA) with bovine serum albumin as standard. Samples (30 μg protein per lane) were separated using SDS-polyacrylamide gels and transferred electrophoretically to Immobilon-PSQ transfer membranes (Millipore), as reported previously [18]. Band intensities were measured using FluorChem™ SP software (Alpha Innotech, San Leandro, CA, USA) and normalized to β-actin or total forms.
Caspase-3/7 Activity Assay
Caspase-3/7 activity was measured by Caspase-Glo assay system (Promega Corporation, WI, USA). SH-SY5Y cells were pretreated with inhibitor for 1 h and with SCE for 6 h and then co-treated with 50 μM etoposide for 24 h. The culture medium was obtained by centrifugation and mixed with an equal volume (100 μl) of Caspase-Glo 3/7 reagent and incubated at room temperature (RT). The luminescence of each sample was measured in a luminescence microplate reader (SpectraMax i3).
Mitogen-Activated Protein Kinase Activity Assay
Mitogen-activated protein kinase (MAPK) activity was measured using MAPK Family Activation InstantOne™ ELISA assay (Affymetrix eBioscience, CA, USA). Detection reagent was added to each well. Cells were incubated for 30 min at RT, and the reaction was stopped with stop solution. Absorbance was measured at 450 nm using a microplate reader.
Flow Cytometry Analysis
Fluorescein isothiocyanate (FITC) Annexin-V Apoptosis Detection Kit I (BD Biosciences, CA, USA) was used to detect cell death, as reported previously [19]. Flow cytometry analysis was performed on a GALLIOS™ (GALLIOS, Beckman Coulter, CA, USA) and analyzed using Kaluza Analysis Software (Kaluza Analysis Version 1.3).
Mitochondrial Membrane Potential Assay
Mitochondrial membrane potential depolarization, an early process in the apoptotic cell death, was measured using a cationic carbocyanine dye JC-1, as reported previously [19]. JC-1 aggregates in mitochondria (red; high mitochondrial membrane potential (MMP) accumulation in the mitochondria) or free JC-1 monomers (green; low MMP in the mitochondria; mitochondrial depolarization) measured by Nikon ECLIPSE Ti microscope (Nikon, NY, USA). Images were analyzed using NIS-Elements Basic Research Imaging Software (Version 4.50, Nikon, NY, USA).
Animals
Adult male Sprague-Dawley (SD) rats (7–8 weeks, weighing 240–260 g) were purchased from Samtako Bio Korea (Osan, Korea) and maintained under temperature- and light-controlled conditions (20–23 °C, 12 h light/12 h dark cycle) with food and water provided ad libitum. All animals were acclimatized for 7 days before the surgical procedure and drug administration in the specific pathogen-free (SPF) facilities. The institutional animal care committee of the Korea Institute of Oriental Medicine approved the experimental protocol (#D-16-006), and all studies were performed according to the guidelines of the Animal Care and Use Committee at KIOM.
Surgical Procedure
The focal ischemic stroke by transient MCAO-reperfusion model was generated as described previously [20, 21]. Rats were anesthetized with 2.5% isoflurane (Hana Pharm. Co., Ltd., Gyeonggi-do, Korea) and provided with supplemental oxygen. A 4-0 monofilament nylon suture (2–3 mm silicon coating length, 0.39 mm tip diameter; Doccol Corporation, MA, USA) was inserted into the internal carotid arteries (ICA) to the origin of the middle cerebral arteries (MCA) via a small incision of the external carotid arteries (ECA). After 45 min MCAO induction, the monofilament was carefully removed to restore blood flow (reperfusion) and tightly tied on the ECA. The incision was sutured, and the rats were returned to their cages with warm light to prevent hypothermia. The rats in the control group did not undergo surgery. All experiments were performed on the 7th day after reperfusion.
Drug Administration
Male SD rats were used in this study and were randomly divided into five groups according to body weight: (1) control; (2) MCAO-reperfusion (MCAO) + saline; (3) MCAO + SCE 100 mg/kg; (4) MCAO + SCE 200 mg/kg; or (5) MCAO + donepezil hydrochloride (Don; Aricept) 5 mg/kg. The rats in the control, MCAO, SCE, or Don group were administered via oral gavage by flexible plastic feeding tubes for three consecutive days prior to MCAO surgery and once a day for 7 days after reperfusion. The oral gavage needle was entered into the mouth of the rats until it reached the stomach, where the drugs were directly injected. Each rat was dosed with 5 ml/kg body weight. SCE or Don was dissolved in 0.9% physiological saline, and rats in the control group and MCAO group were administered an equal volume of 0.9% physiological saline.
Tissue Preparation
For biochemical analyses, the ischemic frontoparietal cortical tissues from the ischemic core and the penumbra site (approximately from −3.8 to +1.7 mm bregma) were harvested and homogenized in RIPA buffer (Millipore), protease inhibitor cocktail, and phosphatase inhibitor cocktail (Roche, Mannheim, Germany) and centrifuged (12,000 rpm for 20 min at 4 °C). The supernatant was stored at −80 °C until required for biochemical analysis. For histological analyses, rats were anesthetized and perfused intracardially with 4% PFA in 0.1 M PBS (pH 7.4). After fixative perfusion, the brains were removed and placed in the same fixative at 4 °C overnight. The brain tissues were embedded in paraffin and sectioned into coronal sections at 5 μm thickness (Leica Biosystems, Wetzlar, Germany). For staining, brain sections were deparaffinized and rehydrated with xylene and ethanol.
Nissl Staining
Brain sections were stained by 0.1% cresyl violet solution for 30 min to determine the histopathological features of neurodegeneration in the MCAO-reperfusion model. Sections were imaged using a Nikon ECLIPSE Ti microscope (Nikon, NY, USA). Images were analyzed using NIS-Elements Basic Research Imaging Software (Version 4.50, Nikon, NY, USA).
Immunohistochemistry
For double-label immunohistochemistry, brain sections were blocked with TBS/0.1% Triton X-100/3% goat serum (Gibco, Grand Island, NY, USA) and incubated with primary antibodies overnight at 4 °C. In staining for immunohistochemistry, primary antibodies were used: doublecortin (DCX), glial fibrillary acidic protein (GFAP), neuronal nuclei (NeuN) Millipore (Millipore, Billerica, MA, USA), β-III-tubulin (microtubule element of the tubulin family, Sigma-Aldrich Co.), and ionized calcium-binding adapter molecule 1 (Iba-1; Wako, Osaka, Japan). Brain sections were then washed with TBS and incubated for 3 h in the presence of IgG-labeled secondary antibodies with Alexa Fluor. Images were acquired using a Nikon ECLIPSE Ti microscope (Nikon, NY, USA). Images were analyzed using NIS-Elements Basic Research Imaging Software (Version 4.50, Nikon, NY, USA). The quantitative analysis in histological data was performed by investigators blinded to the results of all images.
Measurement of ROS
DCFDA is a fluorescence-based probe for ROS detection. DCFDA is hydrolyzed by intracellular esterase to nonfluorescent DCF. Upon oxidation by ROS, nonfluorescent DCF became fluorescent DCF. Five microliters of cortex homogenates from each group was mixed with 95 μl of phosphate buffer (pH 7.4) in a black 96-well plate and then treated with 125 μM DCFDA to each well. Fluorescence was detected using a fluorescence microplate reader (SpectraMax i3, Molecular Devices, CA, USA) with excitation and emission of 495 and 529 nm, respectively.
Brain-Derived Neurotrophic Factor ELISA Assay
Brain-derived neurotrophic factor (BDNF) protein levels were quantified using a commercially available kit (Promega), as reported previously [22]. Absorbance was measured at 450 nm using a plate reader. Duplicate determinations per sample were averaged, and levels of BDNF protein were determined using a standard curve.
Chromatographic System
Separation was performed in an HPLC system (Dionex Ultimate 3000—Thermo Fisher Scientific) comprising a pump, an autosampler, a column oven, and a diode array UV/VIS detector. Chromatograms from the detector were recorded by the Chromeleone software (Ver. 7) system. Procyanidin B2, (−)-gallocatechin (GC), (−)-epigallocatechin (EGC), and (−)-epicatechin (EC) were purchased from Sigma-Aldrich and (+)-catechin (C) hydrate (Tokyo Chemical Industry Co., Tokyo, Japan) was used as the standard component [23]. All reagents were at least of an analytical grade, and the purity of all reference standards was above 98.0%. The components of SC were separated on a Luna phenyl-hexyl C18 column (4.6 × 250 mm, 5 μm, Agilent, CA, USA) at 40 °C. The injection volume was 3 μl, and the detective wavelength was set at 203 nm. The mobile phase, consisting of ultrapure water with 0.1% trifluoroacetic acid (A) and acetonitrile (B), was run at a flow rate of 1.0 ml/min. The following gradient elution program was used: 0–30 min, 5–10% (B); 30–40 min, 10–10.1% (B); and 40–60 min, 10.1–50% (B).
Statistical Analysis
All results were evaluated using a one-way analysis of variance (ANOVA) with Dunnett’s test. The analyses were performed using GraphPad PRISM software® (GraphPad PRISM Software Inc., Version 5.02, CA, USA). The results are expressed as means ± standard error of the means (SEMs), and p values of <0.05 were considered significant.
Results
SCE Preserved Cell Viability Against Etoposide-Induced Apoptotic Cell Death
SH-SY5Y cells are human neuroblastoma-derived cell line and can differentiate into neurons by induction of RA. These cells have been widely used as a neuronal cell model in studies of neurotoxicity and ischemic stroke [24,25,26]. Therefore, SH-SY5Y cells were used to investigate the protective effects of SCE against etoposide-induced cytotoxicity. To determine the effect of etoposide and SCE treatment on cellular viability, SH-SY5Y cells were treated with etoposide (10–200 μM) or SCE (5–200 μg/ml) for 24 h. Etoposide decreased cell viability at concentrations above 50 μM (Fig. 1a). Therefore, we chose 50 μM of etoposide to study the protective effect of SCE because this concentration decreased cell viability to values close to 60% of vehicle. The MTT assay showed no effect of SCE on cell viability at less than 50 μg/ml (Fig. 1b). However, high concentrations (above 100 μg/ml) of SCE decreased cell viability. To examine the protective effect of SCE against etoposide-induced cytotoxicity, SH-SY5Y cells were pretreated with 25 or 50 μg/ml SCE for 6 h and then treated with 50 μM etoposide for 24 h. As shown in Fig. 1c, etoposide-treated cells exhibited obvious morphological changes, such as the presence of shiny, floating, and round-shaped cells and cell shrinkage, which are typical characteristics of apoptotic cell death. However, pretreatment with SCE effectively blocked etoposide-induced morphological changes (Fig. 1c). SCE significantly protected cells against etoposide-induced cytotoxicity, suggesting that SCE may be effective in reversing the cytotoxicity of etoposide (Fig. 1d). The anti-apoptotic effect of SCE against etoposide-induced apoptosis was assessed by expression of cleaved PARP and caspase-3, markers for the induction of apoptosis. SCE significantly suppressed etoposide-induced PARP and caspase-3 cleavage and phosphorylated p53 (Fig. 2a). Also, etoposide increased the activity of caspase-3/7, whereas Z-VAD-FMK, a pan-caspase inhibitor, inhibited etoposide-induced caspase-3/7 activation (Fig. 2b). SCE, especially 50 μg/ml of SCE, completely reduced caspase-3/7 activation (Fig. 2b). To further clarify the effect of SCE on etoposide-induced apoptosis, flow cytometry analysis after Annexin-V and PI staining was performed, as shown by the representative dot plots and quantitative graph in Fig. 2c. Etoposide-only-treated cells were shifted to the lower and upper right quadrant (Annexin-V+/PI−) compared with vehicle-treated cells, indicating that etoposide induced early apoptosis in SH-SY5Y cells (Fig. 2c). However, SCE inhibited etoposide-induced apoptosis in a concentration-dependent manner. Etoposide-induced early apoptosis was also blocked by Z-VAD-FMK, a pan-caspase inhibitor. We further confirmed the effect of SCE on etoposide-induced apoptosis using TUNEL staining. TUNEL staining also indicated that etoposide-treated cells exhibited TUNEL-positive cells, but Z-VAD-FMK decreased TUNEL-stained cells (Fig. 2d). Pretreatment with SCE significantly decreased TUNEL-positive cells. In the apoptotic cell death process, cells have been shown to induce the disruption of MMP and mitochondrial membrane depolarization [27, 28]. To explore the protective effects of SCE against etoposide-induced MMP dysfunction, cells were pretreated with SCE for 6 h and then treated with 50 μM etoposide for 2 h. Cells were stained with JC-1 dye as a MMP indicator, which exhibits MMP-dependent accumulation by a fluorescence emission change from red to green. We also performed the quantitative analysis for red/green fluorescence ratio in JC-1 staining. In vehicle-treated cells (nonapoptotic cells), JC-1 dye accumulated as a J-aggregative form in the mitochondria (MMP accumulation in the mitochondria), as shown by red fluorescence, whereas etoposide-treated cells (apoptotic cells) exhibited decreased red fluorescence and increased green fluorescence (low MMP in the mitochondria) (Fig. 2e). By contrast, pretreatment of SCE significantly inhibited etoposide-induced MMP dysfunction. Z-VAD-FMK also inhibited MMP depolarization by etoposide. These results show that SCE has a protective effect against etoposide-induced apoptotic cell death that is associated with the inhibition of MMP disruption.

Protective effects of Spatholobi Caulis extract (SCE) against etoposide-induced cytotoxicity in SH-SY5Y neuroblastoma cells. SH-SY5Y cells were seeded in 96-well plates (5 × 104 cells/ml) and cultured for 24 h. Cells were treated with the indicated concentrations of etoposide (a) or SCE (b) for 24 or 48 h. The values shown are means ± SEM (n = 8). **p < 0.01, compared to vehicle (ANOVA with Dunnett’s test). Cells were pretreated with SCE for 6 h and then co-treated with SCE and 50 μM etoposide for 24 h. c Inverted microscope images showing representative cell morphology of SH-SY5Y cells after treatment with SCE for 6 h and co-treated with 50 μM etoposide for 24 h (×100 magnification, scale bar = 50 μm). d Cell viability was performed by MTT assay. The values shown are means ± SEM (n = 8). ##p < 0.01, compared to vehicle without etoposide; **p < 0.01, compared to vehicle with etoposide (ANOVA with Dunnett’s test)

Effects of SCE on etoposide-induced apoptosis in SH-SY5Y cells. a PARP, caspase-3, and p-p53 were examined using western blotting. β-Actin was used as a protein loading control. A representative blot is shown from four independent experiments that yielded similar results. Quantification of band densities for cleaved PARP and caspase-3 and p-p53 was measured. Data are expressed as means ± SEM (n = 4). ##p < 0.01, compared to vehicle without etoposide; *p < 0.05, **p < 0.01, compared to vehicle with etoposide (ANOVA with Dunnett’s test). b Caspase-3/7 activity was measured in SH-SY5Y cells by Caspase-Glo assay. Cells were treated with 50 μM Z-VAD-FMK for 1 h before treatment with SCE or etoposide. Data are expressed as means ± SEM (n = 4). ##p < 0.01, compared to vehicle without etoposide; **p < 0.01, compared to vehicle with etoposide (ANOVA with Dunnett’s test). c Cells treated with vehicle, etoposide, or etoposide + SCE (25 or 50 μg/ml) for 24 h were subjected to flow cytometry analysis after PI and Annexin-V staining. Representative images indicated healthy (lower left quadrant), early apoptotic (lower right quadrant), late apoptotic (upper right quadrant), and necrotic cells (upper left quadrant). The quantitative graph showed the percentage of healthy, early apoptotic, late apoptotic, and necrotic cells according to treatment. Values are reported as means ± SEM (n = 3). ##p < 0.01, compared to vehicle without etoposide; **p < 0.01, compared to vehicle with etoposide (ANOVA with Dunnett’s test). d Representative images showed that TUNEL-positive cells were detected in SH-SY5Y cells (×100 magnification, scale bar = 100 μm). The bar graph indicated the TUNEL-positive cells in each treatment group. Data are presented as the mean ± SEM (n = 4). ##p < 0.01, compared to vehicle without etoposide; **p < 0.01, compared to vehicle with etoposide (ANOVA with Dunnett’s test). e Mitochondrial membrane potential (MMP) was assessed by fluorescence microscopy using JC-1 staining. Representative images showing red fluorescence (aggregated form) and green fluorescence (monomeric form) (×200 magnification, scale bar = 50 μm). The graph showed the red/green fluorescence intensity ratio. Values are reported as means ± SEM (n = 3). ##p < 0.01, compared to vehicle without etoposide; **p < 0.01, compared to vehicle with etoposide (ANOVA with Dunnett’s test)
JNK and p38 MAPK Activation Were Implicated in the Protective Effect of SCE Against Etoposide-Induced Apoptotic Cell Death
We investigated which intracellular signaling pathways, including the MAPK signaling pathway, were involved in the protective effect of SCE against etoposide-induced apoptotic cell death in SH-SY5Y cells. After pretreatment with 25 or 50 μg/ml SCE for 6 h, SH-SY5Y cells were treated with 50 μM etoposide for 2 h. Etoposide significantly phosphorylated JNK and p38 MAPK, but pretreatment with SP600125 or SB203580 significantly blocked etoposide-induced JNK and p38 MAPK activation. SCE completely inhibited etoposide-induced JNK and p38 phosphorylation (Fig. 3a, b). Thus, phosphorylation of JNK and p38 MAPK was shown to be involved in etoposide-induced cytotoxicity, whereas SCE inhibited JNK and p38 MAPK activation.

Protective effects of SCE on etoposide-induced cytotoxicity are associated with reduced JNK and p38 MAPK activation. a Cells were pretreated with SCE for 6 h and then co-treated with SCE and 50 μM etoposide for 2 h. Western blotting indicated the expression of phosphorylated (p)-JNK and p-p38 in SH-SY5Y cells. β-Actin was used as a protein loading control. A representative blot is shown from four independent experiments that yielded similar results. Quantification of band densities was normalized to each total form expression. Data are expressed as means ± SEM (n = 4). ##p < 0.01, compared to vehicle without etoposide; **p < 0.01, compared to vehicle with etoposide (ANOVA with Dunnett’s test). b Cells were pretreated with SP600125 (JNK inhibitor, 5 μM) or SB203580 (p38 MAPK inhibitor, 5 μM) for 1 h, with 25 or 50 μg/ml SCE for 6 h, and with 50 μM etoposide for 2 h, and then subjected to Western blotting. β-Actin was used as a protein loading control. A representative blot is shown from three independent experiments that yielded similar results. Quantification of band densities was normalized to each total form expression. Values are shown as means ± SEM (n = 3). ##p < 0.01, compared to vehicle without etoposide; **p < 0.01, compared to vehicle with etoposide (ANOVA with Dunnett’s test). SH-SY5Y cells were pretreated with SP600125 (JNK inhibitor, 5 μM) or SB203580 (p38 MAPK inhibitor, 5 μM) for 1 h, with 25 or 50 μg/ml SCE for 6 h, and then 50 μM etoposide for 24 h. c Cells were observed under an inverted microscope, and cell viability was examined using MTT assay (×100 magnification, scale bar = 50 μm). Values are shown as means ± SEM (n = 6). ##p < 0.01, compared to vehicle without etoposide; **p < 0.01, compared to vehicle with etoposide (ANOVA with Dunnett’s test). d Representative images showed that TUNEL-positive cells were detected in SH-SY5Y cells (×100 magnification, scale bar = 100 μm). The bar graph indicated the TUNEL-positive cells in each treatment group. Data are presented as the mean ± SEM (n = 3). ##p < 0.01, compared to vehicle without etoposide; **p < 0.01, compared to vehicle with etoposide (ANOVA with Dunnett’s test). e Apoptosis-related protein expression in SH-SY5Y cells was detected by Western blotting. A representative blot is shown from three independent experiments that yielded similar results. Quantification of band densities for cleaved PARP and caspase-3 and p-p53 was measured. Data are expressed as means ± SEM (n = 3). ##p < 0.01, compared to vehicle without etoposide; *p < 0.05, **p < 0.01, compared to vehicle with etoposide (ANOVA with Dunnett’s test)
To examine whether JNK and p38 MAPK modulation were related to the protective effect of SCE against etoposide-induced apoptotic cell death, cells were preincubated with SP600125 or SB203580 for 1 h and with SCE for 6 h and then treated with etoposide for 24 h. As indicated in Fig. 3c, etoposide-treated cells showed the characteristics of apoptotic cell death; by contrast, pretreatment with SCE, SP600125, or SB203580 significantly recovered these cellular patterns (Fig. 3c). To determine cell viability, we used the MTT assay. Etoposide-only treatment showed a decline in cell viability, whereas pretreatment of cells with SP600125 or SB203580 significantly preserved cell viability. SCE also showed a remarkable protective effect against etoposide-induced reductions in cell viability. TUNEL staining also indicated that etoposide-treated cells exhibited TUNEL-positive cells, but treatment with SP600125 or SB203580 blocked etoposide-induced apoptotic cell death (Fig. 3d). SCE significantly inhibited etoposide-induced TUNEL-stained cells. Pretreatment of cells with SP600125 or SB203580 dramatically inhibited etoposide-induced cleavage of PARP and caspase-3 and phosphorylated p53, indicating that activation of JNK and p38 MAPK is crucial to etoposide-induced apoptotic cell death in SH-SY5Y cells (Fig. 3e). Pretreatment with SCE also protected against PARP and caspase-3 cleavage and phosphorylation of p53. These results indicate that the inhibitory effect of SCE on etoposide-induced apoptotic cell death was effectively attenuated through inhibition of JNK and p38 MAPK activation.
SCE Reduced Ischemic Damage and Neuronal Cell Loss in Focal Ischemia/Reperfusion Injury
We investigated the protective effects of SCE in vivo with the rat focal cerebral ischemia/reperfusion model. SCE (100 and 200 mg/kg) was administered orally for 3 days before MCAO surgery and for 7 days after reperfusion. Don is generally prescribed for the treatment of Alzheimer’s disease and dementia [29, 30]. Don has been reported to lead to recovery after ischemic stroke [31,32,33,34]. Therefore, in this study, Don (5 mg/kg) was used as a positive control. Nissl staining using cresyl violet was applied to evaluate the pathologic and morphologic changes in neurons within the cortex and striatum after MCAO-reperfusion. As shown in Fig. 4a, in the MCAO-reperfusion injured group, neurons mostly exhibited an irregular shape, dark-stained nuclei, and disordered arrangement (Fig. 4a). SCE administration dramatically improved these pathological deficits in a dose-dependent manner. Quantitative data showed that the numbers of intact neurons were decreased by neurological injury after MCAO-reperfusion for 7 days in the ipsilateral cortex, but SCE treatment protected against this neuronal damage. Administration of Don also significantly decreased neuronal damage and neuronal cell loss after MCAO-reperfusion for 7 days.

SCE protected MCAO-induced neuronal damage in the brain. a To determine the neuropathological changes, Nissl bodies within neurons of brain section were stained by cresyl violet. Representative images showed Nissl bodies in the striatum and cortex of ipsilateral brain after MCAO-reperfusion injury for 7 days (×200 magnification, scale bar = 100 μm). Quantitative analysis indicated the number of intact neurons in the ipsilateral cortex. Values are shown as means ± SEM (n = 5 rats/group). ##p < 0.01, compared to the control group; **p < 0.01, compared to the MCAO group (ANOVA with Dunnett’s test). b Images showing double-labeled immunostaining with anti-NeuN (red) and anti-β-III-tubulin (green) antibodies in the cortex of the rat brain following 7 days reperfusion after 45 min MCAO (×200 magnification, scale bar = 100 μm). The graph shows that the cell number of NeuN or β-III-tubulin-positive cells was normalized to DAPI. Values are shown as means ± SEM (n = 5 rats/group). ##p < 0.01, compared to the control group; **p < 0.01, compared to the MCAO group (ANOVA with Dunnett’s test). c Levels of cortical BDNF from each group were determined quantitatively using the ELISA assay. Data are presented as the mean ± SEM (n = 5 rats/group). ##p < 0.01, compared to the control group; **p < 0.01, compared to the MCAO group (ANOVA with Dunnett’s test)
To investigate the protective neuronal survival effects of SCE against ischemia/reperfusion injury, we evaluated the neuronal cell expression and neurogenic protein levels in the cortex after MCAO-reperfusion for 7 days. We performed immunostaining with neuron-specific markers, NeuN (a mature neuron marker) and β-III-tubulin (a neuron marker). As shown in the immunostaining and quantitative graph, NeuN- and β-III-tubulin-positive cells were markedly decreased after MCAO compared with the control group (Fig. 4b). By contrast, SCE or Don administration significantly reduced MCAO-reperfusion-induced neuronal loss in the cortex. MCAO-reperfusion-induced neuronal loss was associated with a decrease in the level of BDNF in the cortex, but administration with SCE or Don effectively restored the neurogenic protein level (Fig. 4c). These results indicate that SCE and Don protect cortical neurons against MCAO-reperfusion-induced ischemic damage and neuronal cell loss.
SCE Has Protective Effects That Are Mediated by Decreased Glial Activation and Apoptosis in Focal Ischemia/Reperfusion Injury
In response to MCAO-induced ischemia/reperfusion injury, activated glia induce oxidative stress, which results in neuronal damage. We assessed glial activation using immunostaining with Iba-1 (a microglia marker) and GFAP (an astrocyte marker). Iba-1- and GFAP-positive cells were dramatically accumulated in the infarct region after MCAO-reperfusion injury for 7 days (Fig. 5a). In the MCAO-reperfusion group, the expression of Iba-1- and GFAP-positive cells exhibited activated glia morphology, such as enlarged cell bodies and round/amoeboid-shaped cells. However, SCE or Don administration clearly reduced glial activation in the injured field. Quantitative values of Iba-1 and GFAP intensity in the MCAO-reperfusion group exhibited a 3.0- and 2.2-fold increase, respectively, compared with the control group. The SCE-treated group showed significantly reduced activated glial intensity by MCAO. Don treatment also attenuated MCAO-reperfusion-induced glial activation.

SCE ameliorated the glial activation and apoptosis in the MCAO-reperfusion injury. a Images showing double-labeled immunostaining with anti-Iba-1 (red) and anti-GFAP (green) antibodies in the cortex of the rat brain following 7 days reperfusion after 45 min MCAO (×200 magnification, scale bar = 100 μm). The graph shows that the cell number of Iba-1 or GFAP-positive cells was normalized to DAPI. Values are shown as means ± SEM (n = 5 rats/group). ##p < 0.01, compared to the control group; **p < 0.01, compared to the MCAO group (ANOVA with Dunnett’s test). b To determine the anti-inflammatory actions of SCE in MCAO-reperfusion injury for 7 days, ROS generation was measured by DCFDA dye. Data are presented as the mean ± SEM (n = 5 rats/group). ##p < 0.01, compared to the control group; **p < 0.01, compared to the MCAO group (ANOVA with Dunnett’s test). c Representative images showed that TUNEL-positive cells were detected in the ipsilateral ischemic cortex (×200 magnification, scale bar = 100 μm). The bar graph indicated the TUNEL-positive cells in each group. Data are presented as the mean ± SEM (n = 5 rats/group). ##p < 0.01, compared to the control group; **p < 0.01, compared to the MCAO group (ANOVA with Dunnett’s test)
During ischemia/reperfusion injury, glial activation and neuronal cell death is accompanied by induction of inflammatory responses, such as ROS generation, and has been shown to be involved in the pathogenesis of ischemic injury [35,36,37]. To investigate whether the protective effects of SCE in the MCAO-reperfusion model are associated with its anti-inflammatory activities and anti-apoptotic actions, we analyzed ROS generation and apoptosis. As shown in Fig. 5b, MCAO-reperfusion-induced neuronal injury for 7 days significantly elevated ROS production in the ipsilateral cortex; however, this event can be reduced by the administration of SCE or Don (Fig. 5b). Next, TUNEL staining was used to detect apoptotic cell death caused by MCAO-reperfusion in the cortex. The fluorescence and number of TUNEL-positive cells were significantly increased in the MCAO-reperfusion group compared with the control group (Fig. 5c). By contrast, SCE or Don administration significantly alleviated MCAO-reperfusion-mediated apoptotic cell death in the cortex. Therefore, these results indicated that the protective effects of SCE against MCAO-reperfusion-induced brain injury are mediated by reduced glial activation through anti-inflammatory activity and the anti-apoptotic action of SCE.
SCE Exerted Protective Effects Through Suppression of MCAO-Reperfusion-Induced JNK/p38 MAPK Activation and Apoptosis
The representative band and quantitative graph show that MCAO-reperfusion injury for 7 days remarkably activated GFAP expression and reduced β-III-tubulin expression in the ipsilateral cortex (Fig. 6). We found that MCAO-reperfusion increased the cleavage of PARP and caspase-3 in the ipsilateral cortex. SCE or Don administration resulted in a decline of PARP and caspase-3 cleavage, which is mediated by their anti-apoptotic actions. Next, to understand how administration of SCE blocked MCAO-reperfusion-induced neuronal damage, we examined whether SCE could inhibit JNK and p38 MAPK activation following MCAO-reperfusion injury for 7 days. Numerous studies have reported that activation of JNK and p38 MAPK is implicated in ischemic stroke-induced inflammatory response and apoptotic cell death [38, 39]. Our findings are consistent with the results of previous studies that report that cortex injured by MCAO-reperfusion exhibits markedly increased phosphorylation of JNK and p38 MAPK. MCAO-induced p-JNK and p-p38 activation were not reduced by SCE 100 mg/kg administration. SCE 200 mg/kg or Don administration significantly inhibited MCAO-induced p-JNK and p-p38 activation. Therefore, these results suggested that SCE decreased the glial activation and apoptosis in MCAO-reperfusion rats by attenuating JNK and p38 MAPK activation.

Effects of SCE on protein expression in MCAO-reperfusion injury. a Representative bands of protein expression in the ipsilateral cortex following 7 days reperfusion after 45 min MCAO were analyzed by Western blotting. b Bar graph for each protein expression quantified by densitometric analysis and expressed fold changes in ipsilateral cortex from the control group. Data are presented as the mean ± SEM (n = 5 rats/group). ##p < 0.01, compared to the control group; **p < 0.01, compared to the MCAO group (ANOVA with Dunnett’s test)
Identification of Major Components in SCE Using HPLC-DAD Analysis
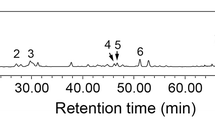
Previous research has demonstrated the identification and quantification of active components, including phenolic compounds, from SCE [23, 40]. In the present study, the UV wavelength of chromatograms was adjusted based on the maximum UV absorption of the major standard compounds: GC (1), 13.19 min; EGC (2), 22.17 min; C (3), 26.32 min; EC (4), 38.13 min; and procyanidin B2 (5), 39.36 min. Therefore, the chromatograms were analyzed at 203 nm. The constituents of SCE were determined by HPLC-DAD analysis, and each peak of the UV spectra was compared with that of the representative standard compounds. As shown in Fig. 7a, b, HPLC-DAD analysis revealed that single peaks of each chemical standard contained in the SCE were identified at similar retention times (t R): GC, 13.22 min; EGC, 22.21 min; C, 26.37 min; EC, 38.17 min; and procyanidin B2, 39.40 min (Fig. 7a, b).

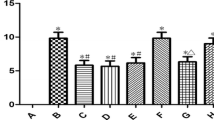
HPLC-DAD analysis of SCE at 203 nm UV wavelength. HPLC-DAD analysis of the components from Spatholobi Caulis extracts. Two chromatograms of five standard mixtures (a) and Spatholobi Caulis extracts (b) were detected at 203 nm, respectively. Retention time of (1) (−)-gallocatechin (GC), (2) (−)-epigallocatechin (EGC), (3) (+)-catechin (C), (4) (−)-epicatechin (EC), and (5) procyanidin B2 was shown in the chromatogram. c Cell viability was performed by MTT assay. The values shown are means ± SEM (n = 6). ##p < 0.01, compared to vehicle without etoposide; **p < 0.01, compared to vehicle with etoposide (ANOVA with Dunnett’s test)
To determine the protective effect of five active compounds within SCE on etoposide-induced cytotoxicity, SH-SY5Y cells were pretreated with 50 μM each compound for 6 h and then treated with 50 μM etoposide for 24 h. GC, EGC, and procyanidin B2 significantly protected the cell viability against etoposide-induced cytotoxicity (Fig. 7c). However, pretreatment with C or EC did not prevent the cell viability on etoposide-treated cells. These phenolic compounds have been reported to confer protective effects against ischemic stroke-associated pathologies [41,42,43,44]. This evidence supports our present findings that active compounds of SCE might contribute to protective effects in etoposide-induced neuronal cell death and MCAO-reperfusion brain injury.
Discussion
SC, the stem of Spatholobus suberectus Dunn, is a traditional herbal medicine and has been shown to have anti-inflammatory activity and cytotoxicity effects. Our result also indicated that high concentrations (above 100 μg/ml) of SCE were cytotoxic in SH-SY5Y cells. There is evidence that high concentrations of SCE may mediate its ability to trigger cell death in cultured tumor cell lines and mouse cancer xenografts [16, 45, 46]. A concentration of 25 or 50 μg/ml SCE, which has not affected the proliferation of SH-SY5Y cells, only exerted protective effects on SH-SY5Y cells against etoposide-induced cytotoxicity. The present findings demonstrate the protective effects of SCE on neuronal cells and the brain under neurotoxic conditions. Our data show that neuronal apoptotic cell death and intracellular ROS production occur following etoposide treatment and MCAO-reperfusion injury. SCE effectively reduced these events, resulting in preservation of cell viability in SH-SY5Y cells and promotion of neuronal cell survival and reduction in neurogenic markers in the infarct region of the brain. Furthermore, MCAO-reperfusion injury-induced glial activation was alleviated by SCE administration. These results reveal that SCE inhibited the activation of JNK and p38 MAPK following etoposide treatment and MCAO-reperfusion injury, which blocked apoptosis.
Ischemic stroke is a severe disease worldwide and is a leading cause of death. It can cause sudden death due to an insufficient supply of blood, oxygen, and nutrients to the brain. The MCA, which is the largest cerebral artery, is the most common infarct site during the occurrence of ischemic stroke [47, 48]. Moreover, reperfusion injury results in secondary damage due to recirculation of the blood supply and restoration of oxygen and nutrients after MCAO. This damage causes enhanced inflammation, oxidative stress, neurotoxicity, and mitochondrial damage, resulting in accelerated brain damage [49, 50]. In the present study, we demonstrated the protective effects of SCE in human neuroblastoma SH-SY5Y cells exposed to cytotoxicity by etoposide and in rats subjected to transient focal ischemic stroke following the subacute phase 7 days after reperfusion with MCAO.
Neuronal apoptosis is one of the main phenotypes of apoptotic cell death during the progression of MCAO-reperfusion injury [51, 52]. Cleavage of caspase-3 is exhibited at the end stage of intrinsic or extrinsic apoptosis cascades, as is cleaved PARP, which is a well-known substrate for caspase-3. These events can cause DNA fragmentation and mitochondrial dysfunction, resulting in the promotion of cell death. The present results indicate that cleavage of PARP and caspase-3 was induced by etoposide-treated SH-SY5Y cells and in the ischemic cortex after MCAO-reperfusion injury, but SCE effectively reduced expression of apoptosis-related protein. Further analysis, such as Annexin-V/PI staining and TUNEL assays, showed that elevation of neuronal apoptosis by etoposide and MCAO-reperfusion injury was alleviated by SCE treatment. Furthermore, etoposide-treated cells caused depolarization of MMP, which was analyzed by JC-1 staining, whereas SCE protected against MMP in SH-SY5Y cells. According to these results, SCE has protective effects against apoptotic cell death in cells and rat brains through its anti-apoptotic activity.
JNK and p38 MAPK are involved in neuronal survival or damage in neurological disorders, including ischemic stroke [53, 54]. Previous research has reported that activation of JNK and p38 MAPK is mediated in neuronal apoptosis, infarcted volume, and neurological deficits in ischemic stroke [55, 56]. Our results were in agreement with previous research, which indicated that phosphorylation of JNK and p38 MAPK was elevated in etoposide-treated cells and MCAO-reperfusion injured cortex. These events were reduced by the inhibitor for JNK and p38 MAPK. These findings were supported by adequate evidence that JNK and p38 MAPK produced antisurvival signaling on neuronal cells during etoposide treatment and MCAO-reperfusion injury. Our findings indicated that SCE inhibited activation of JNK and p38 MAPK during etoposide treatment and MCAO-reperfusion injury. However, unfortunately, SCE 100 mg/kg has not blocked p-JNK and p-p38 activation by MCAO-reperfusion injury. It might be reflected that low dose of SCE was not enough to block MCAO-reperfusion-induced p-JNK and p-p38 activation. In addition, SCE showed protective effects on the MCAO-reperfusion injury in a dose-dependent manner. SCE 100 mg/kg did not completely block MCAO-induced neuronal damage and glial activation. Our in vitro and in vivo results showed that treatment with SCE at the most effective dose (in vitro 50 μM; in vivo 200 mg/kg) dramatically reduced etoposide- or MCAO-induced neuronal damage. Furthermore, the role of JNK and p38 MAPK signaling pathways in the inhibitory effect of SCE was revealed by anisomycin, an activator of JNK and p38 MAPK. Anisomycin-treated cells showed increase of protein levels and activity of JNK and p38 MAPK (Supplementary Fig. S1). By contrast, SCE completely blocked this phosphorylation, suggesting the involvement of JNK and p38 MAPK in the protective effect of SCE. In addition, anisomycin remarkably induced apoptosis and decreased cell viability, and SCE pretreatment reversed these changes (Supplementary Fig. S1). These results suggest that SCE possesses protective effects against apoptosis and cytotoxicity in SH-SY5Y cells via mechanisms involving the activation of JNK and p38 MAPK.
Microglia and astrocytes in the brain are triggered by MCAO-reperfusion injury-induced inflammation and oxidative stress [57, 58]. Their activation contributes to the clearance of dead cells and promotes phagocytosis in the ischemic site of the brain, thereby supporting neuronal survival and maintaining neuronal homeostasis in the brain [36, 59, 60]. Activated glia release pro-inflammatory mediators, such as IL-1β, IL-6, TNF-α, nitric oxide, and superoxide, resulting in neuronal cell death, which leads to exacerbated brain injury [61]. Our results also showed that 7 days following subacute injury after MCAO-reperfusion, both Iba-1 and GFAP were highly expressed at the ischemic site and ROS generation was elevated in the ischemic cortex. SCE effectively ameliorated activation of microglia and astrocytes, indicating that the protective effects of SCE on MCAO-reperfusion injury were partially due to reduction of glial activation and glial activation-mediated oxidative stress.
Don was used as a positive control. It has been reported that Don improves ischemic infarction, neuronal damage, and behavioral deficits in ischemic stroke [31,32,33,34]. In the present study, Don effectively promoted neuronal survival against MCAO-reperfusion injury and reduced apoptosis in the subacute ischemic stroke model. Don also suppressed glial activation and ROS production in the ischemic area. In addition, MCAO-reperfusion injury-induced p38 MAPK activation was remarkably decreased by Don. However, Don slightly inhibited JNK activation, indicating that protective effects of Don are probably mediated by p38 MAPK rather than JNK. Previous research reported that adjuvant treatment or initiated treatment with Don could affect the beneficial effect on acute ischemic stroke and neurological symptoms after ischemic stroke [31, 62,63,64]. Therefore, SCE and Don reduced MCAO-reperfusion-induced damage, with their protective mechanisms involving JNK and p38 MAPK in the brain.
MCAO-reperfusion injury causes neuropathological damage in the ischemic area of the cortex and striatum, but it also affects neuronal damage in the hippocampus [65,66,67]. It is associated with cognitive deficits in vascular dementia because the hippocampus plays a pivotal role in cognitive function, including learning and memory [68, 69]. We examined the neuropathological changes in the hippocampus in our subacute MCAO-reperfusion injury model following 7 days of reperfusion after MCAO for 45 min. MCAO-reperfusion injury impaired the intact neurons, expression of neuronal markers, and BDNF level in the hippocampus, particularly in the CA1 region in the ipsilateral hippocampus, whereas SCE protected against this neuronal damage (Supplementary Fig. S2). Our results are consistent with previous research indicating that neurons in the CA1 region of the hippocampus are more vulnerable to neuronal damage by ischemic stroke injury than neurons in the CA3 region [70, 71]. In addition, SCE effectively decreased MCAO-reperfusion injury-induced glial activation, oxidative stress, and apoptosis in the ipsilateral CA1 and CA3 regions (Supplementary Fig. S3). As shown in Fig. S3, JNK and p38 MAPK was not activated by MCAO-reperfusion injury, indicating that unlike the ischemic cortex, MCAO-reperfusion injury-induced hippocampal neuronal damage seems to be associated with different mechanisms. Western blot analysis showed that expression of neuron markers and apoptosis-related proteins was not changed by MCAO-reperfusion injury (Supplementary Fig. S4). Compared with the ischemic cortex, the hippocampus is more resistant to neuronal damage following MCAO-reperfusion injury. In rats subjected to transient MCAO-reperfusion injury, the neurons in the cortex are more sensitive to ischemic stroke damage. Previous research has shown that neuronal degeneration and functional neurological deficits in the hippocampus might actively progress from 14 days after MCAO-reperfusion injury [49, 72]. Thus, our method of inducing ischemia might have been insufficient to result in neurological deterioration in the hippocampus.
Collectively, we demonstrated that SCE has potent anti-apoptotic and anti-inflammatory activity in neurons, which may be mediated by promoting neuronal survival and preventing glial activation. These observations were found to be associated with the regulation of JNK and p38 MAPK. Our findings are the first to provide evidence that SCE has potential neuroprotective functions in ischemic stroke and neuronal apoptosis-related neurological disorders.
References
Carmichael ST (2005) Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx 2(3):396–409. doi:10.1602/neurorx.2.3.396
Deb P, Sharma S, Hassan KM (2010) Pathophysiologic mechanisms of acute ischemic stroke: an overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology 17(3):197–218. doi:10.1016/j.pathophys.2009.12.001
Espinoza-Rojo M, Iturralde-Rodriguez KI, Chanez-Cardenas ME, Ruiz-Tachiquin ME, Aguilera P (2010) Glucose transporters regulation on ischemic brain: possible role as therapeutic target. Cent Nerv Syst Agents Med Chem 10(4):317–325
Patino P, Parada E, Farre-Alins V, Molz S, Cacabelos R, Marco-Contelles J, Lopez MG, Tasca CI et al (2016) Melatonin protects against oxygen and glucose deprivation by decreasing extracellular glutamate and Nox-derived ROS in rat hippocampal slices. Neurotoxicology 57:61–68. doi:10.1016/j.neuro.2016.09.002
Fukuta T, Ishii T, Asai T, Sato A, Kikuchi T, Shimizu K, Minamino T, Oku N (2015) Treatment of stroke with liposomal neuroprotective agents under cerebral ischemia conditions. Eur J Pharm Biopharm 97(Pt A):1–7. doi:10.1016/j.ejpb.2015.09.020
Nour M, Scalzo F, Liebeskind DS (2013) Ischemia-reperfusion injury in stroke. Interv Neurol 1(3–4):185–199. doi:10.1159/000353125
Quillinan N, Herson PS, Traystman RJ (2016) Neuropathophysiology of brain injury. Anesthesiol Clin 34(3):453–464. doi:10.1016/j.anclin.2016.04.011
Sims NR, Muyderman H (2010) Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta 1802(1):80–91. doi:10.1016/j.bbadis.2009.09.003
Feigin VL (2007) Herbal medicine in stroke: does it have a future? Stroke 38(6):1734–1736. doi:10.1161/STROKEAHA.107.487132
Kim J, Fann DY, Seet RC, Jo DG, Mattson MP, Arumugam TV (2016) Phytochemicals in ischemic stroke. NeuroMolecular Med 18(3):283–305. doi:10.1007/s12017-016-8403-0
Sun K, Fan J, Han J (2015) Ameliorating effects of traditional Chinese medicine preparation, Chinese materia medica and active compounds on ischemia/reperfusion-induced cerebral microcirculatory disturbances and neuron damage. Acta Pharm Sin B 5(1):8–24. doi:10.1016/j.apsb.2014.11.002
Im NK, Lee SG, Lee DS, Park PH, Lee IS, Jeong GS (2014) Spatholobus suberectus inhibits osteoclastogenesis and stimulates chondrogenesis. Am J Chin Med 42(5):1123–1138. doi:10.1142/S0192415X14500700
Li RW, David Lin G, Myers SP, Leach DN (2003) Anti-inflammatory activity of Chinese medicinal vine plants. J Ethnopharmacol 85(1):61–67
Pang J, Guo JP, Jin M, Chen ZQ, Wang XW, Li JW (2011) Antiviral effects of aqueous extract from Spatholobus suberectus Dunn. against coxsackievirus B3 in mice. Chin J Integr Med 17(10):764–769. doi:10.1007/s11655-011-0642-1
Su EY, Fang YH, Chen HS (2012) Clinical observation of treating 62 patients with severe aplastic anemia failing in immunosuppressive therapy by integrative medicine. Zhongguo Zhong Xi Yi Jie He Za Zhi 32(12):1616–1620
Wang ZY, Wang DM, Loo TY, Cheng Y, Chen LL, Shen JG, Yang DP, Chow LW et al (2011) Spatholobus suberectus inhibits cancer cell growth by inducing apoptosis and arresting cell cycle at G2/M checkpoint. J Ethnopharmacol 133(2):751–758. doi:10.1016/j.jep.2010.11.004
Chang CL, Lin CS, Lai GH (2012) Phytochemical characteristics, free radical scavenging activities, and neuroprotection of five medicinal plant extracts. Evid Based Complement Alternat Med 2012:984295. doi:10.1155/2012/984295
Park HR, Lee H, Park H, Cho WK, Ma JY (2016) Fermented Sipjeondaebo-tang alleviates memory deficits and loss of hippocampal neurogenesis in scopolamine-induced amnesia in mice. Sci Rep 6:22405. doi:10.1038/srep22405
Park HR, Lee H, Park H, Jeon JW, Cho WK, Ma JY (2015) Neuroprotective effects of Liriope platyphylla extract against hydrogen peroxide-induced cytotoxicity in human neuroblastoma SH-SY5Y cells. BMC Complement Altern Med 15:171. doi:10.1186/s12906-015-0679-3
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20(1):84–91
Uluc K, Miranpuri A, Kujoth GC, Akture E, Baskaya MK (2011) Focal cerebral ischemia model by endovascular suture occlusion of the middle cerebral artery in the rat. J Vis Exp 48. doi:10.3791/1978
Park HR, Kong KH, Yu BP, Mattson MP, Lee J (2012) Resveratrol inhibits the proliferation of neural progenitor cells and hippocampal neurogenesis. J Biol Chem 287(51):42588–42600. doi:10.1074/jbc.M112.406413
Zhang Y, Guo L, Duan L, Dong X, Zhou P, Liu EH, Li P (2015) Simultaneous determination of 16 phenolic constituents in Spatholobi Caulis by high performance liquid chromatography/electrospray ionization triple quadrupole mass spectrometry. J Pharm Biomed Anal 102:110–118. doi:10.1016/j.jpba.2014.09.006
Qun E, Tang M, Zhang X, Shi Y, Wang D, Gu Y, Li S, Liang X et al (2015) Protection of seven dibenzocyclooctadiene lignans from Schisandra chinensis against serum and glucose deprivation injury in SH-SY5Y cells. Cell Biol Int 39(12):1418–1424. doi:10.1002/cbin.10537
Tucholski J (2010) TG2 protects neuroblastoma cells against DNA-damage-induced stress, suppresses p53 activation. Amino Acids 39(2):523–532. doi:10.1007/s00726-009-0468-8
Wang P, Cao Y, Yu J, Liu R, Bai B, Qi H, Zhang Q, Guo W et al (2016) Baicalin alleviates ischemia-induced memory impairment by inhibiting the phosphorylation of CaMKII in hippocampus. Brain Res 1642:95–103. doi:10.1016/j.brainres.2016.03.019
Galluzzi L, Morselli E, Kepp O, Kroemer G (2009) Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochim Biophys Acta 1787(5):402–413. doi:10.1016/j.bbabio.2008.09.006
Iijima T (2006) Mitochondrial membrane potential and ischemic neuronal death. Neurosci Res 55(3):234–243. doi:10.1016/j.neures.2006.04.005
Doody RS, Cummings JL, Farlow MR (2012) Reviewing the role of donepezil in the treatment of Alzheimer’s disease. Curr Alzheimer Res 9(7):773–781
Geldmacher DS (2004) Donepezil (Aricept) for treatment of Alzheimer’s disease and other dementing conditions. Expert Rev Neurother 4(1):5–16. doi:10.1586/14737175.4.1.5
Fujiki M, Kobayashi H, Uchida S, Inoue R, Ishii K (2005) Neuroprotective effect of donepezil, a nicotinic acetylcholine-receptor activator, on cerebral infarction in rats. Brain Res 1043(1–2):236–241. doi:10.1016/j.brainres.2005.02.063
Kim SH, Chung DK, Lee YJ, Song CH, Ku SK (2016) Neuroprotective effects of Danggui-Jakyak-San on rat stroke model through antioxidant/antiapoptotic pathway. J Ethnopharmacol 188:123–133. doi:10.1016/j.jep.2016.04.060
Lee JH, Park SY, Shin YW, Kim CD, Lee WS, Hong KW (2007) Concurrent administration of cilostazol with donepezil effectively improves cognitive dysfunction with increased neuroprotection after chronic cerebral hypoperfusion in rats. Brain Res 1185:246–255. doi:10.1016/j.brainres.2007.09.016
Nakai M, Akino H, Kaneda T, Matsuta Y, Shiyama R, Tanase K, Ito H, Aoki Y et al (2006) Acetylcholinesterase inhibitor acting on the brain improves detrusor overactivity caused by cerebral infarction in rats. Neuroscience 142(2):475–480. doi:10.1016/j.neuroscience.2006.06.012
Brouns R, De Deyn PP (2009) The complexity of neurobiological processes in acute ischemic stroke. Clin Neurol Neurosurg 111(6):483–495. doi:10.1016/j.clineuro.2009.04.001
Kim JY, Kim N, Yenari MA (2015) Mechanisms and potential therapeutic applications of microglial activation after brain injury. CNS Neurosci Ther 21(4):309–319. doi:10.1111/cns.12360
Lee Y, Lee SR, Choi SS, Yeo HG, Chang KT, Lee HJ (2014) Therapeutically targeting neuroinflammation and microglia after acute ischemic stroke. Biomed Res Int 2014:297241. doi:10.1155/2014/297241
Guo RB, Wang GF, Zhao AP, Gu J, Sun XL, Hu G (2012) Paeoniflorin protects against ischemia-induced brain damages in rats via inhibiting MAPKs/NF-kappaB-mediated inflammatory responses. PLoS One 7(11):e49701. doi:10.1371/journal.pone.0049701
Wang Y, Zhen Y, Wu X, Jiang Q, Li X, Chen Z, Zhang G, Dong L (2015) Vitexin protects brain against ischemia/reperfusion injury via modulating mitogen-activated protein kinase and apoptosis signaling in mice. Phytomedicine 22(3):379–384. doi:10.1016/j.phymed.2015.01.009
Lu D, He H, Wu B, Yao S (2009) Cytotoxic effect on cancer cells and structural identification of phenols from Spatholobi caulis by HPLC-ESI-MS(n). Nat Prod Commun 4(6):809–812
Ashafaq M, Raza SS, Khan MM, Ahmad A, Javed H, Ahmad ME, Tabassum R, Islam F et al (2012) Catechin hydrate ameliorates redox imbalance and limits inflammatory response in focal cerebral ischemia. Neurochem Res 37(8):1747–1760. doi:10.1007/s11064-012-0786-1
Han J, Wang M, Jing X, Shi H, Ren M, Lou H (2014) (−)-Epigallocatechin gallate protects against cerebral ischemia-induced oxidative stress via Nrf2/ARE signaling. Neurochem Res 39(7):1292–1299. doi:10.1007/s11064-014-1311-5
Shah ZA, Li RC, Ahmad AS, Kensler TW, Yamamoto M, Biswal S, Dore S (2010) The flavanol (−)-epicatechin prevents stroke damage through the Nrf2/HO1 pathway. J Cereb Blood Flow Metab 30(12):1951–1961. doi:10.1038/jcbfm.2010.53
Wu S, Yue Y, Li J, Li Z, Li X, Niu Y, Xiang J, Ding H (2015) Procyanidin B2 attenuates neurological deficits and blood-brain barrier disruption in a rat model of cerebral ischemia. Mol Nutr Food Res 59(10):1930–1941. doi:10.1002/mnfr.201500181
Peng F, Meng CW, Zhou QM, Chen JP, Xiong L (2016) Cytotoxic evaluation against breast cancer cells of isoliquiritigenin analogues from Spatholobus suberectus and their synthetic derivatives. J Nat Prod 79(1):248–251. doi:10.1021/acs.jnatprod.5b00774
Wang Z, Wang D, Han S, Wang N, Mo F, Loo TY, Shen J, Huang H et al (2013) Bioactivity-guided identification and cell signaling technology to delineate the lactate dehydrogenase A inhibition effects of Spatholobus suberectus on breast cancer. PLoS One 8(2):e56631. doi:10.1371/journal.pone.0056631
Shanbhag NC, Henning RH, Schilling L (2016) Long-term survival in permanent middle cerebral artery occlusion: a model of malignant stroke in rats. Sci Rep 6:28401. doi:10.1038/srep28401
Smith HK, Russell JM, Granger DN, Gavins FN (2015) Critical differences between two classical surgical approaches for middle cerebral artery occlusion-induced stroke in mice. J Neurosci Methods 249:99–105. doi:10.1016/j.jneumeth.2015.04.008
Butler TL, Kassed CA, Sanberg PR, Willing AE, Pennypacker KR (2002) Neurodegeneration in the rat hippocampus and striatum after middle cerebral artery occlusion. Brain Res 929(2):252–260
Pignataro G, Scorziello A, Di Renzo G, Annunziato L (2009) Post-ischemic brain damage: effect of ischemic preconditioning and postconditioning and identification of potential candidates for stroke therapy. FEBS J 276(1):46–57. doi:10.1111/j.1742-4658.2008.06769.x
Fang L, Gao H, Zhang W, Zhang W, Wang Y (2015) Resveratrol alleviates nerve injury after cerebral ischemia and reperfusion in mice by inhibiting inflammation and apoptosis. Int J Clin Exp Med 8(3):3219–3226
Yasuda N, Ishii T, Oyama D, Fukuta T, Agato Y, Sato A, Shimizu K, Asai T et al (2014) Neuroprotective effect of nobiletin on cerebral ischemia-reperfusion injury in transient middle cerebral artery-occluded rats. Brain Res 1559:46–54. doi:10.1016/j.brainres.2014.02.007
Lennmyr F, Karlsson S, Gerwins P, Ata KA, Terent A (2002) Activation of mitogen-activated protein kinases in experimental cerebral ischemia. Acta Neurol Scand 106(6):333–340
Zhu H, Zhang Y, Shi Z, Lu D, Li T, Ding Y, Ruan Y, Xu A (2016) The neuroprotection of liraglutide against ischaemia-induced apoptosis through the activation of the PI3K/AKT and MAPK pathways. Sci Rep 6:26859. doi:10.1038/srep26859
Lv Y, Qian Y, Fu L, Chen X, Zhong H, Wei X (2015) Hydroxysafflor yellow A exerts neuroprotective effects in cerebral ischemia reperfusion-injured mice by suppressing the innate immune TLR4-inducing pathway. Eur J Pharmacol 769:324–332. doi:10.1016/j.ejphar.2015.11.036
Wu X, Li L, Zhang L, Wu J, Zhou Y, Zhou Y, Zhao Y, Zhao J (2015) Inhibition of thioredoxin-1 with siRNA exacerbates apoptosis by activating the ASK1-JNK/p38 pathway in brain of a stroke model rats. Brain Res 1599:20–31. doi:10.1016/j.brainres.2014.12.033
Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E (2012) Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci 322(1–2):254–262. doi:10.1016/j.jns.2012.05.030
Jha MK, Lee WH, Suk K (2016) Functional polarization of neuroglia: implications in neuroinflammation and neurological disorders. Biochem Pharmacol 103:1–16. doi:10.1016/j.bcp.2015.11.003
Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J (2007) Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 27(10):2596–2605. doi:10.1523/JNEUROSCI.5360-06.2007
Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K (2006) Microglia provide neuroprotection after ischemia. FASEB J 20(6):714–716. doi:10.1096/fj.05-4882fje
Skaper SD (2007) The brain as a target for inflammatory processes and neuroprotective strategies. Ann N Y Acad Sci 1122:23–34. doi:10.1196/annals.1403.002
Barrett KM, Brott TG, Brown RD Jr, Carter RE, Geske JR, Graff-Radford NR, RB MN, Meschia JF et al, Mayo Acute Stroke Trial for Enhancing Recovery Study G (2011) Enhancing recovery after acute ischemic stroke with donepezil as an adjuvant therapy to standard medical care: results of a phase IIA clinical trial. J Stroke Cerebrovasc Dis 20(3):177–182. doi:10.1016/j.jstrokecerebrovasdis.2010.12.009
Martinez-Vila E, Murie-Fernandez M, Gallego Perez-Larraya J, Irimia P (2006) Neuroprotection in vascular dementia. Cerebrovasc Dis 21(Suppl 2):106–117. doi:10.1159/000091710
Pratt RD (2003) Patient populations in clinical studies of donepezil in vascular dementia. Int Psychogeriatr 15(Suppl 1):195–200. doi:10.1017/S1041610203009190
El Amki M, Clavier T, Perzo N, Bernard R, Guichet PO, Castel H (2015) Hypothalamic, thalamic and hippocampal lesions in the mouse MCAO model: potential involvement of deep cerebral arteries? J Neurosci Methods 254:80–85. doi:10.1016/j.jneumeth.2015.07.008
Uchida H, Fujita Y, Matsueda M, Umeda M, Matsuda S, Kato H, Kasahara J, Araki T (2010) Damage to neurons and oligodendrocytes in the hippocampal CA1 sector after transient focal ischemia in rats. Cell Mol Neurobiol 30(7):1125–1134. doi:10.1007/s10571-010-9545-5
Wang CP, Shi YW, Tang M, Zhang XC, Gu Y, Liang XM, Wang ZW, Ding F (2016) Isoquercetin ameliorates cerebral impairment in focal ischemia through anti-oxidative, anti-inflammatory, and anti-apoptotic effects in primary culture of rat hippocampal neurons and hippocampal CA1 region of rats. Mol Neurobiol. doi:10.1007/s12035-016-9806-5
Hattori K, Lee H, Hurn PD, Crain BJ, Traystman RJ, DeVries AC (2000) Cognitive deficits after focal cerebral ischemia in mice. Stroke 31(8):1939–1944
Wahl F, Allix M, Plotkine M, Boulu RG (1992) Neurological and behavioral outcomes of focal cerebral ischemia in rats. Stroke 23(2):267–272
Kreisman NR, Soliman S, Gozal D (2000) Regional differences in hypoxic depolarization and swelling in hippocampal slices. J Neurophysiol 83(2):1031–1038
Wang W, Redecker C, Bidmon HJ, Witte OW (2004) Delayed neuronal death and damage of GDNF family receptors in CA1 following focal cerebral ischemia. Brain Res 1023(1):92–101. doi:10.1016/j.brainres.2004.07.034
Zhang X, Huang G, Liu H, Chang H, Wilson JX (2012) Folic acid enhances Notch signaling, hippocampal neurogenesis, and cognitive function in a rat model of cerebral ischemia. Nutr Neurosci 15(2):55–61. doi:10.1179/1476830511Y.0000000025
Acknowledgments
This research was supported by a grant (No. K16281) from the Korea Institute of Oriental Medicine funded by the Ministry of Science, ICT, and Future Planning, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic Supplementary Material
ESM 1
(DOCX 4182 kb)
Rights and permissions
About this article

Cite this article
Park, H.R., Lee, H., Lee, JJ. et al. Protective Effects of Spatholobi Caulis Extract on Neuronal Damage and Focal Ischemic Stroke/Reperfusion Injury. Mol Neurobiol 55, 4650–4666 (2018). https://doi.org/10.1007/s12035-017-0652-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0652-x