
Abstract
Growing evidences reveal that 17β-estradiol has a wide variety of neuroprotective potential. Recently, it has been shown that 17β-estradiol can limit ethanol-induced neurotoxicity in neonatal rats. Whether it can stimulate SIRT1 signaling against ethanol intoxicity in developing brain remain elusive. Here, we report for the first time that 17β-estradiol activated SIRT1 to deacetylate p53 proteins against acute ethanol-induced oxidative stress, neuroinflammation, and neurodegeneration. A single subcutaneous injection of ethanol-induced oxidative stress triggered phospho c-jun N terminal kinase (p-JNK) and phospho mammalian target of rapamycin (p-mTOR) accompanied by neuroinflammation and widespread neurodegeneration. In contrast, 17β-estradiol cotreatment positively regulated SIRT1, inhibited p53 acetylation, reactive oxygen species (ROS) production, p-JNK, and p-mTOR activation and reduced neuroinflammation and neuronal cell death in the postnatal rat brain. Interestingly, SIRT1 inhibition with its inhibitor, i.e., EX527 further enhanced ethanol intoxication and also abolished the beneficial effects of 17β-estradiol against ethanol in the young rat’s brain. Indeed, 17β-estradiol treatment increased the cell viability (HT22 cells), inhibited ROS production via the SIRT1/Acetyl-p53 pathway, and reduced the nuclear translocation of phospho-nuclear factor kappa B (p-NF-kB) in the BV2 microglia cells. Taken together, these results show that 17β-estradiol can be used as a potential neuroprotective agent against acute ethanol intoxication.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alcohol abuse is the most common custom in advanced countries throughout the world. A lot of research has focused on the chronic nature of alcohol abusing, while a little is known about the mechanism of acute ethanol intoxication [1]. Fetal alcohol syndrome (FAS), a recognized consequence of prenatal alcohol exposure, is characterized by pre- and postnatal growth deficiencies, craniofacial anomalies, and central nervous system (CNS) dysfunction [2]. These deficits are because of the massive stimulation of neuronal and glial apoptosis in the immature brain and in the rodent brain during the first postnatal week [3,4,5]. The parts of the brain that are sensitive to alcohol exposure are the cortex, hippocampus, and thalamus [6]. A signal dose of ethanol lasting for a few hours in developing rodents (pups) causes a significant neuronal loss throughout the forebrain [3]. Ethanol exposure induces neurodegeneration mainly via apoptosis or necrosis through caspase activation [7, 8]. Studies have shown that administration of alcohol increases oxidative stress and raises the ROS level that alters the antioxidant defense system of the brain, resulting in neurodegeneration [9, 10] Also, they may lead to hypoxia, high calcium concentration, and inhibition of the electron transport chain, causing mitochondrial membrane depolarization and mitochondrial dysfunction that leads to the apoptotic pathway [11, 12].
SIRT1 (silent mating type information regulation 2 homolog 1) is a member of Sirtuins family that induces the expression of targeted genes involved in stress protection, and inhibition of cell cycle arrest and apoptosis genes [13]. It has been discovered that these family of enzymes consists of NAD + -dependent histone/protein deacetylases, and a complex of seven homologous proteins regulating cell functions through chromatin remodeling/histone deacetylation and other cellular factors like NF-kB, HSF1, p53, FOXOs, and PGC-1. SIRT1 via deacetylation may either activate or inhibit several protein targets [14,15,16]. SIRT1 is involved in many human physiological functions like DNA repair, aging, gene expression, and apoptosis [17]. Another group reported that SIRT1 through deacetylating some proteins and their expression modulate cellular stress response [18]. Moreover, it also modulates cell death due to exogenous stress, including oxidative damage, interacting with p53, and regulation of other factors/proteins that leads to cell death [19]. In addition, increased sirt1 protein expression, its nuclear translocation, and increased ERK1/2 phosphorylation has been reported with Tryrosol in HepG2 cells [20]. Induction of SIRT1 expression abrogates neuronal degeneration and death in animal models of AD and Huntington’s disease [21].
17β-Estradiol is a steroid hormone mainly produced in the ovary, and also locally synthesized in different tissues including bone, adipose tissues and nervous tissues both in males and females [22]. The enzyme aromatase (estrogen synthase) catalyzed the conversion of testosterone into 17β-estradiol. That is the reason that 17β-estradiol exerts neuroprotection in rodents and birds as a result of acute injury which increase the aromatase activity and expression [22]. It has been widely accepted that 17β-estradiol via its antioxidant activity increases cell survival by inhibiting neurotoxicity, and its free radical scavenging capabilities has been reported to be receptor independent [23,24,25]. The antioxidant effect of 17β-estradiol against ethanol in the developing brain has already been demonstrated, and these authors have shown that it can reduce ethanol intoxication in the young rat’s cerebellum [26].
In the current study, we used 17β-estradiol against acute ethanol intoxication to know its therapeutic efficacy and the underlying mechanism. This study determined for the first time that 17β-estradiol positively regulates SIRT1 to inhibit p53 acetylation which results in the reduction of oxidative stress, neuroinflammation, and neurodegeneration in the young rat’s brain.
Methods
Chemicals
17β-Estradiol, MTT, and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Promega (Madison, WI, USA) respectively.
Rats and Drug Treatment
Postnatal day seven (P7) Sprague-Dawley (SD) rat pups (18–20 g body weight) were used in the present study. The pups were randomly placed in a total of six groups, 1. control, 2. ethanol (5 g/kg), 3. ethanol (5 g/kg) plus 17β-estradiol (10 mg/kg), 4. estradiol (10 mg/kg), 5. ethanol (5 g/kg) plus EX527 (10 mg/kg), and 6. ethanol (5 g/kg) plus EX527 (10 mg/kg) plus 17β-estradiol (10 mg/kg). All the treatments were performed from 4 to 12 h. The control animals received same volume of 0.9% saline solution. The local ethical committee for animals of the Department of Biology, Division of Applied Life Sciences, Gyeongsang National University, South Korea, approved the experimental procedures.
Cell Culturing and Drug Treatment
Mouse hippocampal HT22 and murine BV2 microglia cell line were cultured in 10% FBS and 1% penicillin/streptomycin-supplemented DMEM (Dulbecco’s modified Eagle’s medium) medium in a humidified 5% CO2 incubator at 37 °C. The cells were treated with ethanol (100 mM), ethanol plus three concentrations of 17β-estradiol (100 mM +10, 20, and 30 μg/ml), and ethanol plus EX527 plus 17β-estradiol (100 mM + 80 μM + 20 μg/ml) for 4 h.
Cell Viability Assay
To assess the cell viability after treatment, the MTT assay was performed according to the manufacturer’s instructions (Sigma). The cells were cultured in 96-well plates at a density of 1 × 104 cells per well containing 100 μl DMEM. After the attachment of the cells, the medium was replaced with a fresh medium containing ethanol (100 mM) and different concentrations of 17β-estradiol (10, 20, and 30 μg/ml). The cells were further incubated for another 3 h. The cells were then incubated for 4 h with MTT solution and the medium was replaced with DMSO. The absorbance was measured at 570 nm. The experiments were repeated for three times.
Western Blot Analysis
Western blot analysis details were conducted as previously performed in our lab [27, 28]. Briefly, the animals were euthanized after 4 h following treatment of ethanol with or without 17β-estradiol. Then, the brains were carefully (hippocampus) collected and placed on dry ice for freezing tissue. Similarly, after treatment, BV2 cells were collected in PBS, centrifuged, and the supernatant was removed. The remaining pellet was dissolved in Pro Prep protein extraction solution, according to the manufacturer protocol (iNtRON Biotechnology) to make cell lysates. The brain homogenates and cell lysates were quantified with Bio-Rad protein assay solution. The homogenates (20 μg protein) were fractionated by SDS-PAGE on 4–12% (Bolt ™ Mini Gels, Life Technologies). After transfer, membranes were blocked in 5% skim milk (or BSA), incubated overnight at 4 °C with primary antibody, and cross-reacting proteins were detected by ECL after reaction with horseradish peroxidase-conjugated secondary antibodies. After using the primary antibodies and membrane-derived secondary antibodies, ECL (Amersham Pharmacia Biotech, Uppsala, Sweden) detection reagent was used for visualization according to the manufacturer’s instructions. Densitometry analysis of the bands was performed using the Sigma Gel software (SPSS, Chicago, IL, USA). Density values were calculated in arbitrary units (A.U.) relative to the untreated control.
Antibodies
The following antibodies were used in this study: rabbit-derived anti-Nrf2, anti-p-NF-kB, anti-Bax, anti-SYP, anti-iNOS, anti-p-Akt, anti-P-mTOR, mouse derived anti-HO-1, anti- PARP-1, anti-caspase-3, anti-p-JNK, anti-GFAP, anti-PSD95, and anti-β-actin from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and anti-SIRT1 and anti-Acetyl-p53 from Cell Signaling Technology.
Tissue Collection and Sample Preparation
Animals were euthanized after 12 h of drug treatment to conduct morphological studies. An equal number of animals were kept in each group (n = 5/group) and transcardial perfusion was performed with 4% ice-cold paraformaldehyde in PBS. Additional fixation with 4% paraformaldehyde was performed followed by 20% sucrose solution for 72 h. Brain sections were made with Leica cryostat (CM 3050C, Germany) on plus charged slides (Fisher).
Fluoro-Jade B Staining
Fluoro-Jade B staining was performed as reported earlier [29]. Chamber slides were air-dried overnight. The slides were kept in a solution of 80% ethanol and 1% sodium hydroxide then in 70% alcohol for 5 min and 2 min, respectively, followed by immersion in distilled water for 2 min. Slides were placed for 10 min in 0.06% potassium permanganate solution. The slides were then rinsed with distilled water and immersed in a solution containing 0.1% acetic acid and 0.01% Fluoro-jade B for 20 min. After rinsing with distilled water and applying DAPI, the slides were dried, and glass cover slips were mounted on slides with mounting medium. Images were captured using an FITC filter on a confocal laser-scanning microscope (FV 1000, Olympus, Japan).
Immunofluorescence
Immunofluorescence staining was performed as we previously reported [30]. Briefly, slides containing tissue and cells were washed with 0.01 M PBS two times for 15 min followed by the addition of proteinase K and blocking solution. Primary antibodies, as given in the above paragraph (1:100 in PBS), were added and incubated at 4 °C overnight. After washing with PBS, secondary antibodies (FITC and TRITC conjugated, Santa Cruz Biotechnology, 1:50 in PBS) were then applied at room temperature for an additional 90 min. Slides were twice washed with PBS for 5 min. DAPI (4′,6-diamidino-2-phenylindole) was applied to stain the nucleus and glass cover slips were mounted on slides with mounting medium. Images were captured using a confocal microscope (FluoView FV 1000 Olympus, Japan).
Oxidative Stress (ROS) Detection in Vitro
ROS assay was performed with slight modification as previously described [31]. Briefly, cells were cultured in 96-well plates in 200 μl DMEM medium that was supplemented with 10% FBS and 1% penicillin/streptomycin in every well. Cells were incubated for 24 h at 37 °C in a humidified incubator having 5% CO2. The next day, the media was replaced by fresh media that contained ethanol (100 mM) and ethanol plus 17β-estradiol (100 mM +10 μg/ml, 20 μg/ml) for an additional 4 h. DCFDA (2′, 7′-dichloroflourescin diacetate) 600 μM dissolved in DMSO/PBS was added to each well and incubated for 30 min. Plates were then read in ApoTox-Glo™ (Promega) at 488/530 nm.
Oxidative Stress (ROS) Detection in Vivo
ROS assay was conducted with slight modification as we reported earlier [32]. Briefly, the brain homogenates from the respective groups were diluted 1:20 times with Locke’s buffer (ice-cold) to get 5 mg tissue/ml concentration. Then, the reaction mixture (1 ml) having Locke’s buffer of pH 7.4, 0.2 ml brain homogenate, and 10 ml of DCFH-DA (5 mM) was incubated for 15 min at room temperature to allow the DCFH-DA to be incorporated into any membrane-bound vesicles and the diacetate group cleaved by esterases. After 30 min of further incubation, the conversion of DCFH-DA to the fluorescent product DCF was measured using a spectrofluorimeter with excitation at 484 nm and emission at 530 nm. ROS formation was quantified from a DCF-standard curve and data are expressed as picomole DCF formed/minute/milligram protein.
GSH Assays
The levels of total GSH and GSH/GSSG ratio were determined by using the glutathione assay kit obtained from BioVision (BioVision Incorporated155 S. Milpitas Boulevard, Milpitas, CA, 95035, USA), Fluorometric Assay Kit (catalog #K264–100), according to the manufacturer’s instructions.
Statistical Analysis
A computer-based Sigma Gel System (SPSS Inc., Chicago, IL) and the Image J program were used to analyze the density and integral optical density (IOD) of scanned X-ray films of Western blot and immunofluorescence images. A one-way ANOVA followed by Student’s t test were used to determine the statistical significance (P value) of the obtained data. The density values of the data were expressed as the means ± SEM of three independent experiments. P values less than 0.05 were considered to be statistically significant, a, b, c, d P < 0.01:
-
a Significantly different from the control group
-
b and c Significantly different from the ethanol-treated group
-
d Significantly different from the ethanol and inhibitor-treated group
Results
17β-Estradiol Attenuates Ethanol-Induced Oxidative Stress in the Young Rat’s Brain
In order to examine the effect of 17β-estradiol against ethanol-induced oxidative stress in P7 rat’s brain, ROS assay was conducted. The results showed that a single injection of ethanol induced oxidative stress and significantly increased reactive oxygen species (ROS) production as compared to the saline-treated rats. Conversely, the rat’s pups that received 17β-estradiol treatment along with ethanol showed significantly less ROS generation level in comparison to ethanol-alone-treated rats (Fig. 1a). Similarly, 17β-estradiol treatment markedly enhanced glutathione (GSH) and GSH/GSSG levels in the brain homogenates of ethanol-treated rats pups (Fig. 1b, c).

17β-Estradiol treatment ameliorates ethanol-induced oxidative stress and ROS production in the postnatal rats. The representative histograms showing a ROS level, b GSH level, and c GSH/GSSG ratio levels in postnatal rats (n = 5/group) brain homogenates. GSH levels were measured by a kit method according to the manufacturer instructions, and expressed as nanomole/milligram protein. d The western blot analysis of Nrf2 and HO-1 in the brain homogenates of p7 rats. The bands were quantified using the Sigma Gel software, and the differences are represented by a histogram. β-Actin was used as a loading control. The density values are expressed in arbitrary units (A.U.) as the means ± S.E.M. for the respective indicated protein (n = 5 rats/group). e The immunofluorescence images (green) of 8-oxoguanine (8-Oxo-G) in the P7 rats brains after ethanol and 17β-estradiol administration. The complete procedures are depicted in the material and methods section. DAPI was used to stain the nucleus. Significance: a,b P < 0.01
Previous studies have demonstrated that heme oxygenase-1 (HO-1) plays an important role in protecting cells against oxidative stress as a cellular defense mechanism [33]. Therefore, we evaluated the effects of 17β-estradiol on the activation of Nrf2/HO-1 signaling in the brain of ethanol-treated rats. The Western blot analyses reveal decreased expressions of Nrf2 and HO-1 proteins in ethanol-treated rats, while 17β-estradiol treatment significantly increased protein expression of Nrf2 and HO-1 in the brain of ethanol-treated pups (Fig. 1d).
Ethanol elevates the ROS levels and alters the antioxidant defense system in the brain, leading to neurodegeneration [9,10,11]. This ROS can cause the DNA damage. The 7, 8-dihydro-8-oxoguanine (8-Oxo-G), a prominent form of oxidative DNA damage, is a useful marker of cellular oxidative stress [34]. To understand the effect of 17β-estradiol on ethanol-induced ROS production in vivo, we analyzed the oxidative stress marker 8-oxoguanine (8-Oxo-G) by the immunofluorescence method. The results showed that compared with the saline-treated P7 pups, the expression of 8-Oxo-G were increased in the brain of the ethanol-treated pups. On the other hand, the 17β-estradiol treatment significantly reduced the expression of 8-OxoG against the ethanol-treated P7 rat brain (Fig. 1e).
17β-Estradiol Reduced Ethanol-Induced Neuroinflammation, Neuroapoptosis and Neurodegeneration in the Young Rat’s Brain
Previous studies have demonstrated that ethanol induces inflammation in NF-kB dependent manners [35, 36]. The Western blot results revealed an increased expressions levels of phospho nuclear factor kappa B (p-NF-kB) and its downstream inflammatory markers such as tumor necrosis factor-α (TNF-α) and intrinsic nitric oxide synthase (iNOS) in the brain homogenates of ethanol-treated rat pups as compared to vehicle-treated pups (Fig. 2a). In contrast, 17β-estradiol administration to developing rat pups significantly inhibited the expressions of p-NF-kB, TNF-α, and iNOS levels as shown in Fig. 2a. Additionally, 17β-estradiol treatment also reduced ethanol-activated astrocytes (glial fibrillary acidic protein (GFAP)) as evidenced from the photomicrograph images given in the Fig. 2b.

17β-Estradiol treatment attenuates ethanol-induced neuroinflammation and neuroapoptosis in the postnatal rats. Given are the immunoblots of a phospho-NF-κB, TNF-α, and iNOS protein in the developing rat’s brain. All the respective bands were quantified using Sigma Gel computer-based software, and the differences are depicted in the histograms. b The immunofluorescence images (green) of reactive astrocytes (GFAP) in the brain of treated animals. c The immunoblot analysis of activated Bax, caspase-3, and cleaved PARP-1 in the young rats brain using respective antibodies. β-Actin was used as a housekeeping gene. The density values are expressed in arbitrary units (A.U.) as the means ± SEM for the respective indicated protein (n = 5 animals/group). All the details are given in the “Methods” section. Significance: a,b P < 0.01
Pro-apoptotic Bax and anti-apoptotic Bcl-2 are members of the Bcl-2 family and are the main regulators of the apoptotic pathway in the mitochondria [37]. The brain homogenates from all the treated rats were evaluated for Bax and caspase-3 via immunoblot technique. The results indicate that a single episode of ethanol administration caused the upregulation of Bax and caspase-3 protein expressions while 17β-estradiol cotreatment with ethanol significantly reversed their expression levels in young rats (Fig. 2c).
Poly (ADP-ribose) polymerase-1(PARP-1) is involved in DNA repair, and the overexpression of PARP-1 through different excitotoxic agents induces neurodegeneration [38]. In the current study, the Western blot analysis revealed that PARP-1 cleavage increased in the ethanol-treated young rats while the expression level of cleaved PARP-1 was significantly reduced in 17β-estradiol-treated p7 rats (Fig. 2c).
17β-Estradiol Improved the Pre-and Postsynaptic Dysfunction Induced by Ethanol in the Developing Brain
Fluoro-Jade B is a widely used marker for damaged neuronal cells and was used to determine the neurodegenerative effect of ethanol in the cortex of the P7 rats. The results showed that the ethanol treatment significantly increased the number of degenerated neurons compared with the control animals. Notably, as assessed from the decreased number FJB-positive cells, the cotreatment with 17β-estradiol significantly reduced the extent of ethanol-induced neurodegeneration in young rat pups (Fig. 3a).

The beneficial effects of 17β-estradiol on ethanol-induced neurodegeneration, synaptic dysfunction, and JNK/Akt/mTOR signaling in the young rat’s brain. a The immunofluorescence representation of FJB-positive neurons in the hippocampal CA1 region of P7 rat’s brain following saline, ethanol, and 17β-estradiol administration. DAPI was used to counterstain the nucleus. The immunoblot analysis of b synaptophysin (SYP), PSD95 and c phospho-JNK, phospho Akt, and phospho mTOR proteins in the P7 rat’s brain. The relative density histograms show the bands quantification by using the Sigma Gel software. β-Actin was used as a loading control. The density values are expressed in arbitrary units (A.U.) as the means ± S.E.M. for the respective indicated protein (n = 5 rats/group). Significance: a,b P < 0.01
To assess the synaptic integrity after ethanol treatment, the expression of the presynaptic vesicle membrane protein synaptophysin (SYP) and the postsynaptic marker postsynaptic density protein 95 (PSD95) were quantified in the brain homogenates of the P7 rats. The Western blot analysis revealed a significant reduction in both SYP and PSD95 levels in ethanol-treated postnatal day 7 rat brains compared with the control, indicating that ethanol could induce synaptic dysfunction (Fig. 3b). However, the brains of the animals that received 17β-estradiol either as a co-treatment along with ethanol or alone showed significantly increased expression of both synaptic markers including SYP and PSD95 proteins (Fig. 3b).
The Effect of 17β-Estradiol on JNK/Akt/mTOR Signaling Against Ethanol in the Young Rat’s Brain
The effect of ethanol and 17β-estradiol on JNK/Akt/mTOR signaling was evaluated in the brain homogenates of young rats through the Western blot technique. The immuno blots indicate that ethanol induced the phosphorylation of c-jun N terminal Kinase (p-JNK) which results in phospho-Akt (p-Akt) inhibition and phosphorylation of mTOR (p-mTOR) in the developing rat’s brain. The administration of 17β-estradiol to the developing rat’s reversed the effect of ethanol on p-JNK/p-Akt/p-mTOR signaling molecules as shown in Fig. 3b.
SIRT1/Acetyl-p53 Signaling is Involved in the Anti-Inflammatory Effect of 17β-Estradiol Against Ethanol both In vivo and In vitro
To determine whether the beneficial effects of 17β-estradiol against ethanol is SIRT1 dependent, the pharmacological inhibitor of SIRT1, i.e., EX527, was used both in vivo and in vitro. The immunoblot results indicate that ethanol completely inhibited SIRT1 protein and upregulated the acetylation of p53 (acetyl-p53) protein expressions in the brain homogenates of young rats. In contrast, 17β-estradiol coadministration with ethanol to postnatal day 7 rats not only activated the SIRT1 protein but also significantly inhibited acetyl-p53 and p-NF-kB proteins as shown in Fig. 4a. Interestingly, the coadministration of EX527 along with ethanol to young rats caused further suppression of SIRT1 and upregulation of both acetyl-p53 and p-NF-kB protein levels. More importantly, it abolished the SIRT1 upregulating effect of 17β-estradiol as shown in Fig. 4a. Similarly, EX527 treatment also affected the 17β-estradiol inhibitory effect on acetyl-p53 and p-NF-kB proteins in the developing rat’s brain (Fig. 4a).

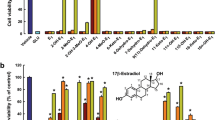
17β-Estradiol reduced ethanol-induced SIRT1/Acetyl-p53 dependent neuroinflammation in vivo and oxidative stress in vitro. Given are the a Western blot results of SIRT1, Acetyl-p53, and phospho-NF-kB after ethanol, 17β-estradiol, and with or without EX527 in the brain homogenates of developing rats. b The MTT and c the ROS assay was conducted in HT22 cells according to the procedure as given in the “Methods” section. These assays were performed in triplicate. The bands were quantified using the Sigma Gel software, and the differences are represented by a histogram. Significance: a,b,c,d P < 0.01
Moreover, 17β-estradiol was nontoxic to HT22 cells in two different concentrations (10 and 20 μg/ml) and all these two concentrations increased the cell viability against ethanol in vitro (Fig. 4b). The second concentration (i.e., 20 μg/ml) completely inhibited ROS production in HT22 cells in vitro (Fig. 4c).
The Beneficial Effects of 17β-Estradiol on SIRT1, p-NF-kB, and Acetyl-p53 Expression
Herein too, 17β-estradiol positively regulates and activates SIRT1 protein signaling. The immunostaining images of SIRT1colocalization either with acetyl-p53 (Fig. 5a) and p-NF-kB (Fig. 5b) in the presence or absence of EX527 reveals that 17β-estradiol can positively regulate SIRT1 and potentially inhibit its downstream signaling such as acetyl-p53 and p-NF-kB. All these beneficial effects of 17β-estradiol against ethanol are depicted in (Fig. 6a, b).

17β-Estradiol stimulates the SIRT1/Acetyl-p53/NF-kB signaling pathway against ethanol in vitro. a The Western blots of Acetyl-p53 and Bax proteins in the cell lysates of BV2 cells after ethanol and 17β-estradiol treatment. The immunofluorescence images of b SIRT1 (green), p-NFkB (red), and c SIRT1 (green), and Acetyl-p53 (red) following ethanol, 17β-estradiol, and with or without EX527 (SIRT1 inhibitor) in BV2 cells. DAPI was used to counterstain the nucleus. Significance: a,b,c P < 0.01

The proposed mechanism of 17β-estradiol against ethanol-induced neurotoxicity. The schematic diagram shows the proposed and underlying mechanism of the neuroprotective effect of 17β-estradiol against the ethanol-induced ROS production, neuroinflammation, and apoptotic neurodegeneration in the developing rat’s brain. This diagram depicts that 17β-estradiol via SIRT1/Acetyl-p53/NF-kB prevents the ethanol-induced ROS production, synaptic deficits, neuroinflammation, and neurodegeneration in the ethanol-treated P7 rats.
Discussion
This current study was conducted to know about the mechanism of 17β-estradiol against acute ethanol intoxication (Fig. 6a, b). It might be the first report which shows that 17β-estradiol can potentially ameliorate ethanol-induced oxidative stress, neuroinflammation, and neurodegeneration in the postnatal day 7 rat’s brain. This mechanistic approach is the first evidence that 17β-estradiol abrogates ethanol-induced toxicity in the SIRT1/Acetyl-p53/NF-kB signaling pathway in a dependent manner in the developing rat brain (Fig. 6a, b).
The existing literature demonstrates that SIRT1 is one of the vital targets of ethanol especially in the liver [39, 40], while its effect on the developing brain is not known yet. The present study provides the direct evidence that SIRT1 activation by 17β-estradiol represents a novel therapeutic mechanism by which SIRT1 inhibits p53 acetylation and prevents oxidative stress, neuroinflammation, and neurodegeneration induced by ethanol in the postnatal day 7 rat’s brain and in the BV2 cell line. Most importantly, the administration of the SIRT1 inhibitor (i.e., EX527) to young rats or treating BV2 cell line further enhanced the ethanol toxic effect (i.e., increased p53 acetylation and phospho-NF-kB activation), while this caused the blockage of 17β-estradiol effects both in vivo and in vitro.
Ethanol is a toxic agent which causes neurotoxicity especially to developing brain as it has been demonstrated that this stage is equivalent to ethanol exposure during the third trimester of pregnancy in humans [41]. In the current study, we employed the same model which states that ethanol exposure to postnatal day 7 rats can evoke apoptotic cascade and induce neurodegeneration [42]. Alcohol-induced oxidative stress is one of the possible and most important mechanisms that lead to neuronal cell death [43]. Due to the large amount of oxygen consumption and reduced antioxidant activities, the brain is extremely susceptible to oxidative damage [44]. Similarly, in contrast to the adult brain, in the immature rat’s brain, there is small number of antioxidant enzymes, which makes it more vulnerable to the neurotoxic effects of oxidative stress. Both the low levels of oxidative stress have a physiological effect on cellular functions including neuronal plasticity while the high levels may cause oxidation/nitrosylation of lipids, proteins, and nucleic acids, resulting in neuronal cell death [45]. That is why to maintain a normal neuronal function the balance between pro- and antioxidant reactions is critical in the brain. Previous reports reveal that prenatal ethanol exposure could cause an increase in oxidative stress in developing organs, including the brain [46, 47]. Our earlier study also supports that ethanol can cause oxidative stress and widespread neurodegeneration in the brain of developing rats [48]. In line with our previous report and other studies here, we too observed that ethanol induces ROS production accompanied by the activation of caspase-dependent neuronal apoptosis both in in vivo and in vitro models. In the current study, we have shown that 17β-estradiol activates SIRT1 expression and exerts its antioxidant effect against ethanol-induced oxidative stress by upregulating endogenous antioxidant systems including both the glutathione levels and Nrf2/HO-1 proteins expressions accompanied by ROS production inhibition. A similar study was conducted in which the authors have shown that 17β-estradiol protected the developing cerebellum against ethanol-induced oxidative stress and neurotoxicity [43]. Another report demonstrates that 17β-estradiol interferes with the ethanol withdrawal-induced alteration of oxidative signaling pathways and thereby protects the neurons, mitochondria, and behaviors [49]. During this study, we conducted the ROS assays both in the postnatal day 7 rat’s brain homogenates and in mouse neuronal and microglial cell lines and all these results have proved that 17β-estradiol can potentially inhibit ethanol-induced oxidative stress.
This study demonstrates for the first time that 17β-estradiol can potentially stimulate SIRT1 to inhibit acetylation of p53 and its downstream signaling. 17β-Estradiol caused the downregulation of activated JNK accompanied by the pro-inflammatory and pro-apoptotic protein markers to rescue the postnatal rat’s brain. Our Western blot and immunofluorescence results reveal that 17β-estradiol completely inhibited the ethanol-induced toxicity via activating endogenous SIRT1. Our hypothesis is that SIRT1 activation can cause the JNK inhibition following its downstream signaling, as the published data indeed supports that FOXO is a substrate of SIRT1 and thus SIRT1 can indirectly (might be via FOXO) inhibit JNK activation against H2O2 and palmitate [50,51,52]. Once JNK is deactivated, it can cause the stoppage of signaling molecules such as various inflammatory and apoptotic proteins hence reduce the toxicity. 17β-Estradiol has been demonstrated as one of the most important neuroprotectant agent since reported. It has been considered as a frontline antioxidant [53] and agent that can ameliorate toxicities induced by various toxins both in in vivo and in vitro models.
In conclusion, the present findings reveal important SIRT1-dependent neuroprotection of 17β-estradiol that reduce the ethanol-induced oxidative damage and subsequent neuroinflammation and apoptosis in the developing rat’s brain. These protective effects of 17β-estradiol include inhibition of ROS production and glutathione upregulation and the deactivation of pro-inflammatory (phospho-NF-kB) and pro-apoptotic proteins (acetylated p53, Bax, caspase-3, and PARP-1) against ethanol intoxication in the postnatal day 7 rats’ brain. A more detailed and mechanistic approach is warranted to explore the therapeutic efficacy of 17β-estradiol against ethanol and other toxins.
Abbreviations
- SIRT1:
-
Silent mating type information regulation 2 homolog 1
- NFκB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- p-JNK:
-
Phospho c-jun N terminal kinase
- p-mTOR:
-
Phospho mammalian target of rapamycin
- ROS:
-
Reactive oxygen species
- FAS:
-
Fetal alcohol syndrome
- CNS:
-
Central nervous system
- NAD:
-
Nicotinamide adenine dinucleotide
- DMSO:
-
Dimethyl sulfoxide
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- PBS:
-
phosphate buffer saline
- SDS-PAGE:
-
Sodium dodecyl sulfate poly acrylamide gel electrophoresis
- BSA:
-
Bovine serum albumin
- FITC:
-
Fluorescein isothiocyanate
- TRITC:
-
Tetramethyl rhodamine isothiocyanate
- DAPI:
-
4′,6-diamidino-2-phenylindole
- FBS:
-
fetal bovine serum
- DCFDA:
-
2′, 7′-dichloroflourescin diacetate
References
Huimin Liu, Wenbin Zheng, Gen Yan, Baoguo Liu, Lingmei Kong, Yan Ding, Zhiwei Shen, Hui Tan, and Guishan Zhang (2014) Acute ethanol-induced changes in edema and metabolite. BioMed Research International 1–8
Jones KL, Smith DW (1973) Recognition of the fetal alcohol syndrome in early infancy. Lancet 2:999–1001
Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V et al (2000) Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 287:1056–1060
Farber NB, Creeley CE, Olney JW (2010) Alcohol-induced neuroapoptosisin the fetal macaque brain. Neurobiol Dis 40:200–206
Creeley CE, Olney JW (2013) Drug-induced apoptosis: mechanism by which alcohol and many other drugs can disrupt brain development. Brain Sci 3:1153–1181
Guerri C (2002) Mechanisms involved in central nervous system dysfunctions induced by prenatal ethanol exposure. Neurotox Res 4:327–335
Obernier JA, Bouldin TW, Crews FT (2002) Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol Clin Exp Res 29:547–557
Valles SL, Blanco AM, Pascual M, Guerri C (2004) Chronic ethanol treatment enhances inflammatory mediators and cell death in the brain and in astrocytes. Brain Pathol 14:365–371
Qin L, Crews FT (2012) NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J Neuroinflammation 9:5
Reddy VD, Padmavathi P, Kavitha G, Saradamma B, Varadacharyulu N (2013) Alcohol-induced oxidative/nitrosative stress alters brain mitochondrial membrane properties. Mol Cell Biochem 375:39–47
Skujal S, Gromal V, Kleina R (2011) Chronic alcohol abuse is implicated in the oxidative stress and the changes in the neurotropic factor receptor expression in the human CNS. Pap Anthropol XX: 368–379
Hoek JB, Cahill A, Pastorino JG (2002) Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology 122:2049–2063
Duan W (2013) Sirtuins: from metabolic regulation to brain aging. Front Aging Neurosci 5:36
Finkel T, Deng CX, Mostoslavsky R (2009) Recent progress in the biology and physiology of sirtuins. Nature 460:587–591
Haigis MC, Sinclair DA (2010) Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5:253–295
Guarente L, Franklin H (2011) Epstein lecture: sirtuins, aging, and medicine. N Engl J Med 364:2235–2244
Grubisha O, Smith BC, Denu JM (2005) Small molecule regulation of Sir2 protein deacetylases. FEBS J 272:4607–4616
Porcu M, Chiarugi A (2005) The emerging therapeutic potential of sirtuin- interacting drugs: from cell death to life span extension. Trends Pharmacol Sci 26:94–103
Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B et al (2004) Calorie restriction promotes mammalian cell survival by inducing the SIRT1deacetylase. Science 305:390–392
Paola S, Maria LB, Concetta PI, Daniela V, Elisa M, Michele C et al (2016) Protective effect of Tyrosol and S-adenosylmethionine against ethanol-induced oxidative stress of Hepg2 cells involves Sirtuin 1, P53 and Erk1/2 signaling. Int J Mol Sci 17(5):622
Donmez G, Outeiro TF (2013) SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Mol Med 5:344–352
Maria AA, Iñigo A, Luis M, Garcia S (2014) The neuroprotective actions of oestradiol and oestrogen receptors. Nat Rev Neurosci. doi:10.1038/nrn3856
Niki E, Nakano M (1990) Estrogens as antioxidants. Methods Enzymol 186:330–333
Ayres SA, Tang M, Subbiah MTR (1996) Estradiol-17B as an antioxidant: some distinct features when compared with common fat-soluble antioxidants. J Lab Clin Med 128:367–375
Moorsmann B, Behl C (1999) The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc Natl Acad Sci U S A 96:8867–8872
Ramezani A, Goudarzi I, Lashkarbolouki T, Ghorbanian MT, Salmani ME, Abrari K (2011) Neuroprotective effects of the 17beta-estradiol against ethanol-induced neurotoxicity and oxidative stress in the developing male rat cerebellum: biochemical, histological and behavioral changes. Pharmacol Biochem Behav 100:144–151
Shah SA, Ali Shah S, Ullah I, Lee HY, Kim MO (2013) Anthocyanins protect against ethanol-induced neuronal apoptosis via GABAB1 receptors intracellular signaling in prenatal rat hippocampal neurons. Mol Neuro 48:257–269
Shah SA, Yoon GH, Chung SS, Abid MN, Kim TH, Lee HY, Kim MO (2016) Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuropathological deficits. Mol Psychiatry:1–10
Shah SA, Yoon GH, Ahmad A, Ullah F, Amin FU, Kim MO (2015) Nanoscale-alumina induces oxidative stress and accelerates amyloid beta (Aβ) production in ICR female mice. Nano 7:15225–15237
Shah SA, Yoon GH, Kim H, Kim MO (2015) Vitamin C neuroprotection against dose-dependent glutamate-induced neurodegeneration in the postnatal brain. Neurochem Res 40:875–884
Shah SA, Hae YL, Ray AB, Dae JY, Kim MO (2014) Novel osmotin attenuates glutamate-induced synaptic dysfunction and neurodegeneration via the JNK/PI3K/Akt pathway in postnatal rat brain. Cell Death and Disease 5:1–10
Shah SA, Khan M, Jo MH, Jo MG, Amin FU, Kim MO (2016) Melatonin stimulates the SIRT1/Nrf2 signaling pathway counteracting lipopolysaccharide (LPS)-induced oxidative stress to rescue postnatal rat brain. CNS Neuroscience & Therapeutics 1–12.
Egea J, Rosa AO, Cuadrado A, García AG, López MG (2007) Nicotinic receptor activation by epibatidine induces heme oxygenase-1 and protects chromaffin cells against oxidative stress. J Neurochem 102:1842–1852
Kasai H, Nishimura S (1984) Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res 12:2137–2145
Tiwari V, Arora V, Chopra K (2012) Attenuation of NF-kB mediated apoptotic signaling by tocotrienol ameliorates cognitive deficits in rats postnatally exposed to ethanol. Neurochem Int 61:310–320
Crews F (2006) BHT blocks NF-kB activation and ethanol-induced brain damage. Alcohol Clin Ex Res 30:1938–1949
Chao DT, Korsmeye SJ (1998) Bcl-2 family: regulators of cell death. Annu Rev Immunol 16:395–419
Berger NA (1985) Poly (ADP-ribose) in the cellular response to DNA damage. Radiat Res 101:4–15
Xiaomei L, Ming H, Christopher Q, Rogers ZS, You M (2011) Role of SIRT1-FoxO1 signaling in dietary saturated fat-dependent upregulation of liver adiponectin receptor 2 in ethanol-administered mice. Antioxid Redox Signal 15:425–435
Shen Z, Liang X, Rogers CQ, Rideout D, You M (2010) Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol 298:G364–G374
Maier SE, Miller JA, Blackwell JM, West JR (1999) Fetal alcohol exposure and temporal vulnerability: regional differences in cell loss as a function of the timing of binge-like alcohol exposure during brain development. Alcohol Clin Exp Res 23:726–734
Olney JW, Tenkova T, Dikranian K, Qin YQ, Labruyere J, Ikonomidou C (2002) Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res 133(2):115–126
Azam R, Iran G, Taghi L, Mohammad TG, Kataneh A, Mahmoud DS (2012) Role of oxidative stress in ethanol-induced neurotoxicity in the developing cerebellum. Iranian Journal of Basic Medical Sciences 15:965–974
Bergamini CM, Gambetti S, Dondi A, Cervellati C (2004) Oxygen, reactive oxygen species and tissue damage. Curr Pharm Des 10:1611–1626
Tadahiro N, Tomoya M, Yumiko N, Misty R, Shigeto Y, Hiroshi K (2011) Protective action of neurotrophic factors and estrogen against oxidative stress-mediated neurodegeneration. Journal of Toxicology Article ID 405194
Chu J, Tong M, Monte SM (2007) Chronic ethanol exposure causes mitochondrial dysfunction and oxidative stress in immature central nervous system neurons. Acta Neuropathol 113(6):659–673
Heaton MB, Paiva M, Mayer J, Miller R (2002) Ethanol-mediated generation of reactive oxygen species in developing rat cerebellum. Neurosci Lett 334:83–86
Shahid AS, Gwang HY, Kim MO (2015) Protection of the developing brain with anthocyanins against ethanol-induced oxidative stress and neurodegeneration. Mol Neurobiol 51:1278–1291
Jung M (2010) Alcohol withdrawal and brain injuries: beyond classical mechanisms. Molecules 15(7):4984–5011
Kawamori D, Kaneto H, Nakatani Y, Matsuoka TA, Matsuhisa M, Hori M, Yamasaki Y (2006) The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J Biol Chem 281:1091–1098
Martinez SC, Tanabe K, Cras-Meneur C, Abumrad NA, Bernal-Mizrachi E, Permutt MA (2008) Inhibition of Foxo1 protects pancreatic islet β-cells against fatty acid and ER-stress induced apoptosis. Diabetes 57:846–859
Longo VD, Kennedy BK (2006) Sirtuins in aging and age-related disease. Cell 126:257–268
El-Mas R (2015) Estrogen modulation of the ethanol-evoked myocardial oxidative stress and dysfunction via DAPK3/Akt/ERK activation in male rats. Toxicol Appl Pharmacol 287(3):284–292
Acknowledgement
This research was supported by the Brain Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning (2016M3C7A1904391).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The local ethical committee for animals of the Department of Biology, Division of Applied Life Sciences, Gyeongsang National University, South Korea, approved the experimental procedures.
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Khan, M., Shah, S.A. & Kim, M.O. 17β-Estradiol via SIRT1/Acetyl-p53/NF-kB Signaling Pathway Rescued Postnatal Rat Brain Against Acute Ethanol Intoxication. Mol Neurobiol 55, 3067–3078 (2018). https://doi.org/10.1007/s12035-017-0520-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0520-8