
Abstract
Emerging evidence has demonstrated the involvement of stromal cell-derived factor 1 (SDF1, also known as CXCL12)-CXCR4 signaling in a variety of pain state. However, the underlying mechanisms of SDF1-CXCR4 signaling leading to the maintenance of chronic pain states are poorly understood. In the present study, we sought to explore the role of SDF1-CXCR4 signaling in the forming of neuroplasticity by applying a model of the transition from acute to chronic pain state, named as hyperalgesic priming. Utilizing intraplantar bee venom (BV) injection, we successfully established hyperalgesic priming state and found that peripheral treating with AMD3100, a CXCR4 antagonist, or knocking down CXCR4 by intraganglionar CXCR4 small interfering RNA (siRNA) injection could prevent BV-induced primary mechanical hyperalgesia and hyperalgesic priming. Moreover, we showed that single intraplantar active SDF1 protein injection is sufficient to induce acute mechanical hyperalgesia and hyperalgesic priming through CXC4. Intraplantar coinjection of ERK inhibitor, U0126, and PI3K inhibitor, LY294002, as well as two protein translation inhibitors, temsirolimus and cordycepin, prevented the development of SDF1-induced acute mechanical hyperalgesia and hyperalgesic priming. Finally, on the models of complete Freund’s adjuvant (CFA)-induced chronic inflammatory pain and spared nerve injury (SNI)-induced chronic neuropathic pain, we observed that knock-down of CXCR4 could both prevent the development and reverse the maintenance of chronic pain state. In conclusion, our present data suggested that through regulating ERK and PI3K-AKT pathways-mediated protein translation SDF1-CXCR4 signaling mediates the transition from acute pain to chronic pain state and finally contributes to the development and maintenance of chronic pain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic pain is a common clinical symptom and produces a large burden of economic losses, which affecting more than 100 million people in the USA [1]. In low-income and middle-income countries, the prevalence rate of chronic pain is even higher, with 33 % in the general adult population, 56 % in the general elderly population, and 35 % in workers [2]. However, to date, the mechanisms underlying chronic pain remain poorly understood, limiting the identification of adequate treatments. In order to improve the management of chronic pain, International Association for the Study of Pain united with WHO and divided the chronic pain into seven classes [3]. During recent decade, emerging evidence has demonstrated that stromal cell-derived factor 1 (SDF1) and its cognate receptor CXCR4 was tightly correlated with various types of chronic pain [4]. In unilateral sciatic nerve injury (CCI)-induced peripheral neuropathic pain model and spinal cord injury-induced central neuropathic pain model, the expression level of SDF1 and CXCR4 was continuously increased [5, 6]. And intrathecal administration of AMD3100, a selective CXCR4 antagonist, or SDF1 antibody significantly alleviated spared nerve injury-induced pain through inhibiting ERK activation in the spinal cord [7]. For diabetic neuropathy, AMD3100 also efficiently reversed the neuropathic pain [8]. Additionally, Shen and colleagues demonstrated that SDF1-CXCR4 signaling contributed to the development and maintenance of bone cancer pain through mitogen-activated protein kinases (MAPKs)-mediated neuroinflammation [9, 10]. Recently, we provided compelling evidence that SDF1-CXCR4 signaling contributes to bee venom (BV)-induced acute inflammatory pain by regulating the excitability of primary nociceptive neurons through ERK-dependent Nav1.8 expression [11]. Usually, chronic pain is observed following the completely recovery from acute inflammation or injuries [3]. Based on the above general roles of SDF1-CXCR4 signaling in both acute and chronic pain, we hypothesized that SDF1-CXCR4 signaling contributed to the process of transition from acute to chronic pain state.
Hyperalgesic priming is a preclinical model which can be induced by prior inflammatory insult and is expressed as a long-lasting prolongation in the response to prostaglandin E2 (PGE2) [12–16]. Briefly, in naïve rats, intraplantar injection of PGE2 only evoked a short-lasting mechanical hyperalgesia (<4 h). However, under primed condition induced by inflammagen, the mechanical hyperalgesia induced by PGE2 is markedly prolonged, lasting >24 h. [17–19]. Nowadays, hyperalgesic priming having served as an important paradigm for probing the neuroplastic mechanism associated with the transition from acute to chronic pain. Thus, in the present study, we firstly established the hyperalgesic priming model by subcutaneous intraplantar injection of BV, which is a frequently used acute inflammatory pain model [20]. Then, we investigated the role of peripheral CXCR4 in BV-induced primary mechanical hyperalgesia and hyperalgesic priming. Moreover, we evaluated whether single intraplantar injection of active SDF1 protein is sufficient to induce mechanical hyperalgesia and hyperalgesic priming. Finally, we examined the intracellular molecular mechanisms underlying SDF1-CXCR4 signaling-induced mechanical hyperalgesia and hyperalgesic priming.
Materials and Methods
Animals
The experiments were performed on male Sprague–Dawley rats (150–250 g, purchased from the Laboratory Animal Center of Fourth Military Medical University, FMMU, Xi’an, People’s Republic of China). Animals were housed in plastic boxes under a 12 h light/dark cycle (with the lights on at 8:00 a.m. to 8:00 p.m.) at room temperature (22–26 °C). Water and food were available ad libitum. All behavioral evaluations were carried out between 9:00 and 17:00. The present experiment protocols were approved by the Institutional Animal Care and Use Committee of FMMU and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80–23) revised 1996. International Association for the Study of Pain (IASP)’s ethical guidelines for pain research in conscious animals was followed. In our present study, total number of 256 rats were used and their suffering were greatly minimized.
Mechanical Pain Sensitivity Testing
The mechanical pain sensitivity testing was performed by experimenters blinded to the experimental treatments. Mechanical stimuli were applied by using ascending graded individual von Frey monofilaments with bending forces of 0.8, 2.0, 4.0, 6.0, 8.0, 10.0, 12.0, 14.0, 16.0, 18.0, 20.0, 25.0, 30.0, 45.0, and 60 g. The rats were placed on a metal mesh floor covered with plastic box and habituated for a minimum of 1 h before the measurement of mechanical withdrawal thresholds. Von Frey filaments were applied from underneath the metal mesh floor to the plantar area of the appropriate hindpaw. Each von Frey filament was applied 10 times (once every several seconds) in order to induce the withdrawal reflex. The bending force value of the von Frey filament that caused an appropriate 50 % occurrence of paw withdrawal was expressed as the paw withdrawal mechanical threshold (PWMT, g).
Drugs
Bee venom solution (200 μg/50 μL, Floret Ltd. and its partner company New Techniques Laboratory Ltd., Tbilisi, Georgia, dissolved in 0.9 % sterile saline) was injected into the plantar surface of the left hind paw to establish acute inflammatory pain and complete Freund’s adjuvant (CFA, 40 μL) was also intraplantar injected to establish chronic inflammatory pain. For evaluating the pro-algesic effect of SDF1-CXCR4 signaling, recombinant active SDF1 protein (10, 100, or 200 ng/10 μL, Abcam, dissolved in 0.9 % sterile saline) was injected into the plantar surface of the left hind paw. To induce hyperalgesic priming, prostaglandin E2 (PGE2, 100 ng/10 μL, Tocris Bioscience, dissolved in 0.9 % sterile saline) was intraplantarly injected following complete resolution of the initial mechanical hypersensitivity (7, 14, or 21 days after BV injection, 5 days after SDF1 injection). The CXCR4 inhibitor AMD3100 (10, 100, or 200 μg/20 μl, Sigma, dissolved in 0.9 % sterile saline) was coinjected into the plantar surface of the left hind paw with BV or SDF1 to evaluate the role of CXCR4 in primary hyperalgesia and hyperalgesic priming. The ERK inhibitor U0126 (10 μg/10 μl, Sigma), the PI3-kinase inhibitor LY294002 (10 μg/10 μl, Abcam), the protein translation inhibitor cordycepin (10 μg/10 μl, Abcam), or the mTOR inhibitor temsirolimus (20 μg/10 μl, Abcam) were coinjected into the same site as SDF1 injection to evaluate the downstream signal of SDF1-CXCR4 signaling. In our present experiments, all the intraplantar administrations of drugs were performed by Hamilton microsyringe.
Dorsal Root Ganglion Microinjection
Dorsal root ganglion microinjection was performed according to the method described previously [21]. Briefly, under sodium pentobarbital anesthesia (50 mg/kg, i.p.), a midline incision was made in the lower lumbar back region, and the L4/5 DRG were exposed. By using in vivo transfection reagent as the delivery vehicle, the CXCR4 small interfering RNA (siRNA) solution (2 μl, 10 μM, sc-270577, Santa Cruz) or no-targeting control scrambled siRNA (2 μl, 10 μM, sc-37007, Santa Cruz) was injected into two sites in the L4 and L5 DRG with a glass Hamilton microsyringe. The microsyringe was remained for 5 min after each injection to prevent spread of the agent. Then, the surgical field was irrigated with 0.9 % sterile saline, and the skin incision was closed using 4.0 sutures. All rats were allowed to recover in individual cages for at least 5 days. In our present experiment, the injected rats showed no signs of paresis or other abnormalities.
Immunohistochemistry
The rats were anesthetized with 1 % pentobarbital sodium (50 mg/kg, i.p.), then perfused with physiological saline, followed by 4 % paraformaldehyde in 0.1 M PB solution. After perfusion, the L4, 5 DRGs, and sciatic nerve as well as skin were removed and postfixed in the same 4 % fixative overnight at 4 °C and cryoprotected by immersion in 30 % sucrose in 0.1 M PB at 4 °C till its sunk on the bottom of the container. Transverse frozen sections (15 μm thick) were cut on CM1900 freezing microtome (Leica, Germany). Sections were blocked with 10 % goat serum in phosphate buffered saline (PBS) for 2 h at room temperature and incubated with goat anti-CXCR4 (Abcam) at 4 °C overnight. After three washes with PBS, the sections were incubated with Cy3-conjugated secondary antibodies (Sigma) for 2–3 h at room temperature. The images were examined under a laser scan confocal fluorescent microscope (Olympus FV1000, Japan), and the immunofluorescence intensity was analyzed by Image J software.
Western Blotting
Rats were sacrificed by decapitation, and the L4, 5 DRGs ipsilateral to i.pl. injection were obtained and homogenized in a RIPA lysis buffer containing protease inhibitors (Applygen Technologies Inc., China). Protein concentrations of the lysate were determined using a BCA Protein Assay kit (Thermo Scientific, Rockford, IL, USA.). Protein samples were heated for 10 min at 95 °C with SDS-PAGE sample buffer and equal amounts of protein were then separated by 10 % separation gels. The resolved proteins were subsequently transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA) followed by the incubation with 5 % nonfat milk (Bio-rad, CA, USA) in PBS with 0.05 % Tween 20 (PBST) for at least 30 min at room temperature. Then, the membranes were incubated with mouse anti-pERK (Santa Cruz) or rabbit anti-pAKT (Cell Signaling) at 4 °C overnight. After washing three times in PBST, the membranes were incubated for 2 h at room temperature with an HRP-conjugated secondary antibody (1:2000, Bio-Rad). The membranes were visualized with enhanced chemiluminescence solution (Alpha Innotech Corp) and the signals were captured with FluorChem FC2 (Alpha Innotech Corp.). The density of specific bands was measured with a computer-assisted imaging analysis system (Bio-rad, CA, USA) and normalized to β-actin intensity.
Spared Nerve Injury
Spared nerve injury (SNI) model of neuropathic pain was carried out as described previously [22]. Briefly, under 1 % pentobarbital sodium (50 mg/kg, i.p.) anesthesia, the sciatic nerve and its three terminal branches were exposed. The common peroneal and the tibial nerves were tight-ligated twice with 5.0 silk, and a 2–4 mm of nerve stump between the two ligations was removed. Then, the muscle and skin were closed layer by layer.
Statistical Analysis
Data were analyzed using GraphPad Prism version 5 (GraphPad, San Diego, CA) and all data were expressed as mean ± SEM. Differences in changes of values of each group were tested using t tests and one-way or two-way repeated ANOVA, followed by individual post hoc comparisons (Bonferroni or Tukey test). A level of P < 0.05 was accepted as significant.
Results
Hyperalgesic Priming Persists for Weeks After Intraplantar Bee Venom Injection
In consistent with our previous report, single intraplantar injection of BV (200 μg/50 μL) in the rat induced a short-term mechanical hyperalgesia in the ipsilateral hindpaw [23]. The PWMT decreased to peak at 2 h after BV injection and then progressively increased to reach baseline value, with resolving within 96 h (Fig. 1a). However, the PWMT of contralateral hindpaw showed no significant change (data not shown). After the rats completely recovered from BV-induced mechanical hyperalgesia, the presence of hyperalgesic priming was evaluated by intraplantar injecting inflammatory mediator PGE2 (100 ng/10 μL) at the same site with previous BV injection. As shown in Fig. 1b, at 7 days after BV injection, intraplantar injection of PGE2 resulted in a prolonged mechanical hyperalgesia lasting >24 h. However, in saline injected control rats, intraplantar injection of PGE2 produced a transient (<4 h) mechanical hyperalgesia. This phenomenon indicated that intraplantar injection of BV was able to establish hyperalgesic priming which could prolong the mechanical hyperalgesia induced by PGE2. By extending the window time between BV and PGE2 injection, we found BV-induced hyperalgesic priming persisted for at least 21 days after BV injection (Fig. 1c, d).

Intraplantar BV injection-induced primary mechanical hyperalgesia and hyperalgesic priming. a Mean time courses of changes in PWMT following intraplantar injection of BV. b, d Time course effect of intraplantar PGE2 injection on PWMT at 7 days (b), 14 days (c), and 21 day (d) after BV injection. PWMT paw withdrawal mechanical threshold; arrow heads indicate the time for PGE2 injection; **P < 0.01, ***P < 0.001 BV vs. vehicle (n = 7–9, two-way ANOVA, Bonferroni post hoc)
CXCR4 Mediates the Bee Venom-Induced Primary Mechanical Hyperalgesia and Hyperalgesic Priming
We have previously demonstrated that intraplantar injection of AMD3100, a selectively CXCR4 inhibitor, could efficiently prevent the development of BV-induced primary mechanical and thermal hyperalgesia [11]. Here, we detailedly evaluated the time-dependent effects of AMD3100 on BV-induced primary mechanical hyperalgesia at 2, 5, 24, 48, 72, and 96 h after BV injection. As shown in Fig. 2a, coinjection AMD3100 (200 μg/10 μL) with BV could remarkably suppress the mechanical hyperalgesia at 2 and 5 h after BV injection compared with coinjection vehicle control group. Next, we injected PGE2 in the same site as BV injection 7 days later to explore whether peripheral CXCR4 was involved in BV-induced hyperalgesic priming. We found that coinjection AMD3100 could prevent the development of BV-induced hyperalgesic priming which was manifested as the increased PWMT value at 4 and 24 h after PGE2 injection in AMD3100-treated group compared to vehicle-treated control group (Fig. 2b). To further verify the specific role of CXCR4 in BV-induced primary mechanical hyperalgesia and hyperalgesic priming, we applied intraganglionar CXCR4 siRNA injection 7 days before intraplantar BV injection which could specifically target CXCR4. As shown in Fig. 3a, b, intraganglionar CXCR4 siRNA injection remarkably knocked down CXCR4 expression in DRG and sciatic nerve as well as ipsilateral skin comparing with scrambled siRNA injection group. And the Western blotting data also demonstrated that intraganglionar CXCR4 siRNA injection could efficiently decrease the expression level of CXCR4 in DRG cells (Fig. 3c, d). Compared with scrambled siRNA injection, intraganglionar CXCR4 siRNA injection had no significant effect on the ipsilateral basal PWMT value (Fig. 3e). However, we found selective knock-down of CXCR4 in primary afferent neurons could provide efficient inhibition on the mechanical hyperalgesia and the inhibitory effect lasted for 72 h (Fig. 3f). In rats received intraganglionar CXCR4 siRNA injection 7 days before intraplantar BV injection, PGE2-induced hyperalgesia at 7 days after intraplantar BV injection was also significantly less at 4 and 24 h compared with intraganglionar scrambled siRNA injection group (Fig. 3g). These results demonstrated that peripheral CXCR4 inhibition prevented not only BV-induced primary mechanical hyperalgesia but also the subsequent hyperalgesic priming.

Role of CXCR4 in bee venom-induced primary mechanical hyperalgesia and hyperalgesic priming. a Effect of intraplantar AMD3100 coinjection on BV-induced primary mechanical hyperalgesia. b Effect of intraplantar AMD3100 coinjection on BV-induced hyperalgesic priming. PWMT paw withdrawal mechanical threshold; arrow heads indicate the time for PGE2 injection; **P < 0.01, ***P < 0.001 BV-AMD3100 vs. BV-vehicle (n = 7 for each group, two-way ANOVA, Bonferroni post hoc)

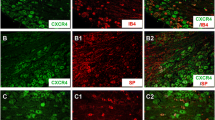
Effect of intraganglionar CXCR4 siRNA injection on the expression of CXCR4 and bee venom-induced primary mechanical hyperalgesia and hyperalgesic priming. a Representative immunofluorescence photomicrographs of CXCR4 in DRG and sciatic nerve as well as skin from scrambled siRNA injection group and CXCR4 siRNA injection group, scale bars, 250 μm. b Quantification of the mean immunofluorescent intensity of CXCR4 showing a decrease in CXCR4 expression following intraganglionar CXCR4 siRNA injection (n = 5/group, ***P < 0.001, t test). c Representative Western blot bands showing the expression of CXCR4 in lumbar DRG from scrambled siRNA injection group and CXCR4 siRNA injection group. d Quantification of the Western blot bands showing the expression of CXCR4 significantly decreased after intraganglionar CXCR4 siRNA injection (n = 4/group, **P < 0.01, t test). e Effect of intraganglionar CXCR4 siRNA injection on the basal PWMT values 7 days later (n = 20 for each group, two-way ANOVA, Bonferroni post hoc). f Effect of intraganglionar CXCR4 siRNA injection on BV-induced primary mechanical hyperalgesia (n = 7 for each group, two-way ANOVA, Bonferroni post hoc). g Effect of intraganglionar CXCR4 siRNA injection on BV-induced hyperalgesic priming (n = 7 for each group, two-way ANOVA, Bonferroni post hoc). PWMT paw withdrawal mechanical threshold; arrow heads indicate the time for PGE2 injection; *P < 0.05, **P < 0.01, ***P < 0.001 CXCR4 siRNA vs. scrambled siRNA
Intraplantar Injection of SDF1 Induces Mechanical Hyperalgesia and Hyperalgesic Priming Through CXCR4
As we know that CXCR4 is a seven transmembrane G protein-coupled receptor and which transduces cellular signals through binding of its solely endogenous ligand, SDF1. So next, we sought to determine whether SDF1 alone was sufficient to induce primary mechanical hyperalgesia and hyperalgesic priming. Compared with vehicle, single intraplantar injection with active SDF1 produced does-dependent acute mechanical hyperalgesia. The pro-algesic effect of intraplantar SDF1 injection began at 0.5 h and remained at a significant level for at least 24 h at higher doses (100 and 200 ng/10 μL) (Fig. 4a). While the lowest dose (10 ng/10 μL) of SDF1 has no significant effect on the baseline value of PWMT over the whole observation period (Fig. 4a). Regardless of the drug does, the primary mechanical hyperalgesia induced by intraplantar SDF1 injection was constricted to the ipsilateral hindpaws, but with the PWMT value being unchanged in the contralateral hindpaw (Fig. 4b). To determine the pro-algesic effect of SDF1 was CXCR4 specific, we coinjected AMD3100 with active SDF1 into the plantar surface of rat hindpaw and found that both the higher doses of AMD3100 (100 and 200 μg/10 μL) could inhibit the SDF1-induced acute mechanical hyperalgesia, while the lowest dose (10 μg/20 μl) had no significant effect (Fig. 4c). Also, the PWMT value in the contralateral side remained unchanged after intraplantar AMD3100 coinjection (Fig. 4d). Similar to the results of intraplantar AMD3100 coinjection, we found that knock-down CXCR4 in the primary afferent neurons by intraganglionar CXCR4 siRNA injection 7 days before intraplantar SDF1 injection could completely prevent SDF1-induced acute mechanical hyperalgesia compared with scrambled siRNA group, while no significant effect was observed on the contralateral baseline PWMT value (Fig. 4e, f).

Intraplantar injection of SDF1 induces mechanical hyperalgesia through CXCR4. a, b Time course recording of the pro-algesic effects of intraplantar SDF1 injection (10, 100, and 200 ng/10 μL). *P < 0.05, ***P < 0.001 SDF1 100 ng vs. vehicle; ### P < 0.001 SDF1 200 ng vs. vehicle (n = 7–9, two-way ANOVA, Bonferroni post hoc). c, d Time course recording of the anti-hyperalgesic effect of AMD3100 (10, 100, and 200 μg/10 μL) coinjection on SDF1 (200 ng/10 μL)-induced primary mechanical hyperalgesia. *P < 0.05, AMD3100 100 μg vs. vehicle; ## P < 0.01, ### P < 0.001 AMD3100 200 μg vs. vehicle (n = 7–9, two-way ANOVA, Bonferroni post hoc). e, f Time course recording of the anti-hyperalgesic effect of intraganglionar CXCR4 siRNA injection on SDF1 (200 ng/10 μL)-induced primary mechanical hyperalgesia. *P < 0.05, **P < 0.01, ***P < 0.001 CXCR4 siRNA vs. scrambled siRNA (n = 6 for each group, two-way ANOVA, Bonferroni post hoc). PWMT paw withdrawal mechanical threshold
At 5 days after intraplantar SDF1 injection when the acute mechanical hyperalgesia was completely restored, intraplantar injecting PGE2 at the same site as SDF1 injection resulted in a prolonged mechanical hyperalgesia lasted more than 24 h, while PGE2 induced hyperalgesia resolved within 4 h in vehicle-treated rats (Fig. 5a). This result indicated that, similar to intraplantar BV injection, single intraplantar SDF1 injection was also sufficient to induce hyperalgesic priming. Not only coinjection AMD3100 with SDF1 but also intraganglionar CXCR4 siRNA injection 7 days before intraplantar SDF1 injection could prevent the development of SDF1-induced hyperalgesic priming (Fig. 5b, c).

Intraplantar injection of SDF1 induces hyperalgesic priming through CXCR4. a Time course effect of intraplantar PGE2 injection on PWMT at 5 days after SDF1 (200 ng/10 μL) injection. ***P < 0.001 SDF1 vs. vehicle (n = 7 for each group, two-way ANOVA, Bonferroni post hoc). b Effect of intraplantar AMD3100 coinjection on SDF1-induced hyperalgesic priming. ***P < 0.001 AMD3100 vs. vehicle (n = 7 for each group, two-way ANOVA, Bonferroni post hoc). c Effect of intraganglionar CXCR4 siRNA injection on SDF1-induced hyperalgesic priming. ***P < 0.001 CXCR4 siRNA vs. scrambled siRNA (n = 6 for each group, two-way ANOVA, Bonferroni post hoc). PWMT paw withdrawal mechanical threshold; arrow heads indicate the time for PGE2 injection
SDF1-Induced Hyperalgesic Priming Is Prevented by Inhibiting Intracellular Second Messengers
Next, we sought to explore the intracellular downstream signaling mechanism underlying SDF1-induced acute mechanical hyperalgesia and hyperalgesic priming. Since a large number of evidence have demonstrated that ERK, a subfamily of mitogen-activated protein kinase (MAPK), and PI3K-AKT pathways are the common intracellular downstream messengers of SDF1–CXCR4 signaling [7, 24–26]. Here, by using Western blot assay, we found that intraplantar active SDF1 (200 ng/10 μL) injection could significantly activate intracellular p-ERK and p-AKT of DRG neurons and which were suppressed by AMD3100 (200 μg/10 μL) (Fig. 6a, b). Then, we coinjected U0126, a potent ERK inhibitor, or LY294002, a highly selective PI3K inhibitor with active SDF1 into the plantar surface of hindpaws and found that both U0126 and LY294002 could prevent SDF1-induced acute mechanical hyperalgesia at first 24 h after SDF1 injection (Fig. 6c, e). For SDF1-induced hyperalgesic priming, the mechanical hyperalgesia induced by intraplantar PGE2 injection at 5 day after SDF1 injection was also partially suppressed by U0126 and LY294002 at 4 and 24 h (Fig. 6d, f).

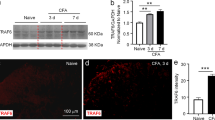
SDF1-induced mechanical hyperalgesia and hyperalgesic priming depends on intracellular second messengers. a Western blot showing intraplantar SDF1 injection activates p-ERK in DRG cells through peripheral CXCR4, *P < 0.05, (n = 3/group, one-way ANOVA, Tukey post hoc). b Western blot showing intraplantar SDF1 injection activates p-AKT in DRG cells through peripheral CXCR4, *P < 0.05, **P < 0.01, (n = 3/group, one-way ANOVA, Tukey post hoc). c Time course effect of U0126 coinjection on SDF1 (200 ng/10 μL)-induced primary mechanical hyperalgesia. **P < 0.01, ***P < 0.001 U0126 vs. vehicle (n = 7 for each group, two-way ANOVA, Bonferroni post hoc). d Effect of U0126 coinjection on SDF1 (200 ng/10 μL)-induced hyperalgesic priming. **P < 0.01, ***P < 0.001 U0126 vs. vehicle (n = 7 for each group, two-way ANOVA, Bonferroni post hoc). e Time course effect of LY294002 coinjection on SDF1 (200 ng/10 μL)-induced primary mechanical hyperalgesia. *P < 0.05, **P < 0.01 LY294002 vs. vehicle (n = 7 for each group, two-way ANOVA, Bonferroni post hoc). f Effect of LY294002 coinjection on SDF1 (200 ng/10 μL)-induced hyperalgesic priming. **P < 0.01, LY294002 vs. vehicle (n = 7 for each group, two-way ANOVA, Bonferroni post hoc). PWMT paw withdrawal mechanical threshold; arrow heads indicate the time for PGE2 injection
Local Protein Translation Is Required in the Induction of SDF1-Induced Hyperalgesic Priming
To test the hypothesis that SDF1–CXCR4 signaling through ERK and PI3K-AKT pathway lead the protein synthesis to result in acute mechanical hyperalgesia and hyperalgesic priming. We firstly coinjected temsirolimus, a selective mTOR inhibitor which has been shown to inhibit translation, in the same site as SDF1 injection and found that not only the SDF1-induced acute mechanical hyperalgesia but also the hyperalgesia priming could be blocked by temsirolimus (Fig. 7a, b). To further demonstrate our above hypothesis, we coinjected a second translation inhibitor, cordycepin, which inhibits translation by a different mechanism than temsirolimus. In contrast with the vehicle-treated group, cordycepin efficiently attenuated SDF1-induced acute mechanical hyperalgesia at 3 and 5 h post SDF1 injection (Fig. 7c). The expression of SDF1-induced hyperalgesic priming reflected as the prolongation of PGE2-induced mechanical hyperalgesia at 5 days after intraplantar SDF1 injection was also partially inhibited at 4 and 24 h post PGE2 injection (Fig. 7d).

Prevention of SDF1-induced mechanical hyperalgesia and hyperalgesic priming by local protein translation. a Time course effect of temsirolimus coinjection on SDF1 (200 ng/10 μL)-induced primary mechanical hyperalgesia. *P < 0.05, ***P < 0.001 temsirolimus vs. vehicle (n = 6 for each group, two-way ANOVA, Bonferroni post hoc). b Effect of temsirolimus coinjection on SDF1 (200 ng/10 μL)-induced hyperalgesic priming. *P < 0.05, ***P < 0.001 U0126 vs. vehicle (n = 6 for each group, two-way ANOVA, Bonferroni post hoc). c Time course effect of cordycepin coinjection on SDF1 (200 ng/10 μL)-induced primary mechanical hyperalgesia. *P < 0.05 **P < 0.01 cordycepin vs. vehicle (n = 7 for each group, two-way ANOVA, Bonferroni post hoc). d Effect of cordycepin coinjection on SDF1 (200 ng/10 μL)-induced hyperalgesic priming. **P < 0.01, ***P < 0.001 cordycepin vs. vehicle (n = 7 for each group, two-way ANOVA, Bonferroni post hoc). PWMT paw withdrawal mechanical threshold; arrow heads indicate the time for PGE2 injection
Knock-Down of CXCR4 Attenuates Chronic Inflammatory Pain and Chronic Neuropathic Pain
To determine the contribution of SDF1-CXCR4 signaling to the development of chronic pain, we used the CFA model as a chronic inflammatory pain and SNI model as a chronic neuropathic pain. As exhibited in Fig. 8a, b, we found knock-down of CXCR4 expression in primary afferent neurons by intraganglionar CXCR4 siRNA injection 7 days before significantly attenuated the chronic mechanical hyperalgesia induced by intraplantar CFA injection and SNI. The anti-hyperalgesic effects persisted over the whole observation period (Fig. 8a, b).

Knock-down of CXCR4 attenuates chronic inflammatory pain and chronic neuropathic pain. a Time course effect of intraganglionar CXCR4 siRNA injection 7 days before on CFA-induced chronic mechanical hyperalgesia. b Time course effect of intraganglionar CXCR4 siRNA injection 7 days before on SNI-induced chronic mechanical hyperalgesia. c Time course effect of intraganglionar CXCR4 siRNA injection 3 days post on CFA-induced chronic mechanical hyperalgesia. d Time course effect of intraganglionar CXCR4 siRNA injection 7 days post on SNI-induced chronic mechanical hyperalgesia. PWMT paw withdrawal mechanical threshold; arrow heads indicate the time for CXCR4 siRNA injection. *P < 0.05, **P < 0.01, ***P < 0.001 CXCR4 siRNA vs. scrambled siRNA (n = 6 or 7 for each group, two-way ANOVA, Bonferroni post hoc)
To further examine the role of SDF1-CXCR4 signaling in the maintenance of chronic pain, we intraganglionar injected CXCR4 siRNA at 3 days post CFA and 7 days post SNI when the mechanical hyperalgesia were stable. The effects of post intraganglionar CXCR4 siRNA injection were evaluated at 3, 5, 7, and 14 days after intraganglionar injection. As shown in Fig. 8c, comparing with scrambled siRNA group, postinjection of CXCR4 siRNA significantly reduced CFA-induced mechanical hyperalgesia at 8, 10, and 17 days post CFA (also 5, 7, and 14 days post CXCR4 siRNA injection). Similarly, on 12, 14, and 21 days post SNI (also 5, 7, and 14 days post CXCR4 siRNA), intraganglionar CXCR4 siRNA injection could persistently reverse the established mechanical hyperalgesia (Fig. 8d).
Discussion
In the present study, we demonstrated that (1) peripheral CXCR4 mediated intraplantar BV injection-induced primary mechanical hyperalgesia and hyperalgesic priming; (2) single intraplantar active SDF1 protein injection is sufficient to induce persistent mechanical hyperalgesia and hyperalgesic priming through CXCR4; (3) intracellular ERK and PI3K-AKT signaling as well as protein translation are required for SDF1-induced primary mechanical hyperalgesia and hyperalgesic priming; and (4) CXCR4 in primary afferent neurons are required for the development and maintenance of chronic inflammatory pain and chronic neuropathic pain. Taken all these together, it is concluded that by regulating the protein synthesis through intracellular ERK and PI3K-AKT signaling pathways SDF1-CXCR4 signaling mediates the transition from acute pain to chronic pain state and finally contributes to the development and maintenance of chronic pain.
SDF1-CXCR4 Signaling Mediates Primary Mechanical Hyperalgesia
In consistent with our previous study, we here demonstrated that blocking peripheral CXCR4 with AMD3100 could remarkably suppress intraplantar BV-induced primary mechanical hyperalgesia [11]. However, the analgesic effect time window of AMD3100 is limited just lasting about 5 h. To address this problem, we applied intraganglionar specific siRNA injection to targeted knocking down CXCR4 in the primary afferent neurons. It is worthy to note that BV-induced acute primary mechanical hyperalgesia was efficiently inhibited over the whole observed time course. Moreover, for CFA-induced chronic inflammatory pain and SNI-induced chronic neuropathic pain, pre-knock-down of CXCR4 persistently prevents the development of mechanical hyperalgesia. More importantly, this novel CXCR4 siRNA treatment could also reverse the established chronic inflammatory pain and chronic neuropathic pain. This therapeutic role of CXCR4 is clinically significant because most of patients come to seek medical treatment with the existing chronic pain like the arthritis pain and sciatica neuralgia [27, 28]. Our results were supported by several other findings that SDF1 and CXCR4 were both increased in the DRG and spinal cord following spared nerve injury in rats and consecutive AMD3100 administration could prevent and reverse the abnormal pain [6]. Since CXCR4 is widely expressed in the body, including nerve system, blood system as well as immune system and participates in a wide physiological processes [29–32]. This severely lowered the translational values of systemic treatments aiming at blocking CXCR4 for analgesia because it is unlikely to completely avoid the unpredictable side effects. The present approach by intraganglionar specific CXCR4 siRNA injection to treat pain may be much more relevant and valid. Another benefit of this approach is that only the pathological pain state is targeted, while the normal beneficial physiological pain is not affected as shown by our data that targeting knock-down of CXCR4 in the DRG of naïve rats had no effects on the basal PWMT.
To date, quite a few findings have demonstrated the involvement of SDF1-CXC4 signaling in pain modulation [4]. In neuropathic pain induced by sciatic nerve injury and spinal cord injury, the expression of SDF1-CXC4 signaling extremely up-regulated in DRG or spinal cord [5–7]. Similarly, the increased expression of SDF1 and CXCR4 was also reported in diabetic neuropathy, opioid-induced hyperalgesia, and bone cancer pain [8–10, 33]. However, all of these studies focused on the evaluation of the anti-hyperalgesic effects of CXCR4 inhibitor, AMD3100, which offering an unclear interpretation of the role of SDF1-CXCR4 signaling in the pain. To the best of our knowledge, no study has detailedly clarified the pro-algesic mechanisms of SDF1-CXCR4 in vivo. In line with previous finding, our present experiments demonstrated that single intraplantar SDF1 injection was sufficient to evoke acute primary mechanical hyperalgesia [34]. The pro-algesic effect of SDF1 was mediated by peripheral CXCR4 as both intraplantar AMD3100 and intraganglionar CXCR4 siRNA could completely block the mechanical hyperalgesia. Likewise, we are the first to provide direct evidence attesting that intraplantar SDF1 injection evoked both ERK and PI3K-AKT signaling activation in primary afferent neurons through peripheral CXCR4. Indeed, emerging evidence has indicated that ERK and PI3K-AKT signaling in primary afferent neurons played an important role in the pain [35]. Our previous work demonstrates that intraplantar BV injection-induced persistent pain and mechanical and thermal hyperalgesia could be suppressed by intraplantar or intrathecal U0126, a selective ERK inhibitor [11, 36]. In oxaliplatin-induced neuropathic pain, the percentage of Akt2 positive primary afferent neurons significantly increased and celecoxib attenuated the cold mechanical hypersensitivity via inhibiting PI3K-Akt2 pathway [37]. Here, we confirmed these results finding that SDF1-induced primary medical hyperalgesia was ERK and PI3K-AKT signaling mediated which could be prevented by coinjection with U0126 or selective PI3K inhibitor, LY294002. As intracellular protein kinases, activated ERK and PI3K-AKT usually contributes to pain through direct phosphorylation of ion channels or through changes in gene expression via translation regulation [38]. By utilizing two protein translation inhibitors, temsirolimus which acts on the initiation of protein biosynthesis by blocking mTOR and cordycepin, a derivative of the nucleoside adenosine which inhibit messenger RNA (mRNA) polyadenylation and consequently preventing the initiation of protein translation [39], we found that protein synthesis also contributed to SDF1-induced mechanical hyperalgesia. Thus, it is conceivable that SDF1-CXCR4 signaling promotes mechanical hyperalgesia via intracellular ERK and PI3K-AKT pathways, leading to peripheral protein translation. Actually, our previous study has shown that peripheral CXCR4 contributed to BV-induced inflammatory pain through ERK-dependent Nav1.8 up-regulation [11]. Although it is now widely accepted that SDF1-CXCR4 signaling participated in both acute and chronic pain, knowledge about the different underlying mechanisms remains limited. Furthermore, some clinical chronic pain can often occur following episodes of acute inflammation and injury [40, 41]. Thus, an important unanswered question arises whether SDF1-CXCR4 signaling participates in this transition process or not.
SDF1-CXCR4 Signaling Participates in Hyperalgesic Priming
Hyperalgesic priming models are a compelling preclinical model of the transition to chronic pain state, allowing to probe the underlying neuroplastic mechanisms. In the present experiments, we demonstrate that, similar to the effect of intraplantar carrageenan injection, the intraplantar bee venom injection also produced a similar plastic changes in primary afferent neurons [13]. Following BV injection, the mechanical hyperalgesia induced by intraplantar PGE2 injection was strikingly prolonged, and this plastic phenomenon was maintained at least for 21 days, suggesting the hyperalgesic priming state was established and maintained. In the last decade, several advances have been made on the researches of hyperalgesic priming. By applying different intraplantar stimuli, such as MCP-1, opioid receptor agonist, and IL-6, various intracellular messengers and membrane receptors have been reported in the development and maintenance of hyperalgesic priming [42–45]. Here, the BV-induced priming model we presented is novel which mimics the process of nature bee sting vividly. Moreover, intraplantar BV injection is a comprehensive inflammatory injury recruiting various membrane receptors and pro-inflammatory mediators as well as second messengers, including P2X receptor, ion channels, MAPKs, PKC, PKA, and cyclooxygenases, which would be helpful to explore and compare the different roles of different molecules in the development of hyperalgesic priming [20]. Importantly, our present study demonstrated the pivotal role of peripheral CXCR4 in pain neuroplasticity for the first time by using BV-induced hyperalgesic priming model. Either blocking or knocking down CXCR4 in the primary afferent neurons could prominently prevent the prolongation of PGE2-induced mechanical hyperalgesia in BV-primed rats. Several in vitro experiments suggested that SDF1-CXCR4 signaling plays an important role in neuronal activity and plasticity in both peripheral and central nerve systems. Hyperexcitability of medium- and small-sized primary nociceptor neurons induced by intraplantar BV injection could be inhibited by CXCR4 antagonist, AMD3100 [11]. Electrophysiological recording revealed that SDF1 increased the action potential frequency and switched the firing pattern from a tonic to a burst firing mode in dopaminergic neurons [46]. Also, in cerebellar slices, SDF1 application increased intracellular Ca2+ level and spontaneous synaptic activity [47]. Under the condition of ischemic brain injury, SDF1 was enriched up for 4 weeks and enhanced GABAergic inputs to the new neurons through CXCR4 [48]. Structurally, SDF1-CXCR4 signaling even regulated axonal elongation and branching in hippocampal neurons [49]. We complemented all these findings, providing strong in vivo evidence that single intraplantar SDF1 injection is sufficient to induce hyperalgesic priming state through peripheral CXCR4. Furthermore, this SDF1-induced hyperalgesic priming is ERK and PI3K-AKT signaling dependent as both U0126 and LY294002 prevented the prolongation of PGE2-induced mechanical hyperalgesia. Given the continuous turnover of most cellular proteins, it is difficult to conceive that simple intracellular second messengers activation were sufficient to explain the maintenance of the plasticity associated with the primed state, the downstream protein translation of these kinases signaling must be amplified [39]. In intraplantar IL-6-induced primed state, Melemedjian and colleagues have demonstrated that CREB was nascently synthesized and transported to the DRG through retrograde axonal trafficking which mediated the BDNF expression and eventually promoted the hyperalgesic priming [50]. In the present study, two protein translation inhibitors, temsirolimus and cordycepin, preventing the development of hyperalgesic priming, also strongly suggested that the local protein synthesis plays an important role in SDF1-induced priming. Our recent data demonstrated that SDF1-CXCR4 signaling contributed to the acute inflammatory pain by regulating the expression of Nav1.8 protein in the DRG soma [11]. Actually, in the axons of sciatic nerve, Nav1.8 mRNA was also identified and which partially contributed the peripheral increases in Nav1.8 protein and eventually induced aberrant nociception after nerve injury [51]. Whether the up-regulated Nav1.8 was involved in the hyperalgesic priming state remains to be determined in the future. Moreover, it also remains to determine whether the present elucidated mechanisms of hyperalgesic priming have broad application in clinical features of chronic pain. At least, we here provided powerful preclinical evidence indicating that targeting SDF1-CXCR4 signaling could help prevent or treat chronic inflammatory or neuropathic pain.
In conclusion, our present data demonstrated a completely novel role for SDF1-CXCR4 signaling in regulating a pathological state of neuroplasticity. Through intracellular ERK and PI3K-AKT signaling mediated protein translation, SDF1-CXCR4 contribute to the transition from acute to chronic pain states which shed light on the development of future pain therapeutics.
References
Gaskin DJ, Richard P (2012) The economic costs of pain in the United States. J Pain 13:715–724
Jackson T, Thomas S, Stabile V, Han X, Shotwell M, McQueen K (2015) Prevalence of chronic pain in low-income and middle-income countries: a systematic review and meta-analysis. Lancet 385:S10
Treede RD, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R et al (2015) A classification of chronic pain for ICD-11. Pain 156:1003–1007
Luo X, Wang X, Xia Z, Chung SK, Cheung CW (2016) CXCL12/CXCR4 axis: an emerging neuromodulator in pathological pain. Rev Neurosci 27:83–92
Knerlich-Lukoschus F, von der Ropp-Brenner B, Lucius R, Mehdorn HM, Held-Feindt J (2011) Spatiotemporal CCR1, CCL3(MIP-1alpha), CXCR4, CXCL12(SDF-1alpha) expression patterns in a rat spinal cord injury model of posttraumatic neuropathic pain. J Neurosurg Spine 14:583–597
Dubovy P, Klusakova I, Svizenska I, Brazda V (2010) Spatio-temporal changes of SDF1 and its CXCR4 receptor in the dorsal root ganglia following unilateral sciatic nerve injury as a model of neuropathic pain. Histochem Cell Biol 133:323–337
Bai L, Wang X, Li Z, Kong C, Zhao Y, Qian JL, Kan Q, Zhang W et al (2016) Upregulation of chemokine CXCL12 in the dorsal root ganglia and spinal cord contributes to the development and maintenance of neuropathic pain following spared nerve injury in rats. Neurosci Bull 32:27–40
Menichella DM, Abdelhak B, Ren D, Shum A, Frietag C, Miller RJ (2014) CXCR4 chemokine receptor signaling mediates pain in diabetic neuropathy. Mol Pain 10:42
Shen W, Hu XM, Liu YN, Han Y, Chen LP, Wang CC, Song C (2014) CXCL12 in astrocytes contributes to bone cancer pain through CXCR4-mediated neuronal sensitization and glial activation in rat spinal cord. J Neuroinflammation 11:75
Hu XM, Liu YN, Zhang HL, Cao SB, Zhang T, Chen LP, Shen W (2015) CXCL12/CXCR4 chemokine signaling in spinal glia induces pain hypersensitivity through MAPKs-mediated neuroinflammation in bone cancer rats. J Neurochem 132:452–463
Yang F, Sun W, Yang Y, Wang Y, Li CL, Fu H, Wang XL, Yang F et al (2015) SDF1-CXCR4 signaling contributes to persistent pain and hypersensitivity via regulating excitability of primary nociceptive neurons: involvement of ERK-dependent Nav1.8 up-regulation. J Neuroinflammation 12:219
Asiedu MN, Tillu DV, Melemedjian OK, Shy A, Sanoja R, Bodell B, Ghosh S, Porreca F et al (2011) Spinal protein kinase M zeta underlies the maintenance mechanism of persistent nociceptive sensitization. J Neurosci 31:6646–6653
Wang H, Heijnen CJ, van Velthoven CT, Willemen HL, Ishikawa Y, Zhang X, Sood AK, Vroon A et al (2013) Balancing GRK2 and EPAC1 levels prevents and relieves chronic pain. J Clin Invest 123:5023–5034
Ferrari LF, Bogen O, Reichling DB, Levine JD (2015) Accounting for the delay in the transition from acute to chronic pain: axonal and nuclear mechanisms. J Neurosci 35:495–507
Araldi D, Ferrari LF, Levine JD (2015) Repeated Mu-opioid exposure induces a novel form of the hyperalgesic priming model for transition to chronic pain. J Neurosci 35:12502–12517
Reichling DB, Levine JD (2009) Critical role of nociceptor plasticity in chronic pain. Trends Neurosci 32:611–618
Aley KO, Messing RO, Mochly-Rosen D, Levine JD (2000) Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci 20:4680–4685
Melemedjian OK, Tillu DV, Asiedu MN, Mandell EK, Moy JK, Blute VM, Taylor CJ, Ghosh S et al (2013) BDNF regulates atypical PKC at spinal synapses to initiate and maintain a centralized chronic pain state. Mol Pain 9:12
Ferrari LF, Araldi D, Levine JD (2015) Distinct terminal and cell body mechanisms in the nociceptor mediate hyperalgesic priming. J Neurosci 35:6107–6116
Chen J, Lariviere WR (2010) The nociceptive and anti-nociceptive effects of bee venom injection and therapy: a double-edged sword. Prog Neurobiol 92:151–183
Li Z, Gu X, Sun L, Wu S, Liang L, Cao J, Lutz BM, Bekker A et al (2015) Dorsal root ganglion myeloid zinc finger protein 1 contributes to neuropathic pain after peripheral nerve trauma. Pain 156:711–721
Decosterd I, Woolf CJ (2000) Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87:149–158
Chen J, Luo C, Li H, Chen H (1999) Primary hyperalgesia to mechanical and heat stimuli following subcutaneous bee venom injection into the plantar surface of hindpaw in the conscious rat: a comparative study with the formalin test. Pain 83:67–76
Luo Y, Lathia J, Mughal M, Mattson MP (2008) SDF1alpha/CXCR4 signaling, via ERKs and the transcription factor Egr1, induces expression of a 67-kDa form of glutamic acid decarboxylase in embryonic hippocampal neurons. J Biol Chem 283:24789–24800
Badr G, Sayed A, Abdel-Maksoud MA, Mohamed AO, El-Amir A, Abdel-Ghaffar FA, Al-Quraishy S, Mahmoud MH (2015) Infection of female BWF1 lupus mice with malaria parasite attenuates B cell autoreactivity by modulating the CXCL12/CXCR4 axis and its downstream signals PI3K/AKT, NFkappaB and ERK. Plos One 10, e125340
Yang P, Wang G, Huo H, Li Q, Zhao Y, Liu Y (2015) SDF-1/CXCR4 signaling up-regulates survivin to regulate human sacral chondrosarcoma cell cycle and epithelial-mesenchymal transition via ERK and PI3K/AKT pathway. Med Oncol 32:377
Gilron I, Jensen TS, Dickenson AH (2013) Combination pharmacotherapy for management of chronic pain: from bench to bedside. Lancet Neurol 12:1084–1095
Dieppe PA, Lohmander LS (2005) Pathogenesis and management of pain in osteoarthritis. Lancet 365:965–973
Tiveron MC, Cremer H (2008) CXCL12/CXCR4 signalling in neuronal cell migration. Curr Opin Neurobiol 18:237–244
Guyon A (2014) CXCL12 chemokine and its receptors as major players in the interactions between immune and nervous systems. Front Cell Neurosci 8:65
Chatterjee M, Rath D, Gawaz M (2015) Role of chemokine receptors CXCR4 and CXCR7 for platelet function. Biochem Soc Trans 43:720–726
Nagasawa T (2015) CXCL12/SDF-1 and CXCR4. Front Immunol 6:301
Wilson NM, Jung H, Ripsch MS, Miller RJ, White FA (2011) CXCR4 signaling mediates morphine-induced tactile hyperalgesia. Brain Behav Immun 25:565–573
Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ (2001) Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J Neurosci 21:5027–5035
Ma W, Quirion R (2005) The ERK/MAPK pathway, as a target for the treatment of neuropathic pain. Expert Opin Ther Targets 9:699–713
Fang D, Kong LY, Cai J, Li S, Liu XD, Han JS, Xing GG (2015) Interleukin-6-mediated functional upregulation of TRPV1 receptors in dorsal root ganglion neurons through the activation of JAK/PI3K signaling pathway: roles in the development of bone cancer pain in a rat model. Pain 156:1124–1144
Jiang SP, Zhang ZD, Kang LM, Wang QH, Zhang L, Chen HP (2016) Celecoxib reverts oxaliplatin-induced neuropathic pain through inhibiting PI3K/Akt2 pathway in the mouse dorsal root ganglion. Exp Neurol 275:11–16
Migliaccio N, Sanges C, Ruggiero I, Martucci NM, Rippa E, Arcari P, Lamberti A (2013) Raf kinases in signal transduction and interaction with translation machinery. Biomol Concepts 4:391–399
Ferrari LF, Bogen O, Chu C, Levine JD (2013) Peripheral administration of translation inhibitors reverses increased hyperalgesia in a model of chronic pain in the rat. J Pain 14:731–738
Kim MH, Nahm FS, Kim TK, Chang MJ, Do SH (2014) Comparison of postoperative pain in the first and second knee in staged bilateral total knee arthroplasty: clinical evidence of enhanced pain sensitivity after surgical injury. Pain 155:22–27
Hutchinson MR, Buijs M, Tuke J, Kwok YH, Gentgall M, Williams D, Rolan P (2013) Low-dose endotoxin potentiates capsaicin-induced pain in man: evidence for a pain neuroimmune connection. Brain Behav Immun 30:3–11
Ferrari LF, Bogen O, Levine JD (2013) Role of nociceptor alphaCaMKII in transition from acute to chronic pain (hyperalgesic priming) in male and female rats. J Neurosci 33:11002–11011
Alvarez P, Green PG, Levine JD (2014) Role for monocyte chemoattractant protein-1 in the induction of chronic muscle pain in the rat. Pain 155:1161–1167
Kim JY, Tillu DV, Quinn TL, Mejia GL, Shy A, Asiedu MN, Murad E, Schumann AP et al (2015) Spinal dopaminergic projections control the transition to pathological pain plasticity via a D1/D5-mediated mechanism. J Neurosci 35:6307–6317
Araldi D, Ferrari LF, Levine JD (2015) Adenosine-A1 receptor agonist induced hyperalgesic priming type II. Pain 157(3):698–709
Skrzydelski D, Guyon A, Dauge V, Rovere C, Apartis E, Kitabgi P, Nahon JL, Rostene W et al (2007) The chemokine stromal cell-derived factor-1/CXCL12 activates the nigrostriatal dopamine system. J Neurochem 102:1175–1183
Limatola C, Giovannelli A, Maggi L, Ragozzino D, Castellani L, Ciotti MT, Vacca F, Mercanti D et al (2000) SDF-1alpha-mediated modulation of synaptic transmission in rat cerebellum. Eur J Neurosci 12:2497–2504
Ardelt AA, Bhattacharyya BJ, Belmadani A, Ren D, Miller RJ (2013) Stromal derived growth factor-1 (CXCL12) modulates synaptic transmission to immature neurons during post-ischemic cerebral repair. Exp Neurol 248:246–253
Pujol F, Kitabgi P, Boudin H (2005) The chemokine SDF-1 differentially regulates axonal elongation and branching in hippocampal neurons. J Cell Sci 118:1071–1080
Melemedjian OK, Tillu DV, Moy JK, Asiedu MN, Mandell EK, Ghosh S, Dussor G, Price TJ (2014) Local translation and retrograde axonal transport of CREB regulates IL-6-induced nociceptive plasticity. Mol Pain 10:45
Thakor DK, Lin A, Matsuka Y, Meyer EM, Ruangsri S, Nishimura I, Spigelman I (2009) Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painful neuropathy. Mol Pain 5:14
Acknowledgments
This work was supported by National Basic Research Development Program of China (2013CB835100), the National Natural Science Foundation of China (81171049, 81571072, 31300919, 31400948), and the National Key Technology R&D Program (2013BAI04B04).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Fei Yang, Wei Sun and Wen-Jun Luo contributed equally to this work.
Rights and permissions
About this article

Cite this article
Yang, F., Sun, W., Luo, WJ. et al. SDF1-CXCR4 Signaling Contributes to the Transition from Acute to Chronic Pain State. Mol Neurobiol 54, 2763–2775 (2017). https://doi.org/10.1007/s12035-016-9875-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-9875-5