
Abstract
Isocitrate dehydrogenase (IDH)1 mutation is one of the most important genetic aberrations in glioma. Even several genetic events have refined its prognostic value, the genome-wide expression alteration has not been systematically profiled. In this work, RNA-seq expression data from 310 patients in the Chinese Glioma Genome Atlas database were included as training set, while another 297 patients with microarray data were used as internal validation set. An independent cohort of GSE16011 (n = 205) constituted an external validation set. Approximately one fifth of the genes were differentially expressed in LGG according to IDH1 mutation status, yielding distinct gene expression profiles. A six-gene risk signature was established for IDH1-mutant LGG to distinguish low- from high-risk cases, which had distinct prognoses. The six-gene signature was an independent prognostic factor for IDH1-mutant LGG and had superior predictive value as compared to traditional clinicopathologic factors. Moreover, we depicted the differential expression pattern in GBM attributing to various IDH1 status, which was similar to that of LGG. It suggested that the effect of IDH1 mutation is conserved across histological classifications. The six-gene signature had equal prognostic value for IDH1-mutant GBM. By combining glioma grade, IDH1 status, and the six-gene signature, all glioma patients could be classified into six subgroups. These six subgroups could be further summarized into three sets with distinct prognosis. Taken together, a gene expression profile associated with IDH1 status was identified in LGG and GBM; a risk signature based on six genes was developed with equal prognostic value for IDH1-mutant LGG and GBM. When combined with clinicopathologic factors, the six-gene signature is a tool that enables precise risk stratification and can improve clinical management.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mutation in isocitrate dehydrogenase (IDH)1 is a stable marker for better prognosis in both lower-grade glioma (LGG) and glioblastoma multiforme (GBM), which have incidences of 75 and 12 %, respectively [1]. As the earliest detectable genetic alteration in gliomagenesis, IDH1 heterozygous missense mutations in codon 132 cause an arginine-to-histidine substitution in 80–90 % of cases (R132H) [2] that leads to a distinct metabolism and hypermethylation phenotype in gliomas [3–6]. Recently, other genetic markers have been used along with the IDH1 mutation to improve the prediction of prognosis, including 1p/19q codeletion and mutations in tumor protein (TP)53, ATP-dependent X-linked helicase (ATRX), and telomerase reverse transcriptase (TERT) promoters [7, 8]. However, there have been few studies investigating genome-wide differences associated with IDH1 status in LGG and GBM. In the present study, whole-genome expression data and biological annotations were compared as a function of IDH1 mutation in LGG and GBM. A six-gene-based risk signature was identified as an independent prognostic factor for patients with IDH1-mutant LGG that was also applicable to IDH1-mutant GBM, underscoring its stability across histological classifications. Combining the six-gene signature with glioma grade and IDH1 status classified patients into subgroups with distinct prognoses, highlighting its potential application in glioma management.
Methods
Patient Samples
Two different cohorts from the Chinese Glioma Genome Atlas (CGGA) database were included in our study: the RNA-seq (n = 310) and messenger RNA (mRNA) microarray (n = 297) cohorts were used as training and internal validation sets, respectively. There are 82 overlapped cases between these two sets. All glioma tissues were diagnosed and independently confirmed histologically by two neuropathologists based on the 2007 World Health Organization classification guidelines [9]. Tumor tissue samples were obtained from patients with newly diagnosed glioma who were treated by the CGGA group. Only samples with 80 % tumor cells were selected for IDH1 mutation status testing and whole-genome expression profiling (details are included in the Supplementary Methods). Overall survival (OS) was calculated from the date of diagnosis until death or the end of follow-up, while progression-free survival (PFS) was defined as the time between diagnosis and the first unequivocal clinical or radiological sign of disease progression. The study protocol was approved by the ethics committees of participating hospitals, and all patients provided written, informed consent. A third glioma cohort (n = 205) with mRNA microarray data from the GSE16011 dataset (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE16011) was used for external validation. The clinicopathologic features of the three cohorts are shown in Table 1 and S1.
Gene Ontology (GO) Analysis and Gene Set Enrichment Analysis (GSEA)
GO analysis was performed using DAVID (http://david.abcc.ncifcrf.gov/home.jsp) as a functional annotation tool [10, 11]. GSEA (http://www.broadinstitute.org/gsea/index.jsp) was performed to determine whether the identified sets of genes showed statistically significant differences between the two biological states (i.e., IDH1 mutant vs. wild type) [11, 12].
Statistical Analysis
Gene expression in patients with RNA-seq data was calculated using the reads per kilobase transcriptome per million reads method [13, 14]. We then filtered genes based on two criteria: (i) >20 % of samples showed a change ≥1.5-fold over the median; and (ii) <10 % of expression data were lacking. A total of 13,181 genes were used for the analysis. Genes differentially expressed between gliomas with wild-type and mutant IDH1 were identified by significance analysis of microarrays (SAM) with a false discovery rate <0.01 and 1000 permutations. A univariate Cox regression analysis was used to assess the prognostic value of each gene in IDH1-mutant LGG samples. A risk signature was formulated according to a linear combination of gene expression levels that was weighted with regression coefficients (β) from the univariate Cox regression analyses [15, 16]. The risk score for each patient was calculated as follows:
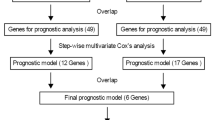
A total of 13 risk signatures were identified based on the top three to 15 genes with significant prognostic value and were evaluated by univariate Cox analysis, which revealed a six-gene risk signature with the greatest prognostic value for IDH1-mutant LGG patients. We separated gliomas into high- and low-risk groups based on the median risk score in corresponding analysis. Cases with higher risk score were classified into high-risk group and cases with lower risk score were classified into low-risk group. Kaplan-Meier survival curves and the log rank test were employed to assess the prognostic significance. Multivariate Cox regression analyses were performed to identify independent prognostic factors. Statistical significance was defined as a two-tailed P value < 0.05.
Results
Whole-Genome Differences Between Wild-Type and Mutant IDH1 LGG
A total of 172 samples were diagnosed as LGG in the CGGA RNA-seq cohort, including 123 samples that were mutant and 49 that were wild type for IDH1. SAM analysis was carried out to compare the expression levels of 13,181 genes in IDH1-mutant vs. IDH1-wild-type cases; 2679 genes were found to be differentially expressed, including 1264 that were upregulated and 1415 that were downregulated in IDH1-mutant LGG (Fig. 1a).

LGG expression profiles and identification of a six-gene signature for IDH1-mutant patients. a Expression patterns of genes differentially expressed in LGG with or without IDH1 mutation. Biological processes associated with upregulated (b) and downregulated (c) genes differentially expressed in IDH1-mutant LGG. Based on median risk score, the six-gene risk signature divided patients with IDH1-mutant LGG in the training (d), internal validation (e), and external validation (f) datasets into two groups with distinct prognoses
A GO analysis was carried out to assess differences in biological processes. Genes that were up- and downregulated in IDH1-mutant LGG were functionally distinct; the former were mainly involved in neurogenesis (Fig. 1b and Supplementary Data 1), with the top three biological processes being neuron differentiation (P = 3.45E − 12), neuron development (P = 3.38E − 10), and neuron projection development (P = 5.14E − 08). On the other hand, downregulated genes were closely associated with the immune response (Fig. 1c and Supplementary Data 2), with the most significant biological processes being immune response (P = 1.23E − 20), response to wounding (P = 1.41E − 15), and defense response (P = 3.68E − 14). A GSEA to validate the variation in immune profiles between LGG cases differing according to IDH1 status showed that the immune-relevant phenotype was enriched in wild-type IDH1 patients (Fig. S1).
Identification of a Six-Gene Risk Signature for IDH1-Mutant LGG
We evaluated the prognostic value of the genes that were differentially expressed in IDH1-mutant LGG by univariate Cox analysis. A correlation was observed between expression level and OS, with 1245 genes showing prognostic significance for IDH1-mutant LGG (Supplementary Data 3). Risky genes (n = 616) were defined as having a hazard ratio (HR) > 1 for death, whereas protective genes (n = 629) had a HR < 1. A GO analysis of risky genes indicated that immune-relevant processes were closely associated with IDH1-mutant LGG prognosis (Table S2).
A risk signature was developed based on genes with the most highly significant prognostic value; 13 risk signatures were formed based on the top three to fifteen genes. A univariate Cox analysis showed that a risk signature based on the top six genes had the greatest prognostic value for IDH1-mutant LGG (Table S3). We therefore selected the six-gene signature—which included cell division cycle (CDC)20, Wiskott-Aldrich syndrome protein family (WASF)3, deleted in breast cancer (DBC)1, engrailed (EN)2, vimentin (VIM), and carboxypeptidase (CP)E—for subsequent analyses. CDC20, EN2, and VIM were risky genes while WASF3, DBC1, and CPE were protective.
IDH1-mutant LGG cases were divided into high- and low-risk groups according to the median six-gene risk score. Survival curves generated for the two risk groups and IDH1 wild-type cases revealed that low-risk patients had longer OS (Fig. 1d) and PFS (Fig. S2) than the other groups. Despite the presence of mutant IDH1, high-risk patients had no survival advantage over those with wild-type IDH1. GSEA patterns indicated a more robust immune response in high-risk than in low-risk patients, confirming the findings from the GO analysis (Fig. S3).
Validation of the Six-Gene LGG Risk Signature
To determine whether the method used to measure gene expression affected patient stratification, the risk score was calculated for each case in the CGGA microarray cohort using the formula described above. IDH1-mutant LGG patients were classified into high- and low-risk groups according to their median risk score. As expected, low-risk patients had the best prognosis, whereas survival was similar in high-risk and wild-type IDH1 patients (Fig. 1e). To determine whether the six-gene risk signature had prognostic value for different populations, we included a cohort from the Netherlands as an external validation set and calculated the risk score. The IDH1-mutant LGG patients were stratified according to their median risk score. A Kaplan-Meier analysis indicated that OS was reduced for high-risk patients to a level comparable to that of wild-type IDH1 patients (Fig. 1f). Therefore, the six-gene signature could reliably identify a group of high-risk LGG patients with IDH1 mutation with a survival time that was similar to that of wild-type IDH1 cases. In addition, gene expression was consistently correlated with risk score, with high-risk patients expressing higher levels of risky genes (CDC20, EN2, and VIM) and low-risk patients expressing higher levels of protective genes (WASF3, DBC1, and CPE).
Six-Gene Signature Is an Independent Prognostic Factor for IDH1-Mutant LGG
Patients with IDH1-mutant LGG were stratified based on several clinicopathologic factors, including age, gender, glioma grade, and histology. The survival curves in all sub-cohorts showed that OS and PFS were shorter in high-risk than in low-risk patients (Fig. 2a–i and S4a–i), suggesting that the six-gene classifier accurately identifies patients with poor prognosis irrespective of clinicopathologic risk factors.

Prognostic significance and predictive accuracy of the six-gene signature for IDH1-mutant LGG. a–i The six-gene signature retained its prognostic significance in different cohorts stratified by age, gender, glioma grade, and histology. j, k Comparison of between the six-gene signature and traditional risk factors such as age, glioma grade, and histology in terms of sensitivity and specificity for predicting 3- and 5-year survival
For IDH1-mutant LGG cases in the training set, uni- and multivariate Cox regression analyses suggested that the six-gene signature was an independent prognostic factor for OS and PFS after adjusting for age, gender, glioma grade, and histology (OS: HR = 1.3315, P < 0.0001; PFS: HR = 1.3248, P < 0.0001) (Table 2). Moreover, the six-gene signature retained its power for independently predicting prognosis after Cox regression analyses in internal and external validation sets (Table S4), confirming the reliability of the six-gene signature as categorical and continuous variable.
Prognostic Validity of the Six-Gene Signature for IDH1-Mutant LGG
We set 3 and 5 years as OS cutoffs for patients with IDH1-mutant LGG. We compared the six-gene signature and traditional prognostic factors (age, glioma grade, and histology) in terms of sensitivity and specificity of survival prediction by analyzing the receiving operator characteristic (ROC) curve. The six-gene signature showed significant prognostic validity, with areas under the ROC curves of 0.938 and 0.956 for 3- and 5-year survival, respectively, which were higher than for the traditional factors (Fig. 2j, k), underscoring the superior predictive value of the six-gene signature.
Differences in Transcriptional Profiles Between GBM Cases with Wild-Type Vs. Mutant IDH1
The CGGA RNA-seq cohort included 138 GBM patients, of which 33 cases were mutant and 105 were wild-type for IDH1. An SAM analysis identified 4344 genes that were differentially expressed between the two groups (Fig. 3a), including 2473 that were upregulated and 1871 that were downregulated in IDH1-mutant GBM. Interestingly, 1031/1264 (82 %) genes that were upregulated in IDH1-mutant LGG were also upregulated in IDH1-mutant GBM, while 1133/1415 (80 %) genes that were downregulated IDH1-mutant LGG were also downregulated in IDH1-mutant GBM.

Transcriptional profiles in GBM and the prognostic value of the six-gene signature in IDH1-mutant GBM. a Profiles of genes differentially expressed in GBM with wild-type vs. mutant IDH1. Biological processes associated with upregulated (b) and downregulated (c) genes differentially expressed in GBM with IDH1 mutation. d–f Application of the six-gene signature to IDH1-mutant GBM classified patients into two groups based on the median risk score. g–i Combining histology grade, IDH1 status, and the six-gene signature in all grades of glioma stratified patients into six subgroups with distinct prognoses
Genes that were differentially expressed between wild-type and mutant IDH1 GBM cases were analyzed with DAVID to identify salient biological processes. The upregulated genes were primarily associated with transcription and neuronal differentiation (Fig. 3b and Supplementary Data 4), whereas the downregulated genes were involved in the immune response (Fig. 3c and Supplementary Data 5). These annotations showed considerable overlap with those of LGG, indicating that the functional relevance of IDH1 mutation status is similar across glioma classifications.
Prognostic Value of the Six-Gene Signature for IDH1-Mutant GBM
Given the common role of the IDH1 mutation in LGG and GBM, we evaluated the prognostic value of the six-gene signature for IDH1-mutant GBM by calculating a risk score using the same formula. Patients were categorized into high- and low-risk groups based on their median risk score. The survival analysis showed that low-risk patients had the longest survival; on the other hand, the survival of high-risk patients was similar to that of patients with wild-type IDH1 (Fig. 3d). This trend was verified with the internal and external validation sets (Fig. 3e, f). These results indicate that the six-gene signature retained its prognostic value for IDH1-mutant GBM.
Subsequently, we sought to combine the survival analyses of LGG and GBM. All cases were classified into six subgroups based on glioma grade, IDH1 status, and the six-gene signature. We observed notable differences in survival trends among these six subgroups (Fig. 3g–i). Moreover, these six subgroups could be further summarized into three sets based on the survival curves (Fig. 3g–i). Set (i) consisted of low-risk LGG subgroup, which had the best prognosis. Set (ii) comprised high-risk LGG, IDH1-wild LGG, and low-risk GBM subgroups, which had similar and intermediate OS. Set (iii) contained high-risk GBM and IDH1-wild GBM subgroups, which suffered the most reduced survival time.
Discussion
Even with the identification of many new molecular markers, the IDH1 mutation is the most stable and widely used one for in glioma study. IDH1 status does not change between primary and recurrent gliomas, suggesting that these tumors are initiated by the clonal expansion of cells with different IDH1 status [17]. Although the role of IDH1 mutations in glioma has been previously investigated [18, 19], their contribution to the development and progression of glioma is not well understood. In our study, 2679 genes were defined as being differentially expressed in LGG, including 1264 that were upregulated and 1415 that were downregulated in IDH1-mutant patients. Those that were upregulated were mainly involved in neuronal differentiation, as previously reported [4, 20, 21], but those that were downregulated revealed a previously unreported association between IDH1 status and immune response. Our results suggest an immunoediting potential of IDH1 mutation that leads to a state of immunosuppression, thereby altering the glioma microenvironment. To date, few studies have compared the role of IDH1 in LGG and GBM. We showed here that the majority of genes that are differentially expressed in LGG (2164/2679, or 83 %) are the same as those in GBM. We therefore propose that the IDH1 mutation is an early driver of gliomagenesis [22] that induces similar transcriptional profiles in LGG and GBM, which was supported by the similarities in biological annotations.
Patients with the same IDH1 status exhibit distinct prognoses, suggesting that other factors contribute to clinical outcome. Whether histopathological grade and classification can predict prognosis based on IDH1 mutation status is controversial [23]. Several molecular markers are now combined with IDH1 status for improved prognostic accuracy [7, 8, 24–26]; however, these are not adequate to fully clarify the exact mechanism determining clinical outcome in equal IDH1 mutation status. We determined that among the 2679 genes differentially expressed in LGG, 1245 had prognostic significance for IDH1-mutant LGG indicating that extensive transcriptional differences involved in the prognostic determination along with IDH1 status. GO analysis of risky genes demonstrated that the immune response was closely associated with patient prognosis, with its positive regulation conferring unfavorable prognosis for IDH1-mutant LGG.
A six-gene risk signature was identified that was shown to have independent prognostic significance for IDH1-mutant LGG and exhibit greater prognostic significance than other gene signatures and its consisting individual genes. The ROC curves confirmed that the predictive value of this signature was superior to that of traditional clinicopathologic factors, highlighting the advantage of using a gene expression-based signature [27, 28]. A positive correlation between risk score and immune phenotype was observed, indicating that an enhanced local immune response contributes to unfavorable prognosis, consistent with other reports that the local immune phenotype in glioma was predominated by the responses supporting glioma invasion, angiogenesis, and proliferation [29]. Because we have little knowledge on what degree of local immune response is required to prolong gliomas’ survival time, our six-gene signature can improve the current glioma classification system so that immune-relevant therapy can be better applied in glioma treatment.
Importantly, the six-gene signature had equal prognostic value for IDH1-mutant GBM, classifying patients into high- and low-risk groups, with the former showing reduced survival that was comparable to that of GBM cases without IDH1 mutation. This suggests that IDH1 mutation affects gene expression patterns to a similar degree irrespective of tumor classification. Our findings also enhance the hypothesis that GBM with IDH1 mutation may rapidly evolve from IDH1-mutant LGG without exhibiting early LGG symptoms [30].
Combining histological grade, IDH1 status, and the six-gene signature enabled the classification of all glioma samples into six subgroups with distinct prognoses. These subgroups could be further summarized into three sets based on survival curves, as follows: (i) low-risk LGG patients, who had the best prognosis; (ii) high-risk LGG, IDH1-wild LGG, and low-risk GBM subgroups had intermediate OS; and (iii) IDH1-wild and high-risk GBM patients, who had the worst prognosis. In both LGG and GBM, the six-gene signature identified IDH1-mutant patients whose prognosis was similar to that of cases with wild-type IDH1, implying that the present treatment strategy is not adequate for high-risk patients. Although there have been significant advances in approaches targeting mutant IDH1 [21, 31], their clinical efficacy—especially in high-risk patients—requires further validation. In addition, the six genes identified in the present study can potentially serve as additional therapeutic targets.
In summary, our findings demonstrate that IDH1 mutation play an extensive and parallel role in the progression of LGG and GBM. A six-gene signature was identified with equal prognostic significance in both IDH1-mutant LGG and GBM. When combined with histological grade and IDH1 status, the six-gene signature can be a useful tool for patient risk stratification. The advantage of our study could originate from the systematical profiling, the robust nature of risk score method [32] and validation across multi-platform and multi-population. However, on the other hand, recent studies have recognized IDH2 mutation with similar function to IDH1 mutation in glioma. Even with an incidence lower than 5 % [19], our study was limited by the deficiency of IDH2 information. Recent studies have incorporated 1p/19q codeletion with IDH mutation for glioma classification [7, 24]. Even the CGGA database did not have information on 1p/19q codeletion, we found that 1p/19q status was similar between different risk groups in IDH1-mutant LGG in the GSE16011 cohort (Fig. S5). Considering that 1p/19q codeletion occurs most often in oligodendroglial histologic types [33], the facts that our signature remained powerful in both oligodendroglioma and oligoastrocytoma enhanced the hypothesis that the six-gene signature could be a prognostic factor for IDH1-mutant LGG independent of 1p/19q status. While, additional studies are still needed to explore the association between 1p/19q codeletion and the six-gene signature, and the frequency of other molecular markers in low- vs. high-risk groups, such as TP53, ATRX, and TERT promoter mutations. Moreover, this study was also limited by its retrospective nature. Although the predictive value of the six-gene signature was validated in different datasets, a prospective study with a larger sample size is required to evaluate the clinical relevance.
References
Kloosterhof NK, Bralten LB, Dubbink HJ, French PJ, van den Bent MJ (2011) Isocitrate dehydrogenase-1 mutations: a fundamentally new understanding of diffuse glioma? The lancet oncology 12(1):83–91. doi:10.1016/S1470-2045(10)70053-X
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116(6):597–602. doi:10.1007/s00401-008-0455-2
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG et al (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462(7274):739–744. doi:10.1038/nature08617
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R et al (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483(7390):474–478. doi:10.1038/nature10860
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW et al (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483(7390):479–483. doi:10.1038/nature10866
Guo C, Pirozzi CJ, Lopez GY, Yan H (2011) Isocitrate dehydrogenase mutations in gliomas: mechanisms, biomarkers and therapeutic target. Curr Opin Neurol 24(6):648–652. doi:10.1097/WCO.0b013e32834cd415
Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, Shimamura T, Niida A et al (2015) Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet 47(5):458–468. doi:10.1038/ng.3273
Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T et al (2015) Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med 372(26):2499–2508. doi:10.1056/NEJMoa1407279
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114(2):97–109. doi:10.1007/s00401-007-0243-4
Huang d W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4(1):44–57. doi:10.1038/nprot.2008.211
Cheng W, Li M, Cai J, Wang K, Zhang C, Bao Z, Liu Y, Wu A (2015) HDAC4, a prognostic and chromosomal instability marker, refines the predictive value of MGMT promoter methylation. J Neuro-Oncol 122(2):303–312. doi:10.1007/s11060-014-1709-6
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL et al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102(43):15545–15550. doi:10.1073/pnas.0506580102
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5(7):621–628. doi:10.1038/nmeth.1226
Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res 7(10):986–995
Lossos IS, Czerwinski DK, Alizadeh AA, Wechser MA, Tibshirani R, Botstein D, Levy R (2004) Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. N Engl J Med 350(18):1828–1837. doi:10.1056/NEJMoa032520
Yu SL, Chen HY, Chang GC, Chen CY, Chen HW, Singh S, Cheng CL, Yu CJ et al (2008) MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell 13(1):48–57. doi:10.1016/j.ccr.2007.12.008
Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S et al (2014) Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343(6167):189–193. doi:10.1126/science.1239947
Hu H, Wang Z, Liu Y, Zhang C, Li M, Zhang W, Wang K, Cai J et al (2015) Genome-wide transcriptional analyses of Chinese patients reveal cell migration is attenuated in IDH1-mutant glioblastomas. Cancer Lett 357(2):566–574. doi:10.1016/j.canlet.2014.12.018
Zhang C, Moore LM, Li X, Yung WK, Zhang W (2013) IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro-Oncology 15(9):1114–1126. doi:10.1093/neuonc/not087
Pirozzi CJ, Reitman ZJ, Yan H (2013) Releasing the block: setting differentiation free with mutant IDH inhibitors. Cancer Cell 23(5):570–572. doi:10.1016/j.ccr.2013.04.024
Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O et al (2013) An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340(6132):626–630. doi:10.1126/science.1236062
Agnihotri S, Aldape KD, Zadeh G (2014) Isocitrate dehydrogenase status and molecular subclasses of glioma and glioblastoma. Neurosurg Focus 37(6):E13. doi:10.3171/2014.9.FOCUS14505
Olar A, Wani KM, Alfaro-Munoz KD, Heathcock LE, van Thuijl HF, Gilbert MR, Armstrong TS, Sulman EP et al (2015) IDH mutation status and role of WHO grade and mitotic index in overall survival in grade II-III diffuse gliomas. Acta Neuropathol 129(4):585–596. doi:10.1007/s00401-015-1398-z
Cancer Genome Atlas Research N, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E et al (2015) Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372(26):2481–2498. doi:10.1056/NEJMoa1402121
Cai J, Yang P, Zhang C, Zhang W, Liu Y, Bao Z, Liu X, Du W et al (2014) ATRX mRNA expression combined with IDH1/2 mutational status and Ki-67 expression refines the molecular classification of astrocytic tumors: evidence from the whole transcriptome sequencing of 169 samples samples. Oncotarget 5(9):2551–2561
Labussiere M, Boisselier B, Mokhtari K, Di Stefano AL, Rahimian A, Rossetto M, Ciccarino P, Saulnier O et al (2014) Combined analysis of TERT, EGFR, and IDH status defines distinct prognostic glioblastoma classes. Neurology 83(13):1200–1206. doi:10.1212/WNL.0000000000000814
Freije WA, Castro-Vargas FE, Fang Z, Horvath S, Cloughesy T, Liau LM, Mischel PS, Nelson SF (2004) Gene expression profiling of gliomas strongly predicts survival. Cancer Res 64(18):6503–6510. doi:10.1158/0008-5472.CAN-04-0452
Nutt CL, Mani DR, Betensky RA, Tamayo P, Cairncross JG, Ladd C, Pohl U, Hartmann C et al (2003) Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res 63(7):1602–1607
Glass R, Synowitz M (2014) CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol 128(3):347–362. doi:10.1007/s00401-014-1274-2
Ohgaki H, Kleihues P (2013) The definition of primary and secondary glioblastoma. Clin Cancer Res 19(4):764–772. doi:10.1158/1078-0432.CCR-12-3002
Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, Menn O, Osswald M et al (2014) A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 512(7514):324–327. doi:10.1038/nature13387
Hayes J, Thygesen H, Tumilson C, Droop A, Boissinot M, Hughes TA, Westhead D, Alder JE et al (2015) Prediction of clinical outcome in glioblastoma using a biologically relevant nine-microRNA signature. Mol Oncol 9(3):704–714. doi:10.1016/j.molonc.2014.11.004
Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T et al (2015) Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. doi:10.1056/NEJMoa1407279
Acknowledgments
The authors conducting this work represent the Chinese Glioma Cooperative Group (CGCG).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study protocol was approved by the ethics committees of participating hospitals, and all patients provided written, informed consent.
Conflict of Interest
We state that all authors have seen the manuscript and approved to submit to your journal. The authors declare that they have no conflict of interest.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant Numbers: 81172409, 81472360, and 81402045) and the Science and Technology Department of Liaoning Province (Grant Number: 2011225034).
Electronic Supplementary Material
ESM 1
(GIF 192 kb)
High Resolution Image
(TIFF 1723 kb)
ESM 2
(GIF 16 kb)
High Resolution Image
(TIFF 169 kb)
ESM 3
(GIF 197 kb)
High Resolution Image
(TIFF 1822 kb)
ESM 4
(GIF 45 kb)
High Resolution Image
(TIFF 482 kb)
ESM 5
(GIF 16 kb)
High Resolution Image
(TIFF 205 kb)
ESM 6
(XLS 937 kb)
ESM 7
(DOC 45 kb)
Table S1
(DOCX 14 kb)
Table S2
(DOCX 14 kb)
Table S3
(DOCX 13 kb)
Table S4
(DOCX 17 kb)
Rights and permissions
About this article

Cite this article
Cheng, W., Ren, X., Zhang, C. et al. Gene Expression Profiling Stratifies IDH1-Mutant Glioma with Distinct Prognoses. Mol Neurobiol 54, 5996–6005 (2017). https://doi.org/10.1007/s12035-016-0150-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0150-6