
Abstract
We investigated whether mild hypothermia combined with sodium hydrosulfide treatment during resuscitation improves neuron survival following cerebral ischemia-reperfusion injury beyond that observed for the individual treatments. Male Sprague-Dawley rats were divided into seven groups (n = 20 for each group). All rats underwent Pulsinelli 4-vessel occlusion. Ischemia was induced for 15 min using ligatures around the common carotid arteries, except for the sham group. Immediately after initiating reperfusion, the mild hypothermia (MH), sodium hydrosulfide (NaHS), hydroxylamine (HA), MH + NaHS, MH + HA, and ischemia-reperfusion (I/R) control groups received an intraperitoneal injection of saline, sodium hydrosulfide, hydroxylamine, sodium hydrosulfide, hydroxylamine, and saline, respectively, and mild hypothermia (32 to 33 °C) was induced in the MH, MH + NaHS, and MH + HA groups for 6 h. The levels of NR2A, NR2B, p-Akt, and p-Gsk-3β in the hippocampus of the MH, NaHS, and MH + NaHS groups were higher than those in the I/R control group, with the highest levels observed in the MH + NaHS group (P < 0.05). Treatment with hydroxylamine reduced the levels of these proteins in the HA and MH + HA groups, compared with the I/R control and MH groups, respectively. The apoptotic index of the CA1 region of the hippocampus was 45.2, 66.5, 63.5, and 84.8 % in the MH + NaHS, MH, NaHS, and I/R control groups, respectively (P < 0.05), indicating that the combination treatment shifted the NR2A/NR2B balance in favor of synaptic neuron stimulation and phosphatidylinositol 3ʹ-kinase (PI3K)/Akt signaling. The combination of mild hypothermia and sodium hydrosulfide treatment for resuscitation following ischemia-reperfusion injury was more beneficial for reducing hippocampal apoptosis and pathology than that of mild hypothermia or hydrogen sulfide treatment alone.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Although the incidence of stroke has decreased in recent years, stroke-related mortality remains high in both developed and developing countries [1], and stroke is the leading cause of permanent disability worldwide, constituting a major global economic burden [2]. The window between an acute ischemic event and reperfusion is critical to the clinical outcome and survival of stroke patients, with longer periods resulting in more severe brain injury and greater risk of death. Clinically feasible treatments are needed that reduce the rate of neuronal apoptosis during acute focal cerebral ischemia to improve the clinical outcomes and survival of stroke patients. Animal models have shown that damage to the blood-brain barrier (BBB) occurs between 1 and 24 h following reperfusion [3, 4], and that additional neuronal apoptosis occurs as late as 7 days after an ischemic event [5]. Therefore, treatments that minimize the delayed effects of cerebral ischemia are also needed to improve patient outcomes.
Endogenous hydrogen sulfide (H2S) functions as a signaling molecule that influences the regulation of vascular smooth muscle relaxation and leukocyte adhesion in the vasculature of the central nervous system in mammals [6, 7], and affects multiple cellular pathways in astrocytes, microglia, and neurons [8]. Hydrogen sulfide has demonstrated protective effects against secondary neuronal injury through the inhibition of malondialdehyde and superoxide dismutase, and suppresses the effects of various reactive oxygen species [7–9]. In rat models of cerebral ischemia-reperfusion (I/R) injury, the administration of sodium hydrosulfide, a hydrogen sulfide donor, reduced infarct volume, and improved markers of neurological function by reducing apoptosis through the inhibition of Bcl-2 and Bax and by reducing the levels of proinflammatory factors in the brain, including tumor necrosis factor-α, interleukin-10, and monocyte chemotactic protein-1 [9, 10].
Hydrogen sulfide enhances N-methyl-D-aspartate receptor (NMDAR)-mediated responses, and facilitates the induction of the long-term potentiation of hippocampal neurons [11]. However, cell culture experiments have shown that high concentrations of hydrogen sulfide induce proapoptotic processes in hippocampal neurons by modulating the excitability of NMDARs. These findings of the effects of hydrogen sulfide on hippocampal neurons can be explained by the selective modulation of the NMDAR subunits, NR2A and NR2B. The activation of NR2A on synaptic neurons promotes the survival of hippocampal neurons via cAMP response element-binding protein (CREB) and phosphatidylinositol 3ʹ-kinase (PI3K)-Akt signaling pathways, whereas the activation of NR2B on extrasynaptic neurons promotes neuronal apoptosis via the downregulation of CREB and the activation neuronal nitric oxide synthase (nNOS)- and JUN-mediated gene expression [12–16]. The phosphorylation of the Akt serine-threonine kinase (also known as protein kinase B), a downstream target of PI3K, upregulates the phosphorylation of BAD, caspase-9, NF-κB, and Gsk-3β, which inhibits neuronal apoptosis [17–19].
Mild hypothermia induced by cooling the brain to between 30 and 33 °C has been shown to reduce brain injury following brief periods (<2 h) of focal or global cerebral ischemia [20–23]. Mild intraischemic hyperthermia improves the outcomes of resuscitation in rat models of cerebral I/R by reducing the oxygen consumption and metabolic rate of brain tissues, postischemic hyperperfusion, delayed postischemic hypoperfusion, infarct volume, BBB disruption, cerebral edema, the destruction of structural proteins in brain cells, the release of endogenous cytotoxic chemicals, neuronal calcium ion influx, and neuronal apoptosis [21, 23–25]. It also sustains neuroendocrine balance [26] and stabilizes the membrane potential of ischemic brain cells by maintaining sodium-potassium ATPase activity [21, 24]. Mild hypothermia has been implemented for the treatment of craniocerebral trauma and stroke in an effort to improve clinical outcomes in humans and animal models [27–30].
We hypothesized that, because hypothermia and hydrogen sulfide affect neuronal survival via different mechanisms, combining these resuscitation treatments might improve neuron survival following I/R injury beyond that of the individual treatments, and thus improve clinical outcomes in patients treated with a combination of hypothermia and hydrogen sulfide following stroke. The aim of our current study was to determine whether the use of hydrogen sulfide in combination with mild hypothermia for cerebral resuscitation would improve hippocampal neuron survival in a rat model of cerebral I/R injury in order to obtain insight into the potential benefits of combination therapy in stroke patients. We also examined the underlying molecular mechanisms that mediate the combined effects of hydrogen sulfide and hypothermia to determine whether PI3K/Akt signaling pathways were induced via the selective activation of synaptic NMDARs by quantifying the levels of NR2A, NR2B, p-Akt, and p-Gsk-3β in rat hippocampal neurons.
Materials and Methods
Experimental Animals
All of our animal experiments were performed in compliance with the Guidelines for the Humane Treatment of Laboratory Animals (Ministry of Science and Technology of the People’s Republic of China, Policy no. 2006 398), and were approved by the Institutional Animal Care and Use Committee at Nanjing General Hospital of the Nanjing Military Command. Male Sprague-Dawley rats, that were 3 months of age and weighed between 250 and 300 g, were obtained from the Rehabilitation Medicine Department of Nanjing General Hospital. The rats were housed under standard husbandry conditions as follows: a room temperature of 22 to 25 °C, a 12-h:12-h light and dark cycle, a relative humidity of 50–60 %, and free access to food and water. The rats were acclimatized to these conditions for 1 week prior to our experiment, and the rats were fasted and deprived of water for 12 h before the commencement of the experiment.
Rat Model of I/R
A modified version of the Pulsinelli 4-vessel occlusion method was used in rats as a model of cerebral I/R injury [31]. Twenty rats were assigned to the sham treatment group. All of the rats were anesthetized by an intraperitoneal injection of 10 % chloral hydrate (0.35 mL/100 g), and placed in the prone position. The cervical posterior midline was incised to expose the foramen alare parvum of the first cervical vertebra, and thermocoagulation was used to permanently occlude the bilateral vertebral arteries. The rats were then placed in the supine position. The lateral cervical regions were dissected, and the right and left common carotid arteries were exposed. A suture line was tied loosely around each common carotid artery for use as a ligature to occlude blood flow for inducing ischemia. Twenty-four hours later, when the rats were in a waking state, both of the ligatures were tightened to block blood flow, initiating cerebral ischemia. Ischemia was not induced in the sham treatment group. After a 15-min period, the ligatures were loosened to restore blood flow, initiating reperfusion. Each rat was examined 30 s after inducing ischemia. Rats with pale eyeballs that did not exhibit the corneal, pupillary, and righting reflexes were included in our analysis, whereas rats that failed to meet these conditions or were spasmodic were excluded from our study.
Resuscitation Treatments
The rats subjected to I/R were randomly divided into the I/R control, mild hypothermia (MH), sodium hydrosulfide (NaHS), hydroxylamine (HA), mild hypothermia plus sodium hydrosulfide (MH + NaHS), and mild hypothermia plus hydroxylamine (MH + HA) groups (n = 20 for each group). Immediately after reperfusion was initiated, the rats in the NaHS and MH + NaHS groups were injected intraperitoneally with sodium hydrosulfide (14 μmol/kg; Sigma-Aldrich, St. Louis, MO, USA), and the rats in the HA and MH + HA groups were injected intraperitoneally with hydroxylamine (5 μmol/kg; Sigma-Aldrich). The rats in the sham, I/R control, and MH groups were injected intraperitoneally with an equivalent volume of normal saline. All of the rats were placed on an ice bag immediately after administering the anesthetic. The rats in the MH, MH + NaHS, and MH + HA groups were cooled until their rectal temperatures were reduced to between 32 and 33 °C, which was achieved within 15 min of initiating reperfusion. Hypothermia was maintained for 6 h by exposing the rat and ice bag to a heat lamp, and the distance between the rat and the lamp was adjusted to maintain the rectal temperature range. The rectal temperatures of the rats in the sham, I/R control, NaHS, and HA groups were also maintained between 36 and 37 °C by warming them with a heat lamp. Six hours after reperfusion was initiated, the hippocampus was surgically removed from 12 rats in each group while the animals remained on the ice bag. The hippocampus specimens were stored at −70 °C. The remaining rats (eight in each group) were removed from the ice bag, and maintained under standard conditions until 72 h after initiating reperfusion.
Quantification of NR2A, NR2B, p-Akt, and p-Gsk-3β Proteins in the Hippocampus
The relative levels of the NR2A, NR2B, p-Akt, and p-Gsk-3β proteins in the hippocampus tissue of 12 rats in each group were measured using western blotting. The specimens were homogenized RIPA lysis buffer on ice, and the homogenates were centrifuged at 17,000×g for 60 min. Western blotting was performed using standard methods, as described previously [9]. The membranes were probed using mouse monoclonal antibodies generated against the NR2A, NR2B, p-Akt, p-Gsk-3β, and β-Actin proteins (1:1000, Cell Signaling Technology, Danvers, MA, USA) as primary antibodies, and primary antibody binding was detected using a horseradish peroxidase-conjugated goat anti-mouse IgG secondary antibody (1:5000, Sigma-Aldrich). The protein bands were visualized using an enhanced chemiluminescence detection kit (Life Technologies, Carlsbad, CA, USA). The x-ray film was scanned using a 5500 Chemi Imager (Alpha Innotech, San Leandro, CA, USA), and the relative quantification of protein levels was performed using the Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA). All protein levels were normalized to that of β-Actin.
Quantification of Hydrogen Sulfide in the Hippocampus
The concentration of hydrogen sulfide in the hippocampus tissue of four rats in each group was measured using a methylene blue colorimetric assay, as described previously [32]. The clarified hippocampal supernatant was collected, and mixed with a solution containing 20 mM N,N-dimethyl-p-phenylenediamine dihydrochloride and 30 mM iron (III) chloride in HCl (Sigma-Aldrich for both). After a 20-min incubation at room temperature, the absorbance of the solution was measured at 670 nm using a model 722S spectrophotometer (Shanghai Analytical Instrument, Shanghai, China). The concentration of hydrogen sulfide was determined as the number of millimoles of hydrogen sulfide per gram of body weight (mmol/g) based on a comparison of the sample absorbance to a standard curve constructed using a methylene blue calibration standard for hydrogen sulfide (Sigma-Aldrich).
Assessment of Apoptosis and Pathology of Hippocampal Neurons
Apoptosis and histopathological changes in pyramidal cells in the CA1 region of the hippocampus were assessed using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and hematoxylin and eosin (H&E) staining, respectively. Seventy-two hours after initiating reperfusion, four rats in each group were perfused transcardially with 200 mL of ice-cold normal saline, followed by 200 mL of 4 % buffered paraformaldehyde, and the brain was removed. Consecutive 40-μm coronal sections of the hippocampus were taken 1 to 4 mm posterior to the optic chiasma in the dentate gyrus plane, and the sections were fixed in 4 % paraformaldehyde for 6 h at 4 °C.
The hippocampal sections were subjected to TUNEL staining, as described previously [33]. Labeling was performed at 37 °C for 60 min using a transferase to nucleotide ratio of 1:9, and visualized by incubation in diaminobenzidine for 5 to 10 min. Control sections were also prepared, in which an equivalent volume of distilled water was used in place of the transferase solution. The sections were mounted on glass microscope slides using rhamsan gum, and examined using light microscopy. The number of TUNEL-positive cells in the CA1 region of the hippocampus was determined, and the apoptotic index (AI) was calculated as the percentage of TUNEL-positive cells relative to the total number of cells. An additional section from each rat was prepared, as described above, and stained with hematoxylin and eosin (H&E) to assess histopathological changes in the pyramidal cells in the CA1 region of the hippocampus.
Statistical Analysis
The statistical analysis was performed using the SPSS, version 13.0, software (IBM, Armonk, NY, USA). The results are reported as the mean ± standard deviation (SD). The intergroup differences were evaluated using an analysis of variance, with the level of statistical significance set at P < 0.05. Adjusted P values were obtained using the Fisher’s least significant difference test.
Results
Exogenous Hydrogen Sulfide Promotes Hippocampal Neuron Survival Following Cerebral Ischemia
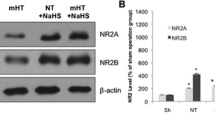
The relative levels of NR2A, NR2B, p-Akt, and p-Gsk-3β in the hippocampus of the rats in the I/R control group were significantly higher than those in the sham treatment group (P < 0.05, Table 1 and Fig. 1). The concentration of hydrogen sulfide in the hippocampus of rats in the I/R control group was also significantly higher than that in the sham group (P < 0.05, Table 2). The concentration of hydrogen sulfide in the hippocampus of rats in the NaHS group was 1.7-fold higher than that of the I/R control group (Table 2), and the relative levels of NR2A, NR2B, p-Akt, and p-Gsk-3β in the hippocampus of rats in the NaHS group were 2.7-, 2.4-, 1.5-, and 1.4-fold higher, respectively, compared with those in the I/R control group (Table 1). The AI of the CA1 hippocampal neurons in the NaHS group was significantly lower (66.5 %) than that in the I/R control group (84.8 %, P < 0.05). These results indicated that treatment with sodium hydrosulfide during resuscitation stimulated the expression of NR2A and NR2B and the phosphorylation of Akt and Gsk-3β, which promoted the survival of CA1 hippocampal neurons.

Western blot analysis of rat hippocampus following cerebral I/R injury. Cerebral ischemia was induced for 15 min using a modified version of the Pulsinelli 4-vessel occlusion method. Upon initiating reperfusion, the NaHS and MH + NaHS groups were treated with sodium hydrosulfide, the HA and MH + HA groups were treated with hydroxylamine, and the sham, I/R control, and MH groups were treated with normal saline. Hypothermia (rectal temperature: 32–33 °C) was induced in the MH, MH + NaHS, and MH + HA groups within 15 min of initiating reperfusion, but was not induced in the sham, NaHS, and HA groups (rectal temperature: 36–37 °C). At 6 h postreperfusion, the hippocampus was removed, and frozen at −70 °C. Hippocampus homogenates were analyzed by western blotting using mouse monoclonal antibodies generated against the NR2A, NR2B, p-Akt, and p-Gsk-3β proteins. Relative quantification of protein levels was performed by normalizing the intensities of the various protein bands to that of β-Actin (see Table 1)
Hydroxylamine inhibits the production of hydrogen sulfide by cystathionine-β-synthase (CBS) in the brain [34]. The levels of NR2A and NR2B in the hippocampus of rats in the HA group were lower (0.5- and 0.8-fold, respectively) than those in the I/R group, whereas the levels of NR2A and NR2B in the HA group were higher (1.9- and 4.7-fold, respectively) than those in the sham group (Table 1). The concentration of hydrogen sulfide in the hippocampus of rats in the HA group was significantly lower than that in the NaHS group (P < 0.05), and was similar to that in the I/R control and sham groups (P > 0.05). The AI of the CA1 hippocampal neurons in the HA group (89.7 %) was similar to that of the I/R control group (84.8 %, P > 0.05), and was significantly greater than that in the NaHS group (66.5 %, P < 0.05). These results confirm that the improved survival of CA1 hippocampal neurons in the NaHS group was caused primarily by treatment with the exogenous source of hydrogen sulfide.
Mild Hypothermia Enhances the Neuroprotective Effects of Hydrogen Sulfide Following Cerebral Ischemia
The induction of mild hypothermia at the initiation of reperfusion had no significant effect on the concentration of hydrogen sulfide in the hippocampus of rats in the MH group, compared with that in the I/R control group. However, the relative levels of p-Akt and p-Gsk-3β in the MH group (1.2 and 1.4, respectively) were significantly higher than those in the I/R control group (0.79 and 1.0, respectively; P < 0.05), and the AI of the CA1 hippocampal neurons in the MH group (63.5 %) was significantly lower than that of the I/R control group (84.8 %, P < 0.05). The levels of hydrogen sulfide, NR2A, NR2B, p-Akt, and p-Gsk-3β in the MH + HA group were similar to those in the I/R control group (P > 0.05). The AI in the MH + HA group was also similar to that of the I/R control group. These results suggest that the beneficial effect of hypothermia on neuronal survival following I/R involves p-Akt and p-Gsk-3β signaling and is influenced by the level of endogenous hydrogen sulfide.
Although the level of hydrogen sulfide in the MH + NaHS group was similar to that in the NaHS group, the combined effects of hypothermia and treatment with sodium hydrosulfide in the MH + NaHS group increased the NR2A/NR2B ratio, and resulted in significantly higher levels of NR2A, NR2B, p-Akt, and p-Gsk-3β (P < 0.05), compared with the I/R control group (Table 1). The H&E-stained sections of the hippocampus of rats in the MH + NaHS group showed three to four layers of loosely arranged neurons exhibiting normal morphology, which contained substantially fewer necrotic cells than the H&E-stained hippocampal sections from the I/R control, MH, and NaHS groups (Fig. 2). The AI of the CA1 region of the hippocampus in the MH + NaHS group (45.2 %) was significantly lower that in the I/R control, MH, and NaHS groups (84.8, 63.5, and 66.5 %, respectively; P < 0.05). These results collectively suggest that the combined effect of mild hypothermia and hydrogen sulfide treatment on the survival of hippocampal neurons following cerebral I/R was more beneficial than the effects of the individual treatments.

Assessment of histopathology in the CA1 region of the rat hippocampus following cerebral I/R injury. Cerebral ischemia was induced for 15 min using a modified version of the Pulsinelli 4-vessel occlusion method. Upon initiating reperfusion, the I/R control and MH groups were treated with normal saline, the NaHS and MH + NaHS groups were treated with sodium hydrosulfide, and the HA and MH + HA groups were treated with hydroxylamine. Hypothermia (rectal temperature: 32–33 °C) was induced in the MH, MH + NaHS, and MH + HA groups within 15 min of initiating reperfusion, but was not induced in the sham, NaHS, and HA groups (rectal temperature: 36–37 °C). At 72 h postreperfusion, rats were perfused transcardially with ice-cold normal saline, followed by buffered paraformaldehyde. Consecutive coronal sections of the hippocampus were taken immediately posterior to the optic chiasma in the dentate gyrus plane, and fixed in paraformaldehyde before staining with H&E to assess histopathology in the pyramidal cells in the CA1 region (×400 magnification)
Discussion
The results of our study support our hypothesis that treatment with sodium hydrosulfide and mild hypothermia immediately following cerebral I/R injury is more beneficial for reducing neuronal apoptosis than the effects of the different treatments used alone. Treatment with sodium hydrosulfide during resuscitation increased the expression of NR2A and NR2B and the activation of Akt and Gsk-3β (Table 1). Although the activation of NR2A in synaptic hippocampal neurons has been shown to promote neuronal survival, the activation of NR2B in extrasynaptic neurons has been shown to promote neuronal apoptosis [14–16, 35]. Our results show that the combined effects of the increased levels of NR2A, NR2B, p-Akt, and p-Gsk-3β had an overall positive effect on the survival of neurons in the CA1 region of the hippocampus (Table 2).
The CREB and PI3K-Akt signaling pathways are associated with the prosurvival effects of synaptic NMDARs, whereas the proapoptotic effects of extrasynaptic NMDARs involve the downregulation of CREB and the activation nNOS- and JUN-mediated gene expression [12, 16, 36]. In our current study, the level of NR2A in the hippocampus of rats in the sham group was higher than the level of NR2B, with an NR2A/NR2B of 1.63 (Table 1). These observations reflect the baseline level of glutamate secretion from presynaptic neurons and the constitutive expression of NR2A in synaptic neurons in healthy brains [14, 16]. In the I/R control group, the levels of NR2A and NR2B in the hippocampus were higher than those in the sham group, and the NR2A/NR2B ratio was less than 1. These data are supported by the findings of previous studies, in which the release of glutamate from presynaptic neurons during cerebral I/R injury was shown to stimulate NR2B and other NMDARs on extrasynaptic neurons, contributing to neuronal apoptosis [14].
Treatment with sodium hydrosulfide increased the NR2A/NR2B ratio in the hippocampus of rats in the NaHS group, compared with that of the I/R control group, indicating that hydrogen sulfide selectively activates the expression of synaptic NMDARs. These results are consistent with the collective findings of previous studies of NMDARs [14–16]. A previous study showed that the influx of calcium ions through activated NMDARs on synaptic neurons caused intranuclear Ca2+/calmodulin dependent protein kinase to phosphorylate Akt, which mediated the expression of downstream prosurvival genes [36]. In our current study, the levels of p-Akt and p-Gsk-3β in the MH and NaHS groups were higher than those in the I/R control group (Table 1). These results showed that both hydrogen sulfide and mild hypothermia activate the PI3K/Akt signaling pathway, which inhibits apoptosis via Gsk-3β phosphorylation. Therefore, it is possible that both hydrogen sulfide and mild hypothermia selectively activate synaptic NMDARs and trigger PI3K/Akt signaling via different upstream regulatory mechanisms.
Endogenous hydrogen sulfide is synthesized by CBS, cystathionine-γ-lyase (CGS), cysteine amino transferase/3-mercaptopyruvate sulfur transferase, and D-amino acid oxidase/3-mercaptopyruvate sulfur transferase [37]. The expression of these enzymes is tissue specific. In the cerebrum, CBS is the primary enzyme involved in endogenous hydrogen sulfide production [34]. Hydrogen sulfide has been shown to be barely detectable in CBS knockout mice, and inhibiting CBS or CGS activity has been shown to substantially reduce the generation of endogenous hydrogen sulfide in mouse cerebral neurons [38, 37]. In our current study, we used the CBS inhibitor, hydroxylamine, to block the generation of endogenous hydrogen sulfide in order to investigate the role of hydrogen sulfide-producing enzymes in hippocampal neuron responses to cerebral I/R injury. We observed that the level of hydrogen sulfide in the hippocampus of rats subjected to I/R injury was lower than that of the I/R control group, but the difference was not statistically significant (Table 2).
We induced mild hypothermia in rats following cerebral I/R injury, and found that hypothermia stimulated the phosphorylation of Akt and p-Gsk-3β and selectively increased the expression of NR2A in the hippocampus of rats in the MH group, whereas the levels of hydrogen sulfide and NR2B were not significantly affected, compared with those in the I/R control group. These results are consistent with the collective findings of previous studies of the effects of hypothermia on PI3K/Akt signaling and NR2B activation in neuronal survival following cerebral I/R injury [39–41]. The TUNEL and H&E analyses showed that the extent of pyramidal cell apoptosis and histopathology in the CA1 region of the hippocampus in the MH group was significantly lower than that in the I/R control group. The results suggested that mild hyperthermia improved neuronal survival via a hydrogen sulfide-independent mechanism. However, treatment with both hypothermia and hydroxylamine substantially reduced the levels of NR2A, NR2B, p-Akt, and p-GDK-3β, compared with those of the MH group, to amounts that were similar to those in the I/R control group (P > 0.05, Table 1), suggesting that the mechanism underlying the prosurvival effect of hypothermia on hippocampal neurons is influenced by the level of endogenous hydrogen sulfide.
The combined use of mild hypothermia and hydrogen sulfide as an emergent treatment to reduce the effects of acute cerebral ischemia in stroke patients is clinically feasible, but it has certain limitations. Markarian et al. (1996) found that mild hypothermia must be induced within 30 min of the onset of cerebral ischemia, and must be continued for a duration of at least 3 h to ensure optimal neuronal outcomes [42]. In our current study, a 6-h hypothermic period induced within 15 min of initiating reperfusion significantly improved hippocampal neuron survival. Whether the combined use of hypothermia and hydrogen sulfide can improve the clinical outcomes and survival of stroke patients requires considerable further study. However, our findings can serve as guidelines for optimizing resuscitation conditions in future translational studies of the combined effects of hypothermia and hydrogen sulfide following focal cerebral ischemia.
Although we investigated the combined effects of hypothermia and hydrogen sulfide using the Pulsinelli 4-vessel occlusion model of cerebral ischemia, both treatments have also been shown to reduce neuronal apoptosis following global cerebral ischemia in rats [9, 43]. Hydrogen sulfide treatment has also been shown to reduce BBB permeability and cerebral edema in a rat model of cardiopulmonary resuscitation [44], which is especially relevant to clinical applications because cerebral edema causes increased intracranial pressure in patients resuscitated following cardiac arrest, further increasing the risk of mortality [45]. Therefore, future studies of the combined effects of hypothermia and hydrogen sulfide following global cerebral ischemia are warranted to assess whether combination therapy might improve clinical outcomes in patients resuscitated after suffering cardiac arrest.
In conclusion, the results of our western blotting and histopathological analyses collectively indicate that sodium hydrosulfide treatment during cerebral resuscitation caused a shift in the NR2A/NR2B balance that favored synaptic neuron stimulation in the rat hippocampus, which mediated prosurvival effects in hippocampal neurons via the PI3K/Akt signaling pathway. Although the prosurvival effects of mild hypothermia were also mediated by PI3K/Akt signaling, the upstream regulatory mechanism differed from that of hydrogen sulfide-induced neuroprotection, as evidenced by lower levels of NR1A and NR2A in the MH group, compared with the NaHS group. The combination of mild hypothermia and sodium hydrosulfide treatment following cerebral I/R injury was more beneficial for reducing apoptosis and histopathology in the hippocampus than that of mild hypothermia or sodium hydrosulfide treatment alone. Our findings suggest that the combined use of hypothermia and sodium hydrosulfide during the resuscitation of stroke patients can improve clinical outcomes and survival.
References
Thrift AG, Cadilhac DA, Thayabaranathan T, Howard G, Howard VJ, Rothwell PM, Donnan GA (2014) Global stroke statistics. Int J Stroke 9(1):6–18. doi:10.1111/ijs.12245
Feigin VL, Forouzanfar MH, Krishnamurthi R, Mensah GA, Connor M, Bennett DA, Moran AE, Sacco RL et al (2014) Global and regional burden of stroke during 1990–2010: findings from the Global Burden of Disease Study 2010. Lancet 383(9913):245–255
Krueger M, Bechmann I, Immig K, Reichenbach A, Hartig W, Michalski D (2015) Blood-brain barrier breakdown involves four distinct stages of vascular damage in various models of experimental focal cerebral ischemia. J Cereb Blood Flow Metab 35(2):292–303. doi:10.1038/jcbfm.2014.199
Belayev L, Busto R, Zhao W, Ginsberg MD (1996) Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res 739(1-2):88–96
Kirino T (1982) Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 239(1):57–69
Liu YH, Lu M, Hu LF, Wong PT, Webb GD, Bian JS (2012) Hydrogen sulfide in the mammalian cardiovascular system. Antioxid Redox Signal 17(1):141–185. doi:10.1089/ars.2011.4005
Yu YP, Chi XL, Liu LJ (2014) A hypothesis: hydrogen sulfide might be neuroprotective against subarachnoid hemorrhage induced brain injury. ScientificWorldJournal 2014:432318. doi:10.1155/2014/432318
Tan BH, Wong PT, Bian JS (2010) Hydrogen sulfide: a novel signaling molecule in the central nervous system. Neurochem Int 56(1):3–10. doi:10.1016/j.neuint.2009.08.008
Yin J, Tu C, Zhao J, Ou D, Chen G, Liu Y, Xiao X (2013) Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain Res 1491:188–196. doi:10.1016/j.brainres.2012.10.046
Gheibi S, Aboutaleb N, Khaksari M, Kalalian-Moghaddam H, Vakili A, Asadi Y, Mehrjerdi FZ, Gheibi A (2014) Hydrogen sulfide protects the brain against ischemic reperfusion injury in a transient model of focal cerebral ischemia. J Mol Neurosci 54(2):264–270. doi:10.1007/s12031-014-0284-9
Kimura H (2000) Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem Biophys Res Commun 267(1):129–133. doi:10.1006/bbrc.1999.1915
Valera E, Sanchez-Martin FJ, Ferrer-Montiel AV, Messeguer A, Merino JM (2008) NMDA-induced neuroprotection in hippocampal neurons is mediated through the protein kinase A and CREB (cAMP-response element-binding protein) pathway. Neurochem Int 53(5):148–154. doi:10.1016/j.neuint.2008.07.007
Hardingham GE, Fukunaga Y, Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5(5):405–414. doi:10.1038/nn835
Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J et al (2007) NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci 27(11):2846–2857. doi:10.1523/JNEUROSCI.0116-07.2007
Luo T, Wu WH, Chen BS (2011) NMDA receptor signaling: death or survival? Front Biol (Beijing) 6(6):468–476. doi:10.1007/s11515-011-1187-6
Hardingham GE, Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11(10):682–696. doi:10.1038/nrn2911
Brunet A, Datta SR, Greenberg ME (2001) Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11(3):297–305
Noshita N, Lewen A, Sugawara T, Chan PH (2002) Akt phosphorylation and neuronal survival after traumatic brain injury in mice. Neurobiol Dis 9(3):294–304. doi:10.1006/nbdi.2002.0482
Franke TF (2008) PI3K/Akt: getting it right matters. Oncogene 27(50):6473–6488. doi:10.1038/onc.2008.313
Karibe H, Zarow GJ, Graham SH, Weinstein PR (1994) Mild intraischemic hypothermia reduces postischemic hyperperfusion, delayed postischemic hypoperfusion, blood-brain barrier disruption, brain edema, and neuronal damage volume after temporary focal cerebral ischemia in rats. J Cereb Blood Flow Metab 14(4):620–627. doi:10.1038/jcbfm.1994.77
Zhao H, Steinberg GK, Sapolsky RM (2007) General versus specific actions of mild-moderate hypothermia in attenuating cerebral ischemic damage. J Cereb Blood Flow Metab 27(12):1879–1894. doi:10.1038/sj.jcbfm.9600540
Busto R, Dietrich WD, Globus MY, Valdes I, Scheinberg P, Ginsberg MD (1987) Small differences in intraischemic brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab 7(6):729–738. doi:10.1038/jcbfm.1987.127
Yenari MA, Han HS (2012) Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat Rev Neurosci 13(4):267–278. doi:10.1038/nrn3174
Erecinska M, Thoresen M, Silver IA (2003) Effects of hypothermia on energy metabolism in Mammalian central nervous system. J Cereb Blood Flow Metab 23(5):513–530. doi:10.1097/01.WCB.0000066287.21705.21
Lee JM, Zipfel GJ, Choi DW (1999) The changing landscape of ischaemic brain injury mechanisms. Nature 399(6738 Suppl):A7–A14
Busto R, Globus MY, Dietrich WD, Martinez E, Valdes I, Ginsberg MD (1989) Effect of mild hypothermia on ischemia-induced release of neurotransmitters and free fatty acids in rat brain. Stroke 20(7):904–910
Duan M, Li D, Xu J (2002) Mechanisms of selective head cooling for resuscitating damaged neurons during post-ischemic reperfusion. Chin Med J (Engl) 115(1):94–98
Qiu L (2011) Hypothermia in traumatic brain injury—a literature review. J Intensive Care Soc 12(3):201–214
van der Worp HB, Sena ES, Donnan GA, Howells DW, Macleod MR (2007) Hypothermia in animal models of acute ischaemic stroke: a systematic review and meta-analysis. Brain 130(Pt 12):3063–3074. doi:10.1093/brain/awm083
Schiefecker AJ, Beer R, Broessner G, Kofler M, Schmutzhard E, Helbok R (2015) Can therapeutic hypothermia be guided by advanced neuromonitoring in neurocritical care patients? A review. Ther Hypothermia Temp Manag. doi:10.1089/ther.2014.0028
Pulsinelli WA, Levy DE, Duffy TE (1983) Cerebral blood flow in the four-vessel occlusion rat model. Stroke 14(5):832–834
Li Z, Wang Y, Xie Y, Yang Z, Zhang T (2011) Protective effects of exogenous hydrogen sulfide on neurons of hippocampus in a rat model of brain ischemia. Neurochem Res 36(10):1840–1849. doi:10.1007/s11064-011-0502-6
Carboni S, Antonsson B, Gaillard P, Gotteland JP, Gillon JY, Vitte PA (2005) Control of death receptor and mitochondrial-dependent apoptosis by c-Jun N-terminal kinase in hippocampal CA1 neurones following global transient ischaemia. J Neurochem 92(5):1054–1060. doi:10.1111/j.1471-4159.2004.02925.x
Kimura H (2014) Production and physiological effects of hydrogen sulfide. Antioxid Redox Signal 20(5):783–793. doi:10.1089/ars.2013.5309
Hardingham GE (2009) Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem Soc Trans 37(Pt 6):1147–1160. doi:10.1042/BST0371147
Papadia S, Hardingham GE (2007) The dichotomy of NMDA receptor signaling. Neuroscientist 13(6):572–579. doi:10.1177/10738584070130060401
Jiang Z, Li C, Manuel ML, Yuan S, Kevil CG, McCarter KD, Lu W, Sun H (2015) Role of hydrogen sulfide in early blood-brain barrier disruption following transient focal cerebral ischemia. PLoS One 10(2), e0117982. doi:10.1371/journal.pone.0117982
Abe K, Kimura H (1996) The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 16(3):1066–1071
Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, Steinberg GK (2005) Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci 25(42):9794–9806. doi:10.1523/JNEUROSCI.3163-05.2005
Shao ZH, Sharp WW, Wojcik KR, Li CQ, Han M, Chang WT, Ramachandran S, Li J et al (2010) Therapeutic hypothermia cardioprotection via Akt- and nitric oxide-mediated attenuation of mitochondrial oxidants. Am J Physiol Heart Circ Physiol 298(6):H2164–H2173. doi:10.1152/ajpheart.00994.2009
Hu B, Friberg H, Wieloch T (2011) Protracted tyrosine phosphorylation of the glutamate receptor subunit NR2 in the rat hippocampus following transient cerebral ischemia is prevented by intra-ischemic hypothermia. Ther Hypothermia Temp Manag 1(3):159–164. doi:10.1089/ther.2011.0013
Markarian GZ, Lee JH, Stein DJ, Hong SC (1996) Mild hypothermia: therapeutic window after experimental cerebral ischemia. Neurosurgery 38(3):542–550, discussion 551
Liu Y, Zhao J, Xiao X, Yin J, Tu C, Ou D, Chen G (2014) P59: the protective effects of exogenous hydrogen sulfide against global cerebral ischemia/reperfusion injury in rats. Nitric Oxide 39:S33–S34
Geng Y, Li E, Mu Q, Zhang Y, Wei X, Li H, Cheng L, Zhang B (2015) Hydrogen sulfide inhalation decreases early blood-brain barrier permeability and brain edema induced by cardiac arrest and resuscitation. J Cereb Blood Flow Metab 35(3):494–500. doi:10.1038/jcbfm.2014.223
Sodha NR, Sellke FW (2015) Attenuation of inflammatory responses by hydrogen sulfide (H(2)S) in ischemia/reperfusion injury. Methods Enzymol 555:127–144. doi:10.1016/bs.mie.2014.11.041
Acknowledgments
This work was supported by funding from the Department of Anesthesiology, Jinling Hospital, School of Medicine, Nanjing University, Nanjing, China.
Conflict of Interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Hai-Bin Dai, Miao-Miao Xu, Jia Lv and Xiang-Jun Ji contributed equally to this work.
Rights and permissions
About this article

Cite this article
Dai, HB., Xu, MM., Lv, J. et al. Mild Hypothermia Combined with Hydrogen Sulfide Treatment During Resuscitation Reduces Hippocampal Neuron Apoptosis Via NR2A, NR2B, and PI3K-Akt Signaling in a Rat Model of Cerebral Ischemia-Reperfusion Injury. Mol Neurobiol 53, 4865–4873 (2016). https://doi.org/10.1007/s12035-015-9391-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9391-z