
Abstract
Rare variants of phospholipase D3 (PLD3) have been identified as Alzheimer’s disease (AD) susceptibility loci, whereas little is known about the potential role of common variants in the progression of AD. To examine the impact of genetic variations in PLD3 on neuroimaging phenotypes in a large non-demented population. A total of 261 normal cognition (NC) and 456 mild cognitive impairment (MCI) individuals from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database are included in our analysis. Multiple linear regression models were applied to examine the association between four single-nucleotide polymorphisms (SNPs; rs7249146, rs4490097, rs12151243, and rs10407447) with the florbetapir retention on florbetapir 18F amyloid positron emission tomography (AV45-PET), the cerebral metabolic rate for glucose (CMRgl) on 18F-fluorodeoxyglucose PET (FDG-PET), and regional volume on magnetic resonance imaging (MRI) at baseline and in the cohort study. We did not detect any significant associations of PLD3 SNPs with florbetapir retention on AV45-PET. In the analysis of FDG-PET, rs10407447 was associated with the CMRgl in the left angular gyrus and bilateral posterior cingulate cortex in the MCI group. Regarding the MRI analysis, rs10407447 was also associated with bilateral inferior lateral ventricle and lateral ventricle volume in MCI group. The main findings of our study provide evidence that support the possible role of PLD3 common variants in influencing AD-related neuroimaging phenotypes. Nevertheless, further work is necessary to explain the functional mechanisms of differences and confirm the causal variants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genetic risk factors have long known to play an important role in the development of Alzheimer’s disease (AD) [1–3]. To date, several genetic variants are known to increase the risk for the late-onset AD including those in the CLU, PICALM, and CR1 genes and most notably the ε4 allele of the APOE gene [4–6]. Recently, rare functional coding variants in phospholipase D3 (PLD3) gene have been identified to be significantly associated with AD risk using whole-exome sequencing method [7]. A single variant rs145999145 (Val232Met) segregates with disease status in two independent families and is validated to increase AD risk in seven independent series of European descent. Subsequently, additional variants in PLD3 are also observed to increase risk for AD through gene-based burden analysis [7]. These evidences strongly support PLD3 as an AD risk gene. In this case, we hypothesize that common variants on commercially available genotyping chips, may also play an important role in AD.
New neuroimaging analysis methods are now being used to clarify the genetics of AD pathology, risk, and variability in human [8]. Unlike the dichotomous clinical diagnosis, neuroimaging phenotypes can capture more of the underlying heritable trait variation and provide a more direct measurement toward characterizing genetic effects on brain structure and function [8]. Thus, genetic analysis of neuroimaging phenotypes may detect novel common variants of PLD3 involved in AD susceptibility.
AD develops following a long pre-clinical phase consisting of mild symptoms, and the pathophysiological processes of AD begin many years prior to the diagnosis of AD [9, 10]. Given that PLD3 is related to AD, it remains unclear whether this gene also plays a role in the preclinical phase of AD. Knowledge on this would provide important confirmation of a role for genetic variations of PLD3 and enhance associations for susceptibility alleles. Additionally, genetic factors might play different roles at different levels of disease severity. In our study, we examined the impact of common variants at the PLD3 loci in normal cognition (NC) and mild cognitive impairment (MCI) population separately, from Alzheimer’s Disease Neuroimaging Initiative (ADNI).
Materials and Methods
Participants
The data used in this article were obtained from the ADNI database. All ADNI studies were carried out in accordance with the Good Clinical Practice guidelines, the Declaration of Helsinki, and the US 21 CFR Part 50 (Protection of Human subjects) and Part 56 (Institutional Review Boards). This study was approved by the Institutional Review Boards of all participating sites at their respective institutions. Written informed consent was obtained from all ADNI participants before the study. ADNI is a longitudinal multisite study launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies, and non-profit organizations with over 50 sites across the USA and Canada (http://adni.loni.usc.edu/). The primary goal of ADNI has been to examine whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. Determination of sensitive and specific markers of very early AD progression is intended to aid researchers and clinicians to develop new treatments and monitor their effectiveness, as well as lessen the time and cost of clinical trials. The initial goal of ADNI was to recruit 800 subjects, but ADNI has been followed by ADNI-GO and ADNI-2. To date, these three protocols have recruited over 1500 adults to participate in the research, consisting of cognitively normal older individuals, individuals with early or late MCI, and individuals with early AD. The follow-up duration of each group is specified in the protocols for ADNI-1, ADNI-2, and ADNI-GO. Subjects originally recruited for ADNI-1 and ADNI-GO had the option to be followed in ADNI-2.
Participants were enrolled based on criteria outlined in the ADNI protocol (http://www.adni-info.org/scientists/adnistudyprocedures.aspx). Participants genotyped in both ADNI-1 and ADNI-2/GO were included. Participants were categorized at baseline as NC, MCI, and AD. More information on diagnostic criteria utilized is available on ADNI website. Here, we restricted the study to NC and MCI subjects whose genotype data of PLD3 single-nucleotide polymorphisms (SNPs; 812 individuals) were available. Population structure was assessed by performing principal component analysis (PCA) to avoid population stratification effects which can lead to spurious genetic associations. We assigned genotype-determined ancestry by comparing ADNI patients and populations form HapMap Phase 3 data. Only individuals clustering with European HapMap samples were retained for analysis. As presented in Supplementary Fig. S1, 48 individuals identified as of non-European ancestry were excluded. The final dataset for the present analysis comprised 713 individuals, including 261 NC and 456 MCI at baseline. Baseline and longitudinal data including structural MRI and PET were collected as parts of this study. Furthermore, all participants underwent a battery of baseline clinical tests including Clinical Dementia Rating Scale Sum of Boxes (CDRSB), the Alzheimer’s Disease Assessment Scale 11 (ADAS11), the Alzheimer’s Disease Assessment Scale 13 (ADAS13), the Mini-Mental State Examination (MMSE), the Rey’ Auditory Verbal Learning Test (RAVLT), and Functional Activities Questionnaire (FAQ).
Genotyping
In our study, we used the results from direct genotyping of SNPs in PLD3 that was conducted as part of the Genome-Wide Association Study (GWAS) of ADNI. The GWAS data was collected from ADNI-1 participants using Illumina Infinium Human610-Quad BeadChip (Illumina, Inc., San Diego, CA), and ADNI-2/GO participants using Illumina Human Omni Express BeadChip. Optimized tag SNP content from all three HapMap phases was strategically selected to capture the greatest amount of common SNP variation [11]. SNP genotypes were generated in Illumina BeadStudio software v3.2 from bead intensity data. The quality control (QC) procedures were performed using PLINK software. We excluded SNPs with the following characteristics: minimum call rates <95 %, minimum minor allele frequencies <0.01, and Hardy–Weinberg equilibrium test p < 1 × 10−6. After QC procedures, four tag SNPs in the intron of PLD3 (rs7249146, rs4490097, rs12151243, rs10407447) remained for data analysis. In addition, we determined the linkage disequilibrium (LD) and detailed other information of the selected SNPs from phase 1 of the 1000 Genomes Project. LD plot containing 22 SNPs for PLD3 was generated from 1000 Genomes genotyping data for the European individuals using Haploview 4.2 software. We also included the previously reported rare variants (rs145999145 and rs4819) in the LD plot for comparison.
Neuroimaging
We used the analyzed data of 18F-AV45-PET and the CMRgl on FDG-PET that had been processed at the Jagust Lab, the University of Berkeley. A detailed description of PET image acquisition and processing can be found at http://adni.loni.usc.edu/data-samples/pet/. For AV45-PET, mean florbetapir uptake within four cortical regions (frontal, angerior/posterior cingulate, lateral parietal, and lateral temporal) was extracted. Cortical standardized uptake value ratios (SUVRs) were calculated by averaging across the four cortical regions and dividing this average by whole cerebellum. Each mean florbetapir uptake of the four main regions and cortical SUVR were used for analysis. The final AV45-PET analyses comprised 619 individuals (220 NC and 399 MCI) with baseline AV45-PET and corresponding genetic data. For longitudinal analysis, we calculated the 2-year changes of AV45-PET in 340 individuals (119 NC and 221 MCI).
The FDG uptake for each subject was determined within a set of pre-defined regions of interest (metaROIs) based on a literature review [12]. PET images were spatially normalized in Statistical Parametric Mapping (SPM) to the MNI PET template. We extracted the mean counts from the five metaROIs (left angular gyrus, right angular gyrus, bilateral posterior cingular, left inferior temporal gyrus, right inferior temporal gyrus) for each subject’s FDG scans at each time point, computing the intensity values with SPM subroutines. Finally, we intensity-normalized each metaROI mean by dividing it by pons/vermis reference region mean. The final FDG-PET analyses comprised 574 individuals (193 NC and 381 MCI) with baseline FDG-PET and corresponding genetic data. We also calculated the 2-year changes of CMRgl on FDG-PET for longitudinal analysis (363 individuals including 127 NC and 236 MCI).
The high-quality data of structural volumetric MRI were available in the ADNI data archive provided by the University of California, San Francisco (UCSF) medical center. Volumes of 119 brain regions were calculated in FreeSurfer (in cubic millimeters) for each subject in the dataset at each time point. In present study, we focused on the most discriminant ROIs that had been found to be strongly associated with AD based on a literature search. These selected regions were the hippocampus, the entorhinal cortex, primary temporal cortex areas, cingulate cortex, amygdala, thalamus, caudate, and lateral ventricle. There were 327 subjects (127 119 NC and 217 208 MCI) with baseline imaging volumes from UCSF and corresponding genetic data included in the MRI analysis. Additionally, it has been demonstrated that atrophy in the hippocampus and entorhinal cortex of the temporal lobes were the earliest sites associated to evidences of AD. Thus, we also calculated 1-year percent volumetric changes of hippocampus, entorhinal cortex, and temporal cortex (including superior temporal gyrus, middle temporal gyrus, inferior temporal gyrus, and temporal pole) using baseline and 1-year imaging data (divide 1-year changes by baseline) for longitudinal analysis (322 individuals, including 116 NC and 206 MCI).
Statistical Analysis
Demographic characteristics of our subjects were presented using means and standard deviations (SDs) for continuous variables and proportions for categorical variables. Demographics and genotypic frequencies were compared using t test or chi-square tests. Each of the four SNPs was examined for associations with the neuroimaging endophenotypes for each diagnostic group separately (NC and MCI). A multiple linear regression model was applied to examine the independent associations and the dosage effect of each minor allele (0 = major allele homozygotes, 1 = heterozygous, 2 = minor allele homozygotes) between SNPs and quantitative phenotypes controlling for the covariates, which were baseline age, gender, years of education, and number of APOE ε4 allele for each diagnostic group separately. We used the IBM SPSS software (version 19.0 for Windows; Chicago, IL, USA) for statistical analyses. We applied correlation matrix analysis to calculate potential association between all phenotypes. As presented in Supplementary Tables S1, S2, and S3, correlation matrix analysis revealed limited association between phenotypes (correlation coefficient range, 0.92 to 0.98 for AV45-PET, 0.6 to 0.85 for FDG-PET, −0.5 to 0.93 for MRI), suggesting that independent analysis was needed for association with genetic variants. All results were reported as nominally significant if a raw P value is <0.05. To account for multiple testing, we performed a Bonferroni correction (P c) for the number of tests applied in each analysis.
Results
Linkage Disequilibrium Analysis
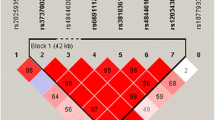
A representation of PLD3 genomic organization and LD patterns, as well as the SNPs evaluated in the present study, is depicted in Fig. 1. SNP rs7249146 was located in the upstream of PLD3 gene, and the other three (rs4490097, rs12151243 and rs10407447) were located in the intron. LD analysis revealed that there were four blocks in the PLD3 gene. The selected SNPs distributed from block 1 to block 3, confirming that these SNPs could capture most of common variants in PLD3. Rs4490097 and rs12151243 were in the same LD block (block 2). Furthermore, we found that these four SNPs were not in LD with the previously reported rare variants (rs145999145 and rs4819) which conferred risk for AD.

PLD3 genomic organization and linkage disequilibrium plot. a PLD3 genomic organization. Exons are presented as gray blocks; linkage disequilibrium blocks are indicated by dotted box. The SNPs analyzed in the present study and the previously reported SNPs are indicated by arrows according to their genomic position. The reported SNPs are also indicated by dotted box. b Linkage disequilibrium plot. The SNPs analyzed in the present study are indicated by black box; the reported SNPs are indicated by dotted box
Demographic and Clinical Characteristics
Subject demographics, cognitive status, and SNP distributions of NC and MCI groups are summarized in Table 1. The MCI individuals were significantly younger than NC (p < 0.01). Gender and education did not vary significantly between the different groups. As expected, frequency of the APOE ε4 allele was significantly higher in MCI patients than NC. CDRSB (P < 0.01), ADAS11 (P < 0.01), ADAS13 (P < 0.01), MMSE (P < 0.01), RAVLT (P < 0.01), and FAQ (P < 0.01) score differed significantly by diagnostic group with better performance in the expected direction of NC > MCI.
The MCI group had hippocampal volumes that were significantly smaller as compared to those of the NC group (P < 0.01). Furthermore, no significant differences in the frequency of four SNPs in the MCI individuals were detected compared with NC in this study.
Impacts of PLD3 Genetic Variants on AV45-PET and FDG-PET
In the analysis of AV45-PET, no SNPs showed significant effects on florbetapir retention in the four main regions (frontal, angerior/posterior cingulate, lateral parietal, lateral temporal), as well as the cortical SUVR (Supplementary Table S4). Likewise, we did not detect any significant associations on the changes over 2 years of these regions in the NC and MCI groups.
Regarding the FDG-PET analyses, only rs10407447 was associated with the CMRgl on FDG-PET in the left angular gyrus and bilateral posterior cingulate cortex in the MCI group (P = 0.042 and P = 0.028, respectively) (Supplementary Table S5). The differences in these traits among individuals with different genotypes are presented in Fig. 2. The minor G allele carriers of rs10407447 had higher CMRgl in both the left angular gyrus and bilateral posterior cingulate cortex, with the dosage effect of G allele (AA < GA < GG). However, no association remained statistically significant after Bonferroni correction. We did not detect any significant association for all SNPs on the CMRgl percent changes over 2 years (Supplementary Table S6).

The association of rs10407447 with baseline CMRgl on FDG-PET in the MCI group. a The association of rs10407447 with CMRgl in the left angular gyrus in the MCI group. b The association of rs10407447 with CMRgl in bilateral posterior cingular in the MCI group. FDG-PET, 18F-fluorodeoxyglucose positron emission tomography; MCI, mild cognitive impairment; CMRgl, cerebral metabolic rate for glucose
Impacts of PLD3 Genetic Variants on Regional Volume
A number of regional volumes, including lateral ventricle, inferior lateral ventricle, and caudate, were identified to have strong associations with two SNPs (rs10407447 and rs12151243), independent of APOE, in the structural MRI analyses (Supplementary Tables S5 and S7). There was a significant association between rs10407447 and baseline right and left lateral ventricle volumes in the MCI group (P c = 0.032 and P c = 0.048, respectively, Fig. 3). Rs10407447 minor G allele carriers had smaller lateral ventricle volume than A allele homozygotes subjects in the MCI group (AA > GA > GG). In addition, rs10407447 was also significantly associated with baseline right and left inferior lateral ventricle volume in MCI group (P c = 0.004 and P c = 0.02, respectively, Fig. 4), with minor G allele carriers showing smaller inferior lateral ventricle volume (AA > GA > GG). We also detected an association between rs10407447 and right hippocampus volume in the MCI group (P = 0.023). However, this finding was not significant after the Bonferroni correction (P c = 0.092).

The association of rs10407447 with baseline lateral ventricle volume in the MCI group. a The association of rs10407447 with right lateral ventricle volume in the MCI group. b The association of rs10407447 with left lateral ventricle volume in the MCI group. MCI, mild cognitive impairment

The association of rs10407447 with baseline inferior lateral ventricle volume in the MCI group. a The association of rs10407447 with right inferior lateral ventricle volume in the MCI group. b The association of rs10407447 with left inferior lateral ventricle volume in the MCI group. MCI, mild cognitive impairment
Both the right (P = 0.018) and left (P = 0.014) caudate volumes were found to be nominally associated with rs12151243 in the NC group (Fig. 5). However, no association remained statistically significant after Bonferroni correction. A trend for a relationship was observed in the left caudate volumes, which did not reach statistical significance (P c = 0.056). The minor T allele carriers showed significant smaller caudate volume with the dosage effect of T allele (GG < TG < TT). We did not detect any significant association for all SNPs on the regional volume percent changes over 1 year from the longitudinal analysis (Supplementary Table S7).

The association of rs12151243 with baseline caudate volume in the NC group. a The association of rs10407447 with right caudate volume in the NC group. b The association of rs10407447 with left caudate volume in the NC group. NC, normal cognition
Discussion
PLD3, also known as Hu-K4, is located at chromosome 19q13.2 and encoded the PLD3 protein which is highly expressed in neurons [13]. Some rare variants of PLD3 have been identified as AD susceptibility loci, whereas little is known about the potential role of common variants in the progression of AD. To our knowledge, this is the first study which finds effects of common variations at the PLD3 locus on neuroimaging phenotypes in non-demented elderly subjects.
Cerebral amyloid deposition, as caused by altered processing of amyloid precursor protein (APP), is considered to be a primary etiologic factor in AD. AV45-PET imaging is correlated with the presence and deposition of Aβ and has been in wide use as a research biomarker of AD [14]. More remarkably, a recent study found that PLD3 appeared to be involved in the processing of APP independently [7]. Therefore, genetic variations of PLD3 might influence the cortical amyloid deposition by modifying the expression of PLD3. In our study, no significant effect was detected for all tested SNPs on florbetapir retention. Nevertheless, these negative results also needed additional validation in larger population samples.
To date, a large body of studies have investigated the effect of APOE ε4 genotype on CMRgl and found significant differences in many regions in APOE ε4 carriers compared to non-carriers [15–17]. From the FDG-PET analysis, our results showed an effect of rs10407447 on CMRgl, independent of APOE ε4 status, in angular gyrus and posterior cingulum of the MCI individuals. The minor G allele carriers of rs10407447 had higher CMRgl relative to non-carriers, and a linear trend in mean CMRgl levels was observed. It should be noted that both angular gyrus and posterior cingulate cortex are typical areas of hypometabolism in early AD [18]. Angular gyrus, within the inferior portion of the posterior parietal lobe, plays important roles in the allocation of visuospatial attention [19]. Posterior cingulate cortex has been shown to play a major role in memory function, and the associations of the posterior cingular with medial temporal lobe (MTL) have been demonstrated in previous studies [20, 21]. However, we just found a nominal impact of PLD3 on CMRgl based on available participants, and the negative results after multiple testing corrections indeed limited the conclusions we can draw. Even so, these results still indicated that rs10407447 may be critical in the CMRgl of angular gyrus and bilateral posterior cingulate cortex and needed to be validated in studies with a larger sample size.
In regional volume analysis, rs10407447 also correlated with bilateral inferior lateral ventricle and lateral ventricle volume, which had been proved to be affected by atrophy process in subjects with AD and MCI [22, 23]. Progressive lateral ventricular expansion could reflect atrophy of the surrounding gray and white matter [24, 25]. Previous studies had indicated that PLD3 was expressed at higher levels in brain, especially in neurons, than in non-nervous tissues [26]. In the brain, PLD3 was also observed to be involved in processes associated with cell innervation, neurotransmission, and neuronal survival. PLD3 expression appeared to coincide with late neuronal development in the hippocampus and the primary somatosensory cortex [26]. Although the common variant rs10407447 is located in the intron of PLD3 gene, this SNP might be in same LD block with functional variants and have the power to affect the function or expression of PLD3 and then affect the neuronal survival and atrophy progression. Of note, rs10407447 was not in LD with the reported risk SNP (rs145999145 and rs4819), indicating that its role cannot be explained by the original rs145999145 or rs4819 association.
Previous MRI-based studies had repeatedly proved that hippocampal atrophy was a recognized biological marker of AD [24, 27]. We also provided weak evidence for the association of rs10407447 and hippocampal volume in MCI individuals. Considering the prior evidence of its involvement in CMRgl, rs10407447 seemed to be a highly associated candidate locus, especially in individuals with MCI. MCI was the most commonly accepted prodromal AD stage. Therefore, our findings provided valuable evidence that rs10407447 may be an important mediator of glucose metabolism and structure especially in these regions in the early stage of AD.
In addition, we identified an effect of one SNP (rs12151243) on bilateral caudate volume in NC individuals. The caudate volume was a highly heritable brain structure, so it was a reasonable point for investigating genetic influences on brain structure [28]. Remarkably, caudate had been found implicated in subjects with AD and was strongly correlated with the formation of new associations to acquire explicit memories and motor learning [29]. Nevertheless, none of these associations survived the multiple testing corrections. It may be due to low-power, small sample sizes of the NC group, further suggesting that it is important to validate the effects of this SNP on these regions.
In our study, we also analyzed changes over 1 or 2 years on florbetapir retention, CMRgl, and regional volume. However, we did not find any significant results for longitudinal analysis. The small number of participants in the longitudinal cohort was especially problematic for the negative results. Another possible explanation could be that the impact of genetic variations might not be detected with a relatively short follow-up. Therefore, future analyses in the growing ADNI cohort, with greater numbers and longer follow-up of longitudinal samples, will allow us to evaluate the effects of PLD3 variations.
Given the marginal P value after correction, additional replication of our findings with lager samples was necessary. However, no available independent population for replication limited the conclusions we can draw. Moreover, some variants may be not covered by genotyping arrays of ADNI. The selected SNPs in our study just distributed from block 1 to block 3 in the PLD3 gene, and none of them covered the fourth LD block; thus, the PLD3 analysis was also limited by the unavailability of more SNPs in ADNI dataset. Batch effects which can be detected by principal components analysis are common for genotyping arrays. However, we cannot acquire sufficient information (e.g., batches for each sample) from the ADNI dataset to evaluate batch effects in our study. Another limitation of our study was that we just provided evidence for association of genetic variants in PLD3, but we were unable to know the mechanistic means by which they may change expression levels or protein structures or how they might affect phenotypes. Unfortunately, no functional and expression data were available in ADNI dataset. Future studies incorporating a detailed expression of PLD3 would be more advantageous.
In summary, the main finding of the current study provide here, for the first time, evidence supporting the possible role of PLD3 common variants in influencing AD-related neuroimaging phenotypes. Therefore, these candidate loci can now serve as a target of study in future large replication samples. Nevertheless, further work is necessary to explain the functional mechanisms of differences and confirm the causal variants.
References
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL (2006) Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 63(2):168–174. doi:10.1001/archpsyc.63.2.168
Jiang T, Yu JT, Tian Y, Tan L (2013) Epidemiology and etiology of Alzheimer’s disease: from genetic to non-genetic factors. Curr Alzheimer Res 10(8):852–867
Jiang T, Yu JT, Tan L (2012) Novel disease-modifying therapies for Alzheimer’s disease. J Alzheimer’s Dis : JAD 31(3):475–492. doi:10.3233/JAD-2012-120640
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V et al (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41(10):1088–1093. doi:10.1038/ng.440
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D et al (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41(10):1094–1099. doi:10.1038/ng.439
Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF et al (1993) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43(8):1467–1472
Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, Harari O, Norton J et al (2014) Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 505(7484):550–554. doi:10.1038/nature12825
Mattay VS, Goldberg TE, Sambataro F, Weinberger DR (2008) Neurobiology of cognitive aging: insights from imaging genetics. Biol Psychol 79(1):9–22. doi:10.1016/j.biopsycho.2008.03.015
Salmon DP, Lange KL (2001) Cognitive screening and neuropsychological assessment in early Alzheimer’s disease. Clin Geriatr Med 17(2):229–254
Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, Wilson RS (2006) Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66(12):1837–1844. doi:10.1212/01.wnl.0000219668.47116.e6
Saykin AJ, Shen L, Foroud TM, Potkin SG, Swaminathan S, Kim S, Risacher SL, Nho K et al (2010) Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: Genetics core aims, progress, and plans. Alzheimers Dement : J Alzheimers Assoc 6(3):265–273. doi:10.1016/j.jalz.2010.03.013
Landau SM, Harvey D, Madison CM, Koeppe RA, Reiman EM, Foster NL, Weiner MW, Jagust WJ (2011) Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging 32(7):1207–1218. doi:10.1016/j.neurobiolaging.2009.07.002
Wang J, Yu JT, Tan L (2014) PLD3 in Alzheimer’s Disease. Mol Neurobiol. doi:10.1007/s12035-014-8779-5
Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, Pontecorvo MJ, Hefti F et al (2011) Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA 305(3):275–283. doi:10.1001/jama.2010.2008
Small GW, Mazziotta JC, Collins MT, Baxter LR, Phelps ME, Mandelkern MA, Kaplan A, La Rue A et al (1995) Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 273(12):942–947
Jagust WJ, Landau SM (2012) Apolipoprotein E, not fibrillar beta-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci : Off J Soc Neurosci 32(50):18227–18233. doi:10.1523/JNEUROSCI.3266-12.2012
Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J (2004) Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci U S A 101(1):284–289. doi:10.1073/pnas.2635903100
Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE (1997) Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann Neurol 42(1):85–94. doi:10.1002/ana.410420114
Cattaneo Z, Silvanto J, Pascual-Leone A, Battelli L (2009) The role of the angular gyrus in the modulation of visuospatial attention by the mental number line. NeuroImage 44(2):563–568. doi:10.1016/j.neuroimage.2008.09.003
Johnson SC, Christian BT, Okonkwo OC, Oh JM, Harding S, Xu G, Hillmer AT, Wooten DW et al (2014) Amyloid burden and neural function in people at risk for Alzheimer’s disease. Neurobiol Aging 35(3):576–584. doi:10.1016/j.neurobiolaging.2013.09.028
Wu TC, Wilde EA, Bigler ED, Yallampalli R, McCauley SR, Troyanskaya M, Chu Z, Li X et al (2010) Evaluating the relationship between memory functioning and cingulum bundles in acute mild traumatic brain injury using diffusion tensor imaging. J Neurotrauma 27(2):303–307. doi:10.1089/neu.2009.1110
Chou YY, Lepore N, de Zubicaray GI, Carmichael OT, Becker JT, Toga AW, Thompson PM (2008) Automated ventricular mapping with multi-atlas fluid image alignment reveals genetic effects in Alzheimer’s disease. NeuroImage 40(2):615–630. doi:10.1016/j.neuroimage.2007.11.047
Nestor SM, Rupsingh R, Borrie M, Smith M, Accomazzi V, Wells JL, Fogarty J, Bartha R (2008) Ventricular enlargement as a possible measure of Alzheimer’s disease progression validated using the Alzheimer’s disease neuroimaging initiative database. Brain : J Neurol 131(Pt 9):2443–2454. doi:10.1093/brain/awn146
Ridha BH, Barnes J, Bartlett JW, Godbolt A, Pepple T, Rossor MN, Fox NC (2006) Tracking atrophy progression in familial Alzheimer’s disease: a serial MRI study. Lancet Neurol 5(10):828–834. doi:10.1016/S1474-4422(06)70550-6
Qiu A, Fennema-Notestine C, Dale AM, Miller MI (2009) Regional shape abnormalities in mild cognitive impairment and Alzheimer’s disease. NeuroImage 45(3):656–661
Pedersen KM, Finsen B, Celis JE, Jensen NA (1998) Expression of a novel murine phospholipase D homolog coincides with late neuronal development in the forebrain. J Biol Chem 273(47):31494–31504
Thompson PM, Hayashi KM, de Zubicaray G, Janke AL, Rose SE, Semple J, Herman D, Hong MS et al (2003) Dynamics of gray matter loss in Alzheimer’s disease. J Neurosci : Off J Soc Neurosci 23(3):994–1005
Kremen WS, Prom-Wormley E, Panizzon MS, Eyler LT, Fischl B, Neale MC, Franz CE, Lyons MJ et al (2010) Genetic and environmental influences on the size of specific brain regions in midlife: the VETSA MRI study. NeuroImage 49(2):1213–1223. doi:10.1016/j.neuroimage.2009.09.043
Madsen SK, Ho AJ, Hua X, Saharan PS, Toga AW, Jack CR Jr, Weiner MW, Thompson PM (2010) 3D maps localize caudate nucleus atrophy in 400 Alzheimer’s disease, mild cognitive impairment, and healthy elderly subjects. Neurobiol Aging 31(8):1312–1325. doi:10.1016/j.neurobiolaging.2010.05.002
Acknowledgments
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). This research was also supported by National Natural Science Foundation of China (81171209, 81371406, 81000544), the Shandong Provincial Outstanding Medical Academic Professional Program, and the Qingdao Key Health Discipline Development Fund.
Conflict of interest
The authors declare that they have no competing interests
Author information
Authors and Affiliations
Consortia
Corresponding authors
Additional information
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 1184 kb)
Rights and permissions
About this article

Cite this article
Wang, C., Wang, HF., Tan, MS. et al. Impact of Common Variations in PLD3 on Neuroimaging Phenotypes in Non-demented Elders. Mol Neurobiol 53, 4343–4351 (2016). https://doi.org/10.1007/s12035-015-9370-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9370-4