
Abstract
Paraquat (PQ) as a Parkinsonian mimetic has been demonstrated to impair dopaminergic (DAergic) neurons and is highly correlated with the etiology of Parkinson’s disease (PD) where the death of DAergic neurons has been mainly attributed to impaired mitochondrial functioning. In this study, PQ-induced cytotoxicity focusing on mitochondrial membrane permeability (MMP), which has been implicated to play a part in neurodegeneration, was investigated. Primarily, PQ-induced cytotoxicity and reactive oxygen species (ROS) were inhibited by an inhibitor of NADPH oxidase (NOX), indicating the toxic effect of PQ redox cycling. Further, dibucaine and cyclosporin A which respectively inhibit mitochondrial apoptosis-induced channels (MAC) and mitochondrial permeability transition pores (mPTP) were used and found to prevent PQ-induced mitochondrial dysfunction, such as decreased mitochondrial membrane potential and increased MMP, mitochondrial ROS, and pro-apoptotic factor release. Knockdown of bax and/or bak blocked PQ-induced mitochondrial clusterization of Bax and/or Bak and cytotoxicity, demonstrating the significance of MAC which is composed of Bax and/or Bak. This clusterization coincided with the release of mitochondrial apoptotic factors before there was an increase in inner MMP, indicating that MAC may precede mPTP formation. Besides, NOX inhibitor but not dibucaine attenuated the earlier PQ-induced cytosolic ROS formation or Bax and/or Bak clusterization indicating PQ redox cycling may account for MAC formation. In this model, we have resolved for the first that PQ cytotoxicity through redox cycling may sequentially result in increased outer (MAC) and inner (mPTP) MMP and suggested MMP could be implicated as a therapeutic target in treating neurodegenerative diseases like PD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Paraquat (1,1′-dimethyl-4,4′- bipyridinium, PQ) has been used as an herbicide worldwide for more than 50 years. Epidemiological studies have indicated a strong correlation between the incidence of Parkinson’s disease (PD) and the level of paraquat exposure [1, 2]. Systemic treatment of rodents with paraquat induces selective dopaminergic neuronal loss and intracellular α-synuclein deposits in the substantia nigra [3, 4], compatible with PD. Other studies have shown that paraquat induces cytotoxicity or cell death in vitro [5, 6], providing a competent cellular model for studying the molecular mechanisms of PD. However, the toxic mechanisms of PQ-induced cytotoxicity are not determined [7, 8]. Although PQ has been shown to inhibit mitochondrial complex I and III activities [9, 10], no direct interaction of PQ with complex I has also been reported [11], indicating an undefined role of PQ targeting on mitochondrial functioning.
Mitochondria have been recognized for their multifunctional roles in metabolism, energy transduction, ion transport, and cell death [12] and are regarded as a therapeutic target for neurodegenerative diseases [13] such as PD. The permeability increase of their membranes has been associated to the release of apoptotic factors and subsequent cell death [14]. Interestingly, the releases of these factors occurs with different kinetics [15], suggesting that different kinetic and mechanical paths are required for permeabilization. Currently, two candidate pathways have been proposed [16], the mitochondrial permeability transition pore (mPTP) in the inner membrane and the mitochondrial apoptosis-induced channel (MAC) in the outer membrane [12]. Both of these channels are critical for programmed cell death.
mPTP is defined as a rapid increase in the nonselective permeability to solutes of molecular mass ≦1.5 kDa across inner mitochondrial membranes [17, 18]. Although a supramolecular complex formed by the voltage-dependent anion channel (VDAC), adenine nucleotide translocase (ANT), peripheral benzodiazepine receptor (PBR), creatine kinase (CK), hexokinase (HK), and/or cyclophilin D (CypD), which can be inhibited by cyclosporin A, have been proposed [19], the molecular structure and identity of the mPTP is not yet determined and remains controversial [20]. Recently, gene inactivation studies have shown that most of the proteins proposed to take part in mPTP formation are not necessary for its occurrence, except for CypD [21, 22]. Therefore, other models have also been proposed such as F0F1 ATP synthase [23] or phosphate carrier [24] as alternative components in the formation of mPTP.
MAC is a high-conductance channel with an estimated mean diameter of >5 nm, large enough to allow the release of the ∼3 nm diameter cytochrome c (Cyto. C) from mitochondria [25, 26]. MAC activity occurs early in the intrinsic apoptotic pathway at about the time that Cyto. C is released [27] or corresponds to a late apoptotic event [28]. Although Bax and/or Bak are presumably required for MAC formation because double knockouts of these proteins fail to display mitochondrial MAC activity or the release of Cyto. C [27], the exact molecular composition of this channel remains undetermined. Besides, the role and significance of MAC in mediating PQ-induced pathology are unknown.
On the other hand, PQ as a divalent cation (PQ2+) undergoes redox cycling, being reduced by diaphorases, such as NADPH oxidase (NOX), by oxidizing an electron donor such as NAD(P)H to yield the monovalent cation PQ+ before being oxidized by an electron receptor such as dioxygen to produce superoxide anions [29]. NOX inhibitor prevents PQ-induced reactive oxygen species (ROS) formation and microglial [30] and DAergic [31] cell death, indicating the lethal redox cycling of PQ [32]. However, the significance of NOX in PC12 cells during PQ intoxication is not known.
Taken together, these articles indicate that unconfirmed molecular entities are not only significant in regulating mitochondrial membrane permeability (MMP) but also in the unresolved mechanisms of PQ impairment of mitochondrial functioning. Therefore, since our previous study showed that PQ can induce endoplasmic reticulum stress and mitochondrial dysfunction [33], in this study, we examined PQ-induced cytotoxicity focusing on mitochondrial dysfunction and permeability.
Results
Cyclosporin A and Dibucaine Prevented PQ-Induced Apoptosis in PC12 Cells
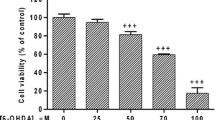
PQ dose-dependently induced significant cell death as measured by neutral red assay (Fig. 1a). The EC50 of PQ is 1.2 mM (Supplement 1); however, for simplification of manipulation and consistency with our previous findings [33], PQ at 1 mM was used for the below-described experiments. The toxicity of PQ at 1 mM was also confirmed by MTT and trypan blue exclusion assays (Fig. 1a). Cyclosporin A and dibucaine both significantly attenuated PQ-induced cell death as estimated by either MTT or neutral red assays (Fig. 1b). Cyclosporin A and dibucaine at 15 μM both tended to give the best protection against PQ-induced apoptosis and were adopted in this study. Cyclosporin A and dibucaine both blocked the PQ-induced cleaving of caspase 3 and PARP (Fig. 1c) and suppressed PQ-induced increases in the fluorescence of Annexin V-FITC (Fig. 1d, upper and lower panel). Cyclosporin A (1 μM) and dibucaine (3 μM) combined tended to prevent PQ-induced cell death synergistically (Fig. 1e).

Cyclosporin A and dibucaine both attenuate PQ-induced apoptosis in PC12 cells. a After treatment with different doses of PQ for 24 h, cells were subjected to MTT, neutral red, or trypan blue exclusion assay. Viability is expressed as a percentage of controls. Data is presented as the mean ± SD. *p < 0.05, compared to controls (n = 3∼6). b After pretreatment with different doses of cyclosporin A or dibucaine for 1 h combined with or without the addition of 1 mM PQ for another 24 h, cells were subjected to MTT or neutral red assay. Cell viability is expressed as a percentage of the results of the assay measured in the control group. Data are presented as the mean ± SD. *p < 0.05, compared to controls (n = 3∼6). # p < 0.05, compared to its PQ-only treated group (n = 3 ∼ 6). c Cells with indicated treatments were harvested and analyzed by Western blotting. d With or without PQ treatments for 24 h, Annexin V-FITC-stained cells were subjected to either imaging or flow cytometric analysis. The bar represents 50 μm. FITC fluorescence intensity was quantified using the FL-1 channel and are expressed as a percentage of controls. Data are presented as the mean ± SD. *p < 0.05, compared to controls (n = 3 ∼ 6). e After pretreatment with the indicated dosages of cyclosporin A and/or dibucaine for 1 h and followed with or without the addition of 1 mM PQ for another 24 h, cells were subjected to neutral red assay. Cell viability is expressed as a percentage of controls. Data are presented as the mean ± SD. *p < 0.05, compared to the PQ-only treated group (n = 6∼12). # p < 0.05, compared to cyclosporin A (1 μM) and PQ-treated groups (n = 3∼6). These data represent one out of three independent experiments that gave similar results
Cyclosporin A and dibucaine prevented PQ-induced decreased mitochondrial membrane potential (∆Ψ), increased mPTP and cytosolic and mitochondrial ROS, and released mitochondrial pro-apoptotic factors
PQ time-dependently resulted in decreased TMRE and increased MitoSOX, HE, and CO3-DCF fluorescence (Fig. 2a, upper panel). Among them, TMRE and MitoSOX fluorescence was significantly decreased and increased, respectively, after 6 h of PQ treatment (Fig. 2a, lower panel). The fluorescence of HE and CO3-DCF were significantly increased after 3 h of PQ treatment (Fig. 2a, lower panel). Cyclosporin A significantly attenuated PQ-induced H2O2 release from the mitochondrial fraction, which was extracted from either naïve or PQ-pretreated cells (Fig. 2b). However, the inhibitory effects of dibucaine on PQ-induced mitochondrial H2O2 release could only be found in PQ-pretreated cells (Fig. 2b). Cyclosporin A and dibucaine significantly blocked PQ-induced decreased fluorescence intensities of TMRE (Fig. 2c and Supplement 2a) and calcein (Fig. 2d and Supplement 2b) and increased fluorescence intensities of MitoSox (Fig. 2e and Supplement 2c), HE, and CO3-DCF (Fig. 2f and Supplement 2d). Besides, cyclosporin A and dibucaine also significantly blocked the PQ-induced releases of mitochondrial HtrA2/Omi and Smac/Diablo (Fig. 3a, b) but not apoptosis-inducing factor (AIF) and endonuclease G (Endo G) (Supplement 3a).

Cyclosporin A and dibucaine prevent PQ-induced increased ROS, mitochondrial dysfunction, and permeability. a Cells treated with PQ for different intervals were stained with TMRE, MitoSOX, DHE, or CO3-DCFDA. Cells were then subjected to fluorescence image acquisition or flow cytometry analysis. The bar represents 50 μm. Mean fluorescence intensity was quantified using the FL-1 or FL-2 channel and are expressed as a percentage of controls. Data are presented as the mean ± SD. *p < 0.05, compared to its respective control group (n = 3∼6). b Mitochondrial fractions extracted from cells pretreated with or without PQ for 24 h were used for H2O2 formation assay. After pretreating with cyclosporin A, dibucaine, or catalase for 10 min, PQ and the Amplex® Red reagent/substrate were added to mitochondrial fractions. Fluorescence intensity was expressed as a percentage of control group levels. c After pretreatment with or without cyclosporin A or dibucaine for 1 h, PQ was added for another 24 h. Cells were then stained with TMRE and subjected to flow cytometry. TMRE fluorescence is expressed as a percentage of controls. Data are presented as the mean ± SD. *p < 0.05, compared to controls (n = 3∼6). # p < 0.05, compared to its PQ-treated group (n = 3∼6). d Before experiments, calcein was stained for 1 day. After pretreatment with or without cyclosporin A or dibucaine for 1 h, PQ was added for another 24 h. Cells were subjected to flow cytometry. Calcein fluorescence is expressed as a percentage of controls. e After pretreatment with or without cyclosporin A or dibucaine for 1 h, PQ was added for another 24 h. Cells were then stained with MitoSOX and subjected to flow cytometry. MitoSOX fluorescence is expressed as a percentage of controls. f Cells pretreated with cyclosporin A or dibucaine for 1 h were treated with PQ for another 24 h. After staining with DHE and CO3-DCFDA, cells were then subjected to fluorescence image acquisition or flow cytometry analysis. Mean fluorescence intensity was quantified using the FL-1 or FL-2 channel and are expressed as a percentage of controls. Data are presented as the mean ± SD. *p < 0.05, compared to controls (n = 3∼6). # p < 0.05, compared to its respective PQ-treated group (n = 3∼6). These data represent one out of three independent experiments that gave similar results

Cyclosporin A and dibucaine prevent the PQ-induced releases of mitochondrial pro-apoptotic factors. a Cells growing on cover slides were pretreated with or without cyclosporin A or dibucaine for 1 h, followed by PQ for another 24 h and subsequently subjected for immunocytochemical analysis. The bar represents 5 μm. b After pretreatment with or without cyclosporin A or dibucaine for 1 h, PQ was added to cells for another 24 h. Cell fractions were collected and subjected to Western blot analysis. These data represent one out of three independent experiments that gave similar results
Diphenylene Iodonium Attenuated PQ-Induced Cytotoxicity
Diphenylene iodonium (DPI) (an inhibitor of NOX) and calpeptin (an inhibitor of calpain which can be activated by calcium overload) significantly attenuated PQ-induced cell death (Fig. 4a, upper panel) and cleaved-caspase 3 and -PARP formations (Fig. 4a, middle panel). U74389G and other known antioxidants, such as MCI-186 and N-acetyl cysteine (NAC), were also protective against PQ-induced cell death (Supplement 4a). DPI also attenuated PQ-induced H2O2 formation from the mitochondrial fraction (Fig. 4a, lower panel). Cyclosporin A and dibucaine failed to inhibit the increased HE fluorescence after PQ treatment for 3 and 6 h (Fig. 4b and Supplement 5a). DPI and U74389G prevented an increase in HE fluorescence after PQ treatment for 3 h; however, it was not U74389G but DPI that attenuated the increase of HE fluorescence after PQ treatment for 6 h (Fig. 4b and Supplement 5a). On the other hand, cyclosporin A, dibucaine, and U74389G significantly prevented PQ-induced increased MitoSOX fluorescence at the sixth hour (Fig. 4c, modified from Supplement 5b). However, DPI only partially attenuated the PQ-induced increase in MitoSOX fluorescence at the sixth hour (Fig. 4c). Instead, cyclosporin A, dibucaine, DPI, and U74389G prevented the PQ-induced decreased calcein fluorescence (Fig. 4c, modified from Supplement 5c). Cyclosporin A, dibucaine, and DPI attenuated the releases of mitochondrial Cyto. C, HtrA2/Omi, and Smac/Diablo after PQ treatment for 24 h (Fig. 4d and Supplement 5d).

DPI attenuates PQ-induced cytotoxicity. a After pretreatment with the indicated reagents for 1 h, PQ was added or not to cells for another 24 h and then subjected to neutral red assays (upper panel). Viability is expressed as a percentage of controls. Data are presented as the mean ± SD. *p < 0.05, compared to the PQ-treated group (n = 3∼6). Cells pretreated with DPI (1 μM) or calpeptin (20 μM) for 1 h were treated with or without PQ for 24 h and then subjected to Western blot analysis (middle panel). The mitochondrial fraction was pretreated with or without DPI and with or without PQ and then with Amplex® Red reagent/substrate. Fluorescence intensity was measured and expressed as a percentage of control group levels (lower panel). Data are presented as the mean ± SD. *p < 0.05, compared to controls (n = 3∼6). # p < 0.05, compared to the PQ-treated group (n = 3∼6). b Cells pretreated with the indicated reagents were treated with or without PQ for different intervals. Cells were stained with HE and then subjected to flow cytometry or imaging. c After pre-staining with or without calcein/CoCl2 for 1 day, PQ was added or not to cells pretreated with the indicated reagents and left to stand for 6 h. The calcein/CoCl2-pre-stained cells were then subjected to flow cytometry or imaging. Cells without calcein/CoCl2 staining were stained with HE and subjected to flow cytometry or imaging. Mean fluorescence intensity was quantified using FL-1 or FL-2 channels and is expressed as a percentage of control group levels. Data are presented as the mean ± SD. *p < 0.05, compared to controls (n = 3∼6). # p < 0.05, compared to its respective PQ-only treated group (n = 3∼6). The bar represents 50 μm. d After co-transfecting with pCytochrome c-EGFP and pDsRed-Mito, cells were pretreated with the indicated reagents and treated with PQ for 24 h. Cells were then fixed, stained (DAPI), and subjected to confocal imaging. The bar represents 5 μm. These data represent one out of three independent experiments that gave similar results
PQ-Induced Mitochondrial Translocation of Bax and Formations of Bax- and Bak-Oligomers and Bax/Bak-Heteromers
PQ but not cyclosporin A and dibucaine time-dependently elevated the expression of Bax instead of Bak (Fig. 5a, upper panel and Supplement 6a). PQ time-dependently increased the formation of Bax- and Bak-oligomers and Bax/Bak-heteromers (Fig. 5a, lower panel); however, pretreatment with cyclosporin A and dibucaine failed to prevent these phenomena (Fig. 5b). Regardless of the presence or absence of cyclosporin A and dibucaine, PQ treatment resulted in Bax mitochondrial translocation (Fig. 5c and Supplement 6b). Normally, at the same exposure time, the signal of Bak on the cytosolic fraction was always absent. Besides, starting from the third hour, PQ resulted in the earlier release of pro-apoptotic factors (Supplement 6b, c). In pDsRed-Mito/pGFP-Bax- and pDsRed-Mito/pGFP-Bak-co-transfected cells, PQ gradually induced Bax (Fig. 5d, left panel) and Bak (Fig. 5d, right panel) aggregates on mitochondria. Cyclosporin A or dibucaine failed to reverse these phenomena (Fig. 5d). In pGFP/pDsRed-Mito-co-transfected cells, PQ failed to alter the translocation of GFP (Supplement 6d).


PQ alters Bax and Bak distributions. a After the indicated treatments, cells were harvested and subjected to Western blot analysis (upper panel). The optical density of the bands were quantified by ImageJ relative to actin and normalized to the levels in control group which was set as 1.0. Cells treated with PQ for different intervals were fixed for the immunocytochemistry assay (lower panel). The bar represents 5 μm. b After cyclosporin A or dibucaine pretreatment for 1 h, cells were treated with PQ for another 24 h and then fixed for the immunocytochemistry assay. The bar represents 5 μm. c After cyclosporin A or dibucaine pretreatment for 1 h, cells were treated with PQ for another 24 h. Cells were then harvested, fractionated, and subjected to Western blot analysis. After co-transfections (1:1) with pEGFP-Bax/pDsRed-Mito (c, left panel) and pEGFP-Bak/pDsRed-Mito (d, right panel) for 24 h, cells with or without cyclosporin A or dibucaine treatment were treated with PQ at the indicated intervals and then fixed for confocal analysis. The bar represents 5 μm. These data represent one out of three independent experiments that gave similar results
Bax and Bak Gene Silencing Prevented PQ-Induced Cell Death
Bax and Bak gene silencing (Fig. 6a, upper panel) promoted cell viability after PQ treatment (Fig. 6a, lower panel) and the protective effects of cyclosporin A and dibucaine in preventing PQ-induced cell death were equivalent in naïve and Bax-, Bak-, and Bax/Bak-silenced cells (Fig. 6a, lower panel). Further, PQ induced less cell death in PC12-shRNA-Bax-1, -shRNA-Bak-1, and -shRNA-Bax/Bak-3 permanent cell lines (Fig. 6b, upper panel and Supplement 7a) in comparison to naïve PC12 cells (Fig. 6b, lower panel). Similarly, cyclosporin A and dibucaine exerted equivalent protection among these cell lines (Fig. 6b, lower panel).

Silencing of bax and bak prevents PQ-induced cell death. a After transfection of non-targeting (NT) siRNA or siRNAs targeting bax and/or bak for 48 h, cells were subjected to Western blot analysis or MTT assay. In MTT assays, cells with or without gene silencing were pretreated with or without cyclosporin A or dibucaine for 1 h, and then PQ was added for another 24 h. Cell viability is expressed as a percentage of controls. Data were presented as the mean ± SD. *p < 0.05, compared to the PQ-only treated group (n = 3∼6). # p < 0.05, compared to controls (n = 3–6). b Cells were transfected with pshRNA-Bax-RFP and/or pshRNA-Bax-GFP, and appropriate antibiotics were continuously presented for single colony selection. The expressions of Bax and Bak in several amplified colonies analyzed by Western blot are shown as examples. Some stable cell lines were used and pretreated with or without cyclosporin A or dibucaine for 1 h, and then PQ was added for another 24 h. Cell viability is expressed as a percentage of controls. Data are presented as the mean ± SD. *p < 0.05, compared to controls (n = 3∼6). # p < 0.05, compared to its respective PQ-treated group (n = 3∼6). These data represent one out of three independent experiments that gave similar results
PQ Resulted in Retarded Behavioral Performance and the DAergic System
PQ significantly and dose-dependently induced cell death in primary cultured midbrain cells (Fig. 7a, upper panel) that could also be attenuated by cyclosporine A and dibucaine (Fig. 7a, lower panel). Similarly, PQ increased the formation of Bax- and Bak-oligomers and Bax/Bak-heteromers in primary TH-positive cells (Fig. 7b). Again, cyclosporin A and dibucaine did not block these phenomena (Fig. 7b). During PQ treatment, rat body weights were decreased by the fourth week (Fig. 7c). In addition, traverse times and the number of slips while walking across a balance beam for PQ-treated rats were significantly elevated in comparison to controls (Fig. 7c). Striatal DA contents (Fig. 7d, upper panel) and TH staining in the substantia nigra pars compacta (SNpc) (Fig. 7d, lower panel, modified from Supplement 8) were both decreased in PQ-treated rats. In addition, local PQ injections tended to result in decreases in TH staining in SNpc (Fig. 7e). However, in PQ-treated groups, the intensity of SNpc TH staining in pshRNA-RFP/pshRNA-GFP-pre-injected areas was lighter than that in pshRNA-Bax-RFP/pshRNA-Bak-GFP-injected areas (Fig. 7e).

PQ induces cytotoxicity in midbrain primary cultures, behavioral retardation, and decreases in striatal DA content in rats. a After treatment with different doses of PQ for 24 h, midbrain primary cultured cells were subjected to MTT assay (upper panel). After pretreatment with cyclosporin A or dibucaine for 1 h in combination with or without the addition of 100 μM PQ for another 24 h, midbrain primary cultured cells were subjected to MTT assay (lower panel). Cell viability is expressed as a percentage of controls. Data are presented as the mean ± SD. *p < 0.05, compared to controls (n = 3). # p < 0.05, compared to its PQ-only treated group (n = 3–6). b After cyclosporin A (10 μM) or dibucaine (10 μM) pretreatment for 1 h, cells were treated with 100 μM PQ for another 24 h and then fixed for immunocytochemistry assay. The bar represents 5 μm. These data represent one out of three independent experiments that gave similar results. c During experiments, the body weights of rats with or without PQ treatment were recorded (upper panel). At the fourth week, traverse time and number of slips on a balance beam were measured (lower panel). Data are presented as the mean ± SD. *p < 0.05, compared to controls (n = 8–12). d After the end of experiments, rat brains were excised and striatum dissected for DA content analysis (upper panel). Data are presented as the mean ± SD. *p < 0.05, compared to controls (n = 4–6). In addition, rats were fixed with paraformaldehyde and brain slices subjected to immunohistochemistry (lower panel). The bar represents 10 μm. e After shRNA silencing and with or without in situ PBS (n = 8) or PQ (n = 12) infusions, rats were fixed and brain slices subjected to immunohistochemistry (lower panel). The bar represents 500 μm
Discussions
Our previous article shows that PQ can induce mitochondrial dysfunction [33], and the detailed mechanisms of PQ-induced cell death are further elucidated and discussed in this study.
Cyclosporin A and Dibucaine Prevented PQ-Induced PC12 Cell Death
PQ-induced cytotoxicity was primarily reproduced by a neutral red assay (Fig. 1a). For the purposes of consistency with our previous study and ease of manipulation, 1 mM PQ was adopted and reconfirmed herein by MTT and trypan blue exclusion assays (Fig. 1a). PQ-induced apoptotic cell death was demonstrated by identifying the cleavages of caspase 3 and PARP (Fig. 1c) and increased staining of Annexin V-FITC (Fig. 1d), consistent with previous findings [33]. Since PQ is known to induce mPTP opening [34], we thus demonstrated that cyclosporin A (a known mPTP inhibitor) can prevent PQ-induced cell death (Fig. 1), indicating the involvement of mPTP in this model. In addition, for some amphiphilic cations, such as dibucaine and propranolol, which block Bax-induced Cyto. C release [35] and MAC [25, 16], we tested and found for the first time that dibucaine attenuated PQ-induced cell death (Fig. 1). This indicates that PQ might activate mPTP and MAC, both of which might account for the induction of apoptotic cell death [36]. Therefore, we further investigated whether applying these two drugs in combination would further prevent PQ-induced cell death. To our surprise, only certain cyclosporin A and dibucaine combinations in low doses further promoted protection, but higher doses failed to further prevent PQ-induced cell death (Fig. 1e). Instead, higher combined dosages (>10 μM) resulted in cell death (data not shown). It is unclear why combined doses have only limited protection. This phenomenon has also been found in Bax/Bak/CypD triple-knockout mice, which fail to show any further reduction in infarct size in Bax/Bak double-knockout mice [37]. Taken together, these data indicate that PQ-induced MAC and mPTP may be interconnected and/or redundant and play important roles in mediating PQ cytotoxicity.
Although mPTP and MAC have been respectively regarded to mediate necrosis and apoptosis [20], recent studies indicate that Bax and/or Bak are also required for necrosis [37] and mPTP could be involve in apoptosis [38]. These extraordinary results indicate the complicated interactions of mPTP and MAC in regulating apoptosis and necrosis. Indeed, their crosstalk has just received extensive attention [39]. For instance, mPTP may results in Bax translocation and multimerization [40], Bax may regulate the opening of mPTP [41], and cyclosporin A that targets mPTP could prevent PQ-induced apoptosis (Fig. 1). Besides, although MAC formation may precede mPTP opening in many cases [20, 26], MAC assembly may also occur after mPTP opening [28]. Therefore, MAC and mPTP opening may act alone or in combination, depending on cell type and death stimulus, to amplify the death signals [39]. Since PQ-induced clustered Bax and/or Bak (Fig. 5) and released Cyto. C, HtrA2/Omi, and Smac/Diablo (Supplement 6b) at early third hour and decreased ∆Ψ (Fig. 2a, lower panel) and increased mPTP (Fig. 4c) [33] at sixth hour, PQ-induced MAC formation may precede mPTP opening.
Cyclosporin A and Dibucaine Prevented PQ-Induced Mitochondrial Dysfunction
Since PQ interferes with the mitochondrial complex I/III activities [9, 10] that normally result in decreased ∆Ψ and increased free-radical formation [14], we examined and found that PQ resulted in decreased ∆Ψ (Fig. 2a) and increased mPTP opening (Fig. 4c) and mitochondrial superoxide formation (Fig. 2f). These phenomena were all attenuated by cyclosporin A and dibucaine (Fig. 2c–f), indicating that mPTP and MAC might be the targeting events of PQ in triggering cell death. This notion was substantiated by cyclosporin A and dibucaine blocking the PQ-induced mitochondrial release of pro-apoptotic factors, including HtrA2/Omi and Smac/Diablo (Fig. 3a, b). In addition, the protection of HtrA2/Omi protease inhibitor in antagonizing PQ-induced toxicity (Supplement 3b, c) further demonstrated that the release of mitochondrial pro-apoptotic factor and mitochondria dysfunction might be lethal.
Bax and Bak Mediated PQ-Induced Cytotoxicity
Since the induction of mPTP opening by PQ was already known [34], we thus focused on MAC formation. Besides, since dibucaine that targets MAC [25] exerted protection against PQ-induced cytotoxicity (Fig. 1 and 3), we examined the significance of Bax and Bak during PQ intoxication and found that PQ time-dependently increased the expression of Bax but not Bak (Fig. 5a). Although PQ toxicity has been shown to involve p53 [42], which is a known direct transcriptional activator of the human bax gene [43], it was not clear if p53 would regulate the PQ-induced Bax expression in this model. Moreover, for the first time, we have shown that PQ concomitantly resulted in increased oligomerization of Bax and Bak (Fig. 5a) and heteromerization of Bax/Bak, indicating the formation of MAC [27]. The failure of cyclosporin A and dibucaine on the clusterization of Bax and/or Bak (Fig. 5b) indicated that their protection did not derive from clusterization interference. This notion was demonstrated by PQ-induced mitochondrial translocation of Bax regardless of the presence or absence of cyclosporin A and dibucaine (Fig. 5c). By gene overexpression experiments, Bax (Fig. 5d, left panel) and Bak (Fig. 5d, right panel) were time-dependently clustered on mitochondria during PQ treatment, further confirming the PQ-induced mitochondrial translocation of Bax and oligomerization of Bax and Bak on mitochondria.
In order to study the significance of Bax and Bak, gene knockdown experiments were performed. Cells that were silenced with Bax or Bak (Fig. 6a, upper panel) exerted protection against PQ cytotoxicity (Fig. 6a, lower panel). Besides, Bax- and/or Bak-silenced permanent cells (Fig. 6b, upper panel and Supplement 7b) also exerted protection against PQ cytotoxicity (Fig. 6b, lower panel and Supplement 7a), demonstrating the pivotal roles of Bax and Bak in mediating PQ-induced cytotoxicity. These results partly consist with the previous findings showing that Bak knockdown in human SH-SY5Y cells provides resistance to PQ treatment [6]. It is unclear why cyclosporin A and dibucaine exerted no further protection against PQ toxicity in Bax and/or Bak knockdown cells (Fig. 6). Since Bax and Bak may have functional redundancy [44] and MAC and mPTP may be interconnected and/or also redundant (Fig. 1e), it is possible that the PQ-induced death effect from MAC formation could be diminished in Bax and/or Bak knockdown cells that hamper protection by cyclosporin A and dibucaine.
PQ, ROS Formation, and MMP
Since cyclosporin A and dibucaine blocked PQ-induced mitochondrial ROS formation (Fig. 2e), suggesting the blockade of PQ from entering into mitochondria, we thus directly measured ROS release from mitochondria. In the naive PC12 cell-derived mitochondrial fraction, cyclosporin A but not dibucaine significantly attenuated PQ-induced H2O2 formation (Fig. 2b), demonstrating that mitochondria could be the targeted organelle for PQ to enter through mPTP. In another mitochondrial fraction deriving from PQ-treated PC12 cells, both cyclosporin A and dibucaine significantly attenuated PQ-induced H2O2 formation (Fig. 2b); further supporting Bax and/or Bak clusterization (MAC formation) after PQ treatment (Fig. 5) and indicating MAC as another channel for PQ entry. Therefore, these findings indicated that PQ may induce a permeability increase of both outer and inner mitochondrial membrane. Although PQ may target mitochondria (such as complexes I and III) to generate ROS [9, 10], there are still other mitochondrial (such as aconitase) and cytosolic (such as nitric oxide synthase, thioredoxin reductase, and NADPH oxidase) enzymes that may also be responsible for ROS generation [45]. In order to get a more detailed time course of ROS formation, we measured PQ-induced ROS formations at different intervals and found that cytosolic ROS significantly increased (at the third hour) prior to elevated mitochondrial ROS formation and decreased ∆Ψ (at sixth hour) during PQ treatment (Fig. 2a), indicating that PQ-induced cytosolic ROS might precede mitochondrial ROS formation.
Further, we examined the involvements of the above described cytosolic enzymes and only found that NADPH oxidase (NOX) inhibitor, diphenylene iodonium (DPI), could prevent PQ-induced cell death (Fig. 4a), consistent with the previous study [30]. DPI also blocked the releases of mitochondrial pro-apoptotic factors (Fig. 4d and Supplement 5d and 6c). NOX has been proposed to play a role in regulating or is regarded as a therapeutic target in treating neurodegeneration [46]. Besides, PQ as a divalent cation (PQ2+) will undergo redox cycling, being reduced by diaphorases (such as NOX) by oxidizing an electron donor such as NAD(P)H to yield the monovalent cation PQ+ before being oxidized by an electron receptor such as dioxygen to produce superoxide anions [29]. Therefore, since DPI attenuated PQ-induced ROS formation in the cytosol (Fig. 4b), mitochondria (Fig. 4c), and mitochondrial fraction (Fig. 4a, lower panel), cytosolic and mitochondrial NOX (or DPI-sensitive enzyme) might mediate PQ redox cycling and then ROS production. Since the cross talk between NOX and mitochondria represents a vicious positive feedback cycle of ROS production [47], it is possible that DPI protection might be due to blocking this cycle. Yet, the detailed mechanisms of this, including subtypes and locations of NOX, require further investigation.
On the other hand, since the study of outer and inner mitochondrial membrane permeabilization is focused on the mitochondria-mediated death decision [48], PQ-induced increased MMP (Fig. 2 and 5) might be crucial for the organized death process. From this study, it is not clear how PQ drives MAC and mPTP formation. Accordingly, when excessive stimulation by endogenous signals (e.g., excessive ROS, Ca2+ overload, and protease activation) or activation of harmful signaling pathways (e.g., kinases/phosphatases, proteases, and Bax/Bid-mediated pathways) occur, mitochondria undergo mPTP formation [14]. Because redox events like quinone production might account for mPTP formation [49] and PQ is known for its redoxing effects in generating superoxide anions or other reactive oxygen species within mitochondria [50], PQ redox cycling may possibly result in mPTP formation. Accordingly, mitochondrially generated ROS are involved in mPTP induction [51]. However, we observed early cytosolic ROS formation (at the third hour, Fig. 2a) and then mitochondrial ROS formation occurred in companion with reduced calcein/CoCl2 fluorescence (at the sixth hour, Fig. 4c) after PQ treatment, suggesting the entry of PQ into mitochondria may pass through MAC and then mPTP. Whether PQ-induced mPTP formation was triggered by cytosolic ROS or mitochondrial ROS remains elusive.
The molecular mechanisms that stimulate Bax and/or Bak clusterization are still being debated and under investigation [52]. Accordingly, at a resting state, Bax interacting with Bcl-xL (or Bcl-2) may translocate back and forth between mitochondria and cytosol [53]. The formation of truncated Bid (tBid) could be mediated by the cleavage of caspase 8 and calpain [52]. However, when stimulated by stress, Bax or Bak can be directly stimulated by BH3-only proteins (tBid, Bim, or Puma) [54] or calpain cleavage to become inserted onto the mitochondria’s outer membrane. In addition, oxidative stress may also stimulate Bax translocation [55]. Thus, PQ-induced ROS formation may also contribute to the clusterization of Bax and/or Bak since DPI attenuated ROS formation (Fig. 4 and Supplement 5). Indeed, as with DPI, we tested an antioxidant, U74389G, which is known to prevent PQ-induced pneumotoxicity in rats [56], and found that it prevented PQ-induced ROS and cytotoxicity (Fig. 4b, c and Supplement 4a and 5) and Bax and/or Bak clusterization (Supplement 4b), indicating PQ redox cycling-induced ROS can trigger MAC formation. We also testified two antioxidants, MCI-186 [57] and NAC [58] which are known to prevent PQ-induced oxidative stress, and found to prevent PQ-induced cleaved-caspase 3 formations (Supplement 4a, lower panel), indicating PQ-induced oxidative stress as a cause of apoptotic cell death. In addition, since calpeptin, an inhibitor of calpain activated during Ca2+ overloading, prevents PQ-induced cytotoxicity (Fig. 4a), tBid cleaving by calpain may stimulate Bax and/or Bak clusterization. However, more experiments are required to demonstrate PQ-induced Ca2+ overloading and tBid formation. Collectively, PQ redox cycling-induced ROS may stimulate the induction of increased MMP.
PQ and In Vivo Neurotoxicity
In order to further strengthen the mechanism of PQ-induced cytotoxicity, several in vivo experiments were manipulated. PQ dose-dependently resulted in cytotoxicity in midbrain primary cultured cells (Fig. 7a, upper panel). PQ at a concentration of 100 μM was adopted. Cyclosporin A and dibucaine were also protective in preventing PQ-induced cytotoxicity (Fig. 7a, lower panel) but failed to prevent PQ-induced clusterization of Bax or Bak (Fig. 7b), consistent with the findings for PC12 cells (Figs. 1 and 4) and indicating that common mechanisms (mPTP and MAC) exist in PC12 and midbrain cells. We successfully reproduced PQ-induced toxic effects in rats (Fig. 7c, d), including retarded behavioral performance and impaired DAergic systems, confirming the neurotoxicity of PQ in vivo [59]. In this model, PQ has been shown to elevate cortical and striatal Bax expression [60] consistent with our findings in PC12 cells (Fig. 5a). In order to further confirm the role of Bax and Bak in regulating PQ cytotoxicity, pshRNA-Bax-RFP and pshRNA-Bak-GFP were locally injected and resulted in protection against a PQ-induced decreased intensity in TH immunostaining in SNpc (Fig. 7e and Supplement 8b), further strengthening the notion that PQ-induced neurotoxicity could at least in part be mediated through Bax- and Bak-dependent pathways.
Taken together, PQ through redox cycling-generated ROS may induce MAC formation (∼3 h) and then mPTP opening (∼6 h). During these processes, Bax and/or Bak clustered on mitochondria, pro-apoptotic factors released from mitochondria, and membrane potential dropped and ROS increased in mitochondria (Fig. 8). Although cyclosporin A and dibucaine tend not to be blood-brain barrier (BBB) permeable, the rationale of targeting MMP in treating PD might still hold true, and the search for BBB-permeable drug(s) that could stabilize MMP is preferred. Finally, MMP might play an important role in regulating PQ-induced cytotoxicity, supporting MMP as a therapeutic target in treating neurodegeneration like PD.

Mechanism of PQ-induced PC12 cell death through impairing mitochondrial membrane permeability. PQ primarily through redox cycling by NOX may generate ROS and induce Bax and/or Bak clusterization (MAC formation) and pro-apoptotic factor release around third hour. Further, after around 6 h, MAC and/or redox cycling PQ (or its native form) may induce mPTP formation, decreased ∆Ψ, and increased mitochondrial ROS (Mito-ROS). Finally, PQ may induce cell death. The green, yellow, and red areas, respectively, represent safe, danger, and lethal status of cells during PQ intoxication. After PQ treatment for 6 h, the morphology of cells was subtly changed, but the cell death was not significant and the death progression was irreversible (data no shown)
Materials and Methods
Reagents
All reagents were purchased from Sigma (St. Louis, MO, USA) except where otherwise specified. HtrA2/Omi protease inhibitor was purchased from EMD Millipore (Billerica, MA, USA). Goat serum (GS) was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Dulbecco’s modified Eagle’s medium (DMEM), Acutase solution, B27 supplement, Neurobasal medium, Hanks’ balanced salt solution (HBSS), fetal bovine serum (FBS), and horse serum were purchased from HyClone (Logan, UT, USA). Lipofectamine 2000, fluorescent dyes (such as tetramethylrhodamine ethyl ester (TMRE), MitoSOX, and Calcein-AM), Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit, and secondary antibodies were purchased from Invitrogen (Grand Island, NY, USA). Anti-PARP (9542), -caspase-3 (9662) and -endonuclease (Endo; 4969) G antibodies were purchased from Cell Signaling (Beverly, MA, USA). Anti-apoptosis-inducing factor (AIF, 1020-1) and -HtrA2/Omi (2544-1) antibodies were purchased from Epitomics (Burlingame, CA, USA). Anti-Bak (bs-1284R) and -Bax (bs-0127M) antibodies were purchased from Bioss (Woburn, MA, USA). Anti-HSP 60 (sc-13966 and sc-271215) and -Tom 20 (sc-11415 and sc-17764) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-tubulin (ab6046) and -tyrosine hydroxylase (TH) antibodies were purchased from Abcam (Cambridge, MA, USA). Anti-actin (MAB1501) antibody was purchased from Millipore (Billerica, MA, USA). All plasmids except pDsRed-Mito (expressing as a red fluorescent mitochondrial marker, purchased from Clontech, Mountain View, CA, USA) were purchased from Addgene (Cambridge, MA, USA). siGENOME SMART pool siRNAs and DharmaFECT transfection reagents were purchased from Thermo Scientifics (Waltham, MA, USA), and shRNA plasmids were purchased from OriGene Technologies (Rockville, MD, USA).
Cell Culture
Neuronal-like rat pheochromocytoma (PC12) cells were maintained in DMEM supplemented with 10 % vol/vol horse serum, 5 % vol/vol FBS, 100 U/ml penicillin, and 0.1 mg/ml streptomycin and incubated in a CO2 incubator (5 %, 37 °C). Plating numbers were 3 × 104 cells/cm2 during experiments.
Survival Assays
Survival was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and neutral red uptake assay as described before [33]. EC50 was analyzed by GraphPad Prism 4 (GraphPad Software).
Annexin V-FITC (Fluorescein Isothiocyanate) Staining
An annexin V-FITC-conjugated apoptosis kit (Imgenex, San Diego, CA, USA) was used to analyze apoptotic cells. The experimental protocol followed manufacturer instructions. The stained cells were subjected to fluorescence images (Axiovert 200 M, Carl Zeiss, Gottingen, Germany) or flow cytometry (FACScan®, Becton Dickinson, Franklin Lakes, NJ, USA).
Transient Gene Silences of Bak and Bax
The non-targeting (NT) siRNA and SMART pools of siRNAs that target rat Bak and Bax were used to silence the expression of Bak and Bax according to standard protocols. In brief, for transfecting 106 cells in 3 cm dishes, 50 nM siRNAs (1.6 μl) and DharmaFECT (10 μl) were added into 38.4 and 90 μl DMEM, respectively, for 5 min and mixed for another 20 min. After supplementing with 660 μl DMEM and adding cells for 6 h, the mixture was replaced with normal culturing medium and incubated for 2 days.
Permanent Cell Lines of Gene-Silenced Bak, Bax, and Bak/Bax
Four 29-mer of Bak- and Bax-specific shRNAs, constructed respectively in red fluorescence protein (RFP) and green fluorescence protein (GFP) vectors, were transfected into 50 % confluent cells [61]. These plasmids were also used in animal experiments. The selections for RFP- and GFP-expressing vectors were puromycin and blasticidin, respectively. Cells were selected by supplementing with puromycin (1 μg/ml) and/or blasticidin (2 μg/ml). Single colonies with dominant green and/or red fluorescence were selected for further propagation in the continuous presence of antibiotics. Western blot analysis was used to verify the expression of Bak and Bax in each colony.
Animals
Male Sprague-Dawley rats weighting 300 ± 25 g were purchased from the National Laboratory Animal Center (Taiwan). Rats were kept in a 12/12 h light/dark cycle and temperature (22 ± 2 °C) controlled room by following the principles and directives of the NIH Guide for the Care and Use of Laboratory Animals. The experiments were reviewed and approved by the Institutional Animal Care and Use Committee at the National Research Institute of Chinese Medicine (Approval No: 102-806-1). PQ dissolving in saline was administered (10 mg/kg) through intraperitoneal (IP) injection. Rats treated with PQ (n = 12) received one injection weekly for 4 weeks in accordance with earlier work [59]. Control animals (n = 10) were treated with saline once a week. Rats were decapitated or perfused at the fifth week for subsequent analyses. Rats were anesthetized, stereotaxic fixed to implant 23-gauge stainless-steel cannulae (15-mm-long) bilaterally into the SN (AP −5.4 mm, ML ±2.4 mm, and DV −7.5 mm), and then recovered for 1 week. The cannulae were affixed on the skull with dental cement. Bax/Bak shRNA (1.5 μg) and scramble shRNA plasmids (1.5 μg) were separately intra-nigral administered (1 μl/min) in each rat. Branched polyethylenimine (25-kDa)/DNA complex (6 equivalent nitrogens per DNA phosphate) was used as the transfection agent. After 48 h of transfection, animals were randomly divided into two groups. One group received bilateral infusion of 1.0 μl PBS and the other group received bilateral infusion of paraquat (5 μg/1.0 μl). After the infusions for 48 h, rats were perfused and the brains were dissected for further analysis.
Measurement of Dopamine
Striatal dissections were weighed, homogenized, and deproteinized with a volume (1/4 w/v) of perchloric acid (0.4 M) containing diethylenetriamine-pentaacetic acid (1 mg/ml) and 1,4-dithioerythritol (0.1 mg/ml) [62]. After centrifugation (10,000g, 10 min), 10 μl of clear perchloric acid extract was used for dopamine (DA) measurement by applying DA ELISA Kit (KA1887; Abnova, Taipei City, Taiwan).
Isolation of Mitochondria
Mitochondrial isolation was carried out as described by Nedergaard and Cannon [63] with modifications [33].
Measurement of PQ-Induced Mitochondria-Generated H2O2
Simplified in vitro techniques for measuring mitochondria-generated H2O2 were adopted as described by Drechsel and Patel [29].
Measurement of Mitochondrial Superoxide
Cells were washed with Hank’s buffer and stained with 5 μM MitoSOXTM for 10 min. Cells were used for taking pictures or running flow cytometry. Mean values were acquired using channel FL-2. Ten thousand live cells were analyzed per sample.
Measurement of ∆Ψ
Cells were stained with 100 nM TMRE for 15 min in the incubator and scraped for flow cytometry analysis or imagined. Mean values were acquired using channel FL-2. Ten thousand live cells were analyzed per sample.
Measurement of mPTP Opening
Cells were preincubated for 15 min at 37 °C with 1 μM Calcein-AM and 1 mM CoCl2 in HBSS supplemented with 1 mM HEPES at pH 7.3. HBSS was replaced with normal medium for 24 h [64]. After treatments, cells were then used for imaging or conducting flow cytometry experiments.
Measurement of Intracellular Reactive Oxygen Species
Normally, the term reactive oxygen species (ROS) includes free oxygen radicals (such as the superoxide anion radical and hydroxyl radical) and non-radical oxidants (such as hydrogen peroxide and singlet oxygen). Intracellular superoxide and peroxide formations stained with dihydroethidium (DHE) and 5-(and 6-)carboxy-2′,7′-dichlorodihydrofluorescein diacetate (CO3-H2DCFDA), respectively, were measured as described previously [65].
Western Blot Analysis
Western blot analysis was performed as described previously [66]. In brief, equal amounts of cell lysates (∼20 μg/well) derived from the filtered assay were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and then electroblotted onto Immobilon polyvinylidene difluoride (PVDF) membranes (Millipore). Membranes were blocked with 5 % skim milk in TBST (100 mM Tris-HC1 and 150 mM NaC1 at pH 7.4 containing 0.05 % Tween 20) for 1 h at room temperature and then incubated with the first antibody (1:1000∼2000) at 4 °C overnight. After three washes with TBST, the blot was incubated with a second antibody (1:5000) conjugated to horseradish peroxidase for 1 h, processed for visualization using an enhanced chemiluminescence system (Pierce, Rockford, IL, USA), and exposed to Fuji medical X-ray film (Super RX-N, FUJIFILM Corporation, Tokyo, Japan) to obtain fluorographic images.
Immunocytochemistry
Cells were seeded and grown on sterile glass coverslips. After various treatments, cells were fixed for 10 min with 4 % paraformaldehyde at room temperature. Following fixation, cells were rinsed three times with phosphate-buffered saline (PBS). Further, cells were permeabilized by using 0.5 % Triton-100 in PBS for 15 min. A blocking agent composed of 10 % GS and 0.3 % Triton-100 in PBS was then applied at room temperature for 90 min to reduce nonspecific antibody activity. After three washes with PBS, cells were labeled with the antibodies (1:200), which were dissolved in 1 % bovine serum albumin (BSA) and 0.3 % Triton-100 in PBS and incubated at 4 °C overnight. After labeling, cells were washed three times with PBS at room temperature. Cells with IgG fluorescein-conjugated secondary antibodies (1:200) were incubated in 1 % BSA/PBS at room temperature for 90 min and then counterstained with 4′,6-diamidino-2-phenylindole (DAPI; 1 μg/ml) at room temperature for 10 min if required. Cells were washed three times with PBS and then mounted on microscope slides with Aqua-Poly/Mount (Polysciences, Warrington, PA, USA).
Immunohistochemistry
Rats were anesthetized (40 mg/ml chlorohydrate, IP) and perfused with 4 % paraformaldehyde/PBS. Brains were immersed in 30 % sucrose and 4 % paraformaldehyde/PBS. After dissection (20 μm/section), brain slices were first heated in antigen retrieval buffer (5 % urea and 100 mM Tris-HCl, pH 9.5) at 95 °C for 10 min. Slices were permeabilized and blocked by using 10 % GS and 0.1 % BSA in immunohistochemistry (IHC) buffer (0.1 % Triton-100, 0.05 % Tween-20, and 0.05 % NaN3 in PBS) at room temperature for 1 h. After three washes with IHC buffer, brain slices were labeled with the antibodies (1:200) dissolved in 1 % GS and 0.05 % BSA/IHC buffer at 4 °C overnight. After three washes with IHC buffer, slices were incubated with IgG fluorescein-conjugated secondary antibodies (1:200) in the presence or absence of DAPI (1 μg/ml) in IHC buffer at room temperature for 60 min. Brain slices were washed three times with IHC buffer and then mounted on a microscope slide with Aqua-Poly/Mount.
Balance Beam
The balance beam was constructed as previously described [67] except for the diameter of the rod being 3.2 cm. The time taken to traverse the beam and the number of hind leg slips were recorded.
Primary Culture
Neural cell cultures from the ventral mesencephalon of E14 fetuses of SD rats were prepared as described previously [68]. At seventh day, cells were subjected to an MTT assay or immunocytochemistry study.
Statistics
The significance of the drug treatments was determined by one- or two-way analyses of variance (ANOVA) or Student’s t test. Post hoc comparisons were carried out between means as appropriate. Statistical significance was set at p < 0.05.
References
Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, Chen RC (1997) Environmental risk factors and Parkinson’s disease: a case-control study in Taiwan. Neurology 48:1583–1588
Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS et al (2011) Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect 119:866–872
McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA (2002) Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis 10:119–127
Manning-Bog AB, McCormack AL, Purisai MG, Bolin LM, Di Monte DA (2003) Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration. J Neurosci 23:3095–3099
Gonzalez-Polo RA, Rodriguez-Martin A, Moran JM, Niso M, Soler G, Fuentes JM (2004) Paraquat-induced apoptotic cell death in cerebellar granule cells. Brain Res 1011:170–176
Fei Q, McCormack AL, Di Monte DA, Ethell DW (2008) Paraquat neurotoxicity is mediated by a Bak-dependent mechanism. J Biol Chem 283:3357–3364
Miller GW (2007) Paraquat: the red herring of Parkinson’s disease research. Toxicol Sci 100:1–2
Cory-Slechta DA, Thiruchelvam M, Di Monte DA (2008) Letter regarding: “Paraquat: the red herring of Parkinson’s disease research”. Toxicol Sci 103:215–216, author reply 217-218
Cocheme HM, Murphy MP (2008) Complex I is the major site of mitochondrial superoxide production by paraquat. J Biol Chem 283:1786–1798
Castello PR, Drechsel DA, Patel M (2007) Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J Biol Chem 282:14186–14193
Richardson JR, Quan Y, Sherer TB, Greenamyre JT, Miller GW (2005) Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol Sci 88:193–201
O’Rourke B (2007) Mitochondrial ion channels. Annu Rev Physiol 69:19–49
Reddy PH, Reddy TP (2011) Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr Alzheimer Res 8:393–409
Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87:99–163
Arnoult D, Parone P, Martinou JC, Antonsson B, Estaquier J, Ameisen JC (2002) Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J Cell Biol 159:923–929
Kinnally KW, Antonsson B (2007) A tale of two mitochondrial channels, MAC and PTP, in apoptosis. Apoptosis 12:857–868
Haworth RA, Hunter DR (1979) The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys 195:460–467
Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ (2009) Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res 83:213–225
Zamzami N, Kroemer G (2001) The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol 2:67–71
Ryu SY, Peixoto PM, Teijido O, Dejean LM, Kinnally KW (2010) Role of mitochondrial ion channels in cell death. Biofactors 36:255–263
Bernardi P (2013) The mitochondrial permeability transition pore: a mystery solved? Front Physiol 4:95
Basanez G, Soane L, Hardwick JM (2012) A new view of the lethal apoptotic pore. PLoS Biol 10, e1001399
Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A et al (2013) Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12:674–683
Leung AW, Varanyuwatana P, Halestrap AP (2008) The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem 283:26312–26323
Martinez-Caballero S, Dejean LM, Kinnally KW (2004) Some amphiphilic cations block the mitochondrial apoptosis-induced channel, MAC. FEBS Lett 568:35–38
Pavlov EV, Priault M, Pietkiewicz D, Cheng EH, Antonsson B, Manon S, Korsmeyer SJ, Mannella CA et al (2001) A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J Cell Biol 155:725–731
Dejean LM, Martinez-Caballero S, Guo L, Hughes C, Teijido O, Ducret T, Ichas F, Korsmeyer SJ et al (2005) Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol Biol Cell 16:2424–2432
Guihard G, Bellot G, Moreau C, Pradal G, Ferry N, Thomy R, Fichet P, Meflah K et al (2004) The mitochondrial apoptosis-induced channel (MAC) corresponds to a late apoptotic event. J Biol Chem 279:46542–46550
Drechsel DA, Patel M (2009) Chapter 21 Paraquat-induced production of reactive oxygen species in brain mitochondria. Methods Enzymol 456:381–393
Miller RL, Sun GY, Sun AY (2007) Cytotoxicity of paraquat in microglial cells: Involvement of PKCdelta- and ERK1/2-dependent NADPH oxidase. Brain Res 1167:129–139
Cristovao AC, Choi DH, Baltazar G, Beal MF, Kim YS (2009) The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxid Redox Signaling 11:2105–2118
Bonneh-Barkay D, Reaney SH, Langston WJ, Di Monte DA (2005) Redox cycling of the herbicide paraquat in microglial cultures. Brain Res Mol Brain Res 134:52–56
Huang CL, Lee YC, Yang YC, Kuo TY, Huang NK (2012) Minocycline prevents paraquat-induced cell death through attenuating endoplasmic reticulum stress and mitochondrial dysfunction. Toxicol Lett 209:203–210
Costantini P, Petronilli V, Colonna R, Bernardi P (1995) On the effects of paraquat on isolated mitochondria Evidence that paraquat causes opening of the cyclosporin A-sensitive permeability transition pore synergistically with nitric oxide. Toxicology 99:77–88
Polster BM, Basanez G, Young M, Suzuki M, Fiskum G (2003) Inhibition of Bax-induced cytochrome c release from neural cell and brain mitochondria by dibucaine and propranolol. J Neurosci 23:2735–2743
Jourdain A, Martinou JC (2009) Mitochondrial outer-membrane permeabilization and remodelling in apoptosis. Int J Biochem Cell Biol 41:1884–1889
Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW et al (2012) Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A 109:6566–6571
Gao J, Sana R, Calder V, Calonge M, Lee W, Wheeler LA, Stern ME (2013) Mitochondrial permeability transition pore in inflammatory apoptosis of human conjunctival epithelial cells and T cells: effect of cyclosporin A. Investig Ophthalmol Visual Sci 54:4717–4733
Kinnally KW, Peixoto PM, Ryu SY, Dejean LM (2011) Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta 1813:616–622
De Giorgi F, Lartigue L, Bauer MK, Schubert A, Grimm S, Hanson GT, Remington SJ, Youle RJ et al (2002) The permeability transition pore signals apoptosis by directing Bax translocation and multimerization. FASEB J 16:607–609
Kubli DA, Gustafsson AB (2012) Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 111:1208–1221
Yang W, Tiffany-Castiglioni E (2008) Paraquat-induced apoptosis in human neuroblastoma SH-SY5Y cells: involvement of p53 and mitochondria. J Toxicol Environ Health, Part A 71:289–299
Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80:293–299
Degli Esposti M (2003) The mitochondrial battlefield and membrane lipids during cell death signalling. Ital J Biochem 52:43–50
Franco R, Li S, Rodriguez-Rocha H, Burns M, Panayiotidis MI (2010) Molecular mechanisms of pesticide-induced neurotoxicity: relevance to Parkinson’s disease. Chem Biol Interact 188:289–300
Gao HM, Zhou H, Hong JS (2012) NADPH oxidases: novel therapeutic targets for neurodegenerative diseases. Trends Pharmacol Sci 33:295–303
Dikalov S (2011) Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med 51:1289–1301
Belizario JE, Alves J, Occhiucci JM, Garay-Malpartida M, Sesso A (2007) A mechanistic view of mitochondrial death decision pores. Braz J Med Biol Res 40:1011–1024
Giorgio V, Petronilli V, Ghelli A, Carelli V, Rugolo M, Lenaz G, Bernardi P (2012) The effects of idebenone on mitochondrial bioenergetics. Biochim Biophys Acta 1817:363–369
Yumino K, Kawakami I, Tamura M, Hayashi T, Nakamura M (2002) Paraquat- and diquat-induced oxygen radical generation and lipid peroxidation in rat brain microsomes. J Biochem 131:565–570
Kowaltowski AJ, Castilho RF, Vercesi AE (1996) Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett 378:150–152
Ghibelli L, Diederich M (2010) Multistep and multitask Bax activation. Mitochondrion 10:604–613
Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, Neutzner A, Tjandra N et al (2011) Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145:104–116
Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH (2009) Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell 36:487–499
D’Alessio M, De Nicola M, Coppola S, Gualandi G, Pugliese L, Cerella C, Cristofanon S, Civitareale P et al (2005) Oxidative Bax dimerization promotes its translocation to mitochondria independently of apoptosis. FASEB J 19:1504–1506
Shopova VL, Dancheva VY, Salovsky PT, Stoyanova AM, Lukanov TH (2007) Protective effect of U-74389G on paraquat induced pneumotoxicity in rats. Environ Toxicol Pharmacol 24:167–173
Zhi Q, Sun H, Qian X, Yang L (2011) Edaravone, a novel antidote against lung injury and pulmonary fibrosis induced by paraquat? Int Immunopharmacol 11:96–102
Unnithan AS, Jiang Y, Rumble JL, Pulugulla SH, Posimo JM, Gleixner AM, Leak RK (2014) N-acetyl cysteine prevents synergistic, severe toxicity from two hits of oxidative stress. Neurosc Lett 560:71–76
Czerniczyniec A, Karadayian AG, Bustamante J, Cutrera RA, Lores-Arnaiz S (2011) Paraquat induces behavioral changes and cortical and striatal mitochondrial dysfunction. Free Radic Biol Med 51:1428–1436
Czerniczyniec A, Lores-Arnaiz S, Bustamante J (2013) Mitochondrial susceptibility in a model of paraquat neurotoxicity. Free Radic Res 47:614–623
Lee YC, Yang YC, Huang CL, Kuo TY, Lin JH, Yang DM, Huang NK (2012) When cytokinin, a plant hormone, meets the adenosine A2A receptor: a novel neuroprotectant and lead for treating neurodegenerative disorders? PLoS One 7, e38865
Mochel F, Durant B, Durr A, Schiffmann R (2011) Altered dopamine and serotonin metabolism in motorically asymptomatic R6/2 mice. PLoS One 6, e18336
Nedergaard J, Cannon B (1979) Overview—preparation and properties of mitochondria from different sources. Methods Enzymol 55:3–28
Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F (1999) Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J 76:725–734
Chen HM, Lee YC, Huang CL, Liu HK, Liao WC, Lai WL, Lin YR, Huang NK (2007) Methamphetamine downregulates peroxiredoxins in rat pheochromocytoma cells. Biochem Biophys Res Commun 354:96–101
Huang NK, Lin YW, Huang CL, Messing RO, Chern Y (2001) Activation of protein kinase A and atypical protein kinase C by A(2A) adenosine receptors antagonizes apoptosis due to serum deprivation in PC12 cells. J Biol Chem 276:13838–13846
Song YN, Li HZ, Zhu JN, Guo CL, Wang JJ (2006) Histamine improves rat rota-rod and balance beam performances through H(2) receptors in the cerebellar interpositus nucleus. Neurosci 140:33–43
Choi WS, Klintworth HM, Xia Z (2011) JNK3-mediated apoptotic cell death in primary dopaminergic neurons. Methods Mol Biol 758:279–292
Acknowledgments
This work was supported by the National Research Institute of Chinese Medicine (NRICM101-DBCM-06), Cardinal Tien Hospital (CTH-100-1-2A13, CTH-101-1-2A03, CTH-101-1-2A12, and CTH-103-1-2A15), and National Science Council, R.O.C. (NSC 101-2320-B-077-007-MY3). We thank Mr. B.C. Pruitt, Jr. for the English language editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Huang, CL., Chao, CC., Lee, YC. et al. Paraquat Induces Cell Death Through Impairing Mitochondrial Membrane Permeability. Mol Neurobiol 53, 2169–2188 (2016). https://doi.org/10.1007/s12035-015-9198-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9198-y