
Abstract
Many patients with systemic immune-inflammatory and neuro-inflammatory disorders, including depression, rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s disease, cancer, cardiovascular disorder, Parkinson’s disease, multiple sclerosis, stroke, and chronic fatigue syndrome/myalgic encephalomyelitis, endure pathological levels of fatigue. The aim of this narrative review is to delineate the wide array of pathways that may underpin the incapacitating fatigue occurring in systemic and neuro-inflammatory disorders. A wide array of immune, inflammatory, oxidative and nitrosative stress (O&NS), bioenergetic, and neurophysiological abnormalities are involved in the etiopathology of these disease states and may underpin the incapacitating fatigue that accompanies these disorders. This range of abnormalities comprises: increased levels of pro-inflammatory cytokines, e.g., interleukin-1 (IL-1), IL-6, tumor necrosis factor (TNF) α and interferon (IFN) α; O&NS-induced muscle fatigue; activation of the Toll-Like Receptor Cycle through pathogen-associated (PAMPs) and damage-associated (DAMPs) molecular patterns, including heat shock proteins; altered glutaminergic and dopaminergic neurotransmission; mitochondrial dysfunctions; and O&NS-induced defects in the sodium-potassium pump. Fatigue is also associated with altered activities in specific brain regions and muscle pathology, such as reductions in maximum voluntary muscle force, downregulation of the mitochondrial biogenesis master gene peroxisome proliferator-activated receptor gamma coactivator 1-alpha, a shift to glycolysis and buildup of toxic metabolites within myocytes. As such, both mental and physical fatigue, which frequently accompany immune-inflammatory and neuro-inflammatory disorders, are the consequence of interactions between multiple systemic and central pathways.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Severe intractable fatigue is endured by many people with a wide range of neuro-inflammatory, neuropsychiatric, and autoimmune disorders and cancer. Pathological fatigue is experienced by between 59 and 100 % of cancer sufferers depending on the clinical status of the disease [1]. Severe chronic fatigue is also experienced by many people with an autoimmune disease with 67 % of people with Sjögren’s disease [2], up to 76 % of patients with systemic lupus erythromatosis (SLE) [3] and upwards of 70 % of people with rheumatoid arthritis [4] suffering incapacitating levels of fatigue. Upwards of 80 % of multiple sclerosis patients suffer from incapacitating fatigue [5]. Beiske and Svensson reported that between 37 and 57 % of patients with Parkinson’s disease experience incapacitating fatigue [6]. Winward et al. reported data which strongly indicates that fatigue in stroke victims is a direct consequence of the cerebral infarct and not merely a consequence of increased effort during rehabilitation [7]. Severe fatigue was experienced by 87 % of people following major stroke 56 % of people following minor stroke and 29 % of people following a TIA [7]. Fatigue is commonplace in people following a myocardial infarction with almost 100 % of patients reporting debilitating levels of exhaustion. Interestingly, many patients reported severe fatigue without any concomitant signs of depression [8]. Numerous studies report a high co-morbidity between severe fatigue and major depression [9]. Fatigue is a hallmark symptom of depression in bipolar disorder, where so-called atypical depression is disproportionally present; this includes fatigue, hypersomnia, and carbohydrate craving [10]. Severe fatigue is also commonplace in people with chronic obstructive pulmonary disease and mild traumatic brain injury with 38 and 39 % of patients, respectively, suffering the fatigue symptom [11, 12]. In the case of inflammatory bowel disease, 40 % of patients suffer severe intractable fatigue even when in remission [13]. Pathological fatigue is also a mandatory element in affording a diagnosis of idiopathic chronic fatigue, chronic fatigue syndrome, and myalgic encephalomyelitis [5].
Mental fatigue involving an impaired capacity for concentration, learning, attention, and disturbance of short-term memory is commonly observed in neurodegenerative and neuro-inflammatory diseases, such as multiple sclerosis, and Alzheimer’s and Parkinson’s disease [14–19]. Mental fatigue presents as an impaired ability to take in and subsequently process information over any given time period. Mental exhaustion is generally precipitated during prolonged incessant cognitive tasks. Symptoms of mental exhaustion are usually absent or trivial when a patient is relatively relaxed and stress free. Mental fatigue predominates following sleep deprivation. Patients with mental fatigue often report extreme sensitivity to noise and light and severe headaches [20].
Activated immune-inflammatory pathways, oxidative and nitrosative stress (O&NS), mitochondrial dysfunctions, and brain metabolic disorders are involved in the pathophysiology of severe fatigue [21]. A chronically activated peripheral or central immune system is a feature of autoimmune disorders, such as Sjögren’s disease, systemic lupus erythematosus and rheumatoid arthritis, and neuro-inflammatory diseases, such as Parkinson’s disease, multiple sclerosis and stroke [22, 9]. It may be no coincidence that elevated levels of oxidative stress are found in patients with systemic lupus erythematosus [23], Sjögren’s disease [24], rheumatoid arthritis [25], Parkinson’s disease [26], multiple sclerosis [5], stroke [27], cancer [28], depression [29], and bipolar disorder [30]. Mitochondrial dysfunction is also a common finding in all these disease states [23, 24, 31, 32, 21]. Elevated levels of O&NS, mitochondrial dysfunction and a chronically activated immune system, together with neuroimaging abnormalities, are found in many individuals with a diagnosis of CFS [5, 21].
This review aims to examine the various pathways capable of generating intractable fatigue of peripheral or central origin. We will review that central fatigue does not only result from elevated levels of pro-inflammatory cytokines, increased O&NS and mitochondrial dysfunctions, but also pathological changes in glutaminergic and dopaminergic neurotransmission, functional or structural abnormalities in brain regions, including the prefrontal cortex and the basal ganglia. We will review that peripheral fatigue can stem from the breakdown of the homeostatic relationship between cortical and spinal neuron activity in striated muscle and increased patterns of gene expression in exercising muscle with or without the buildup of toxic metabolites within myocytes.
Immune-inflammatory and O&NS pathways leading to fatigue
Immune-inflammatory pathways and incapacitating fatigue
It has been proposed that fatigue stems in part from immune dysregulations and consequent cytokine abnormities in diseases such as Parkinson’s disease [33], multiple sclerosis [34], fibromyalgia [35], cancer [36], CFS [37, 38], and depression [39]. There is also strong evidence that cytokines make a considerable contribution to the development of stroke associated fatigue and neuro-inflammation [40, 41]. For example, high levels of interleukin-1 (IL-1) β, a pro-inflammatory cytokine, and depressed levels of IL1-receptor antagonist (IL-1RA), which inhibits IL-1 signaling, are robust predictors of the development of chronic fatigue after acute ischemic stroke supporting the role of chronic inflammation in the genesis and possible long-term maintenance of post stroke fatigue [42].
Several authors have implicated increased levels of pro-inflammatory cytokines in the generation of fatigue in ill and healthy individuals. Chronically sick patients with high underlying levels of inflammation experience markedly greater rates of fatigue than age-sex matched population norms [43, 44]. This association may be explained by invoking the concept of cytokine-induced illness behavior and the effects of IL-1, IL-6, IL-2, and interferon-(IFN) α, acting in the brain to induce symptoms such as fatigue, anhedonia, loss of appetite, weight loss, sleepiness, etc. [45]. There is now overwhelming evidence that the acute response to pathogen is mediated via the actions of pro-inflammatory cytokines, including IL-1β, IL-6, and TNFα, [46–50]. Pro-inflammatory cytokines in the systemic circulation are actively transported to the brain by transporters on endothelial cells or by via diffusion through relatively permeability areas of the blood-brain barrier [51]. Signals relating to systemic immune-inflammatory responses also reach the nuclei of the solitary tract and the caudal medulla [51–53] through afferent vagal signals induced by the presence of pro-inflammatory cytokines in lymph nodes and the spleen. Ultimately, signals from ascending neural pathways conveying information regarding the status of peripheral activation of immune-inflammatory pathways induces the ‘danger pathway’ in the caudal brainstem, leading to attenuated arousal mediated by the hypothalamus [54]. Effects of TNFα, IL-1β, and IL-6 not only result in the activation of microglia and astrocytes inducing neuro-inflammation [55, 56], but also inhibit energy-consuming locomotor, and neurocognitive processes ensuring that energy is diverted from tissues, including peripheral organs and the brain, to be mobilized to counteract pathogen-induced pathology. The energy conserved by this cytokine-regulated process initiates and maintains pyrexia which enhances the performance of immune cells and inhibits viral replication [57–59]. Neuro-inflammation and microglial activation may also be provoked by the presence of lipopolysaccharide (LPS) from gam negative bacteria in the systemic circulation [46]. We recently reviewed the role of increased LPS following increased bacterial translocation in the initiation of fatigue and other illness symptoms [55]. Many of the symptoms associated with the sickness response are conserved by evolution with the express purpose of redirecting and conserving vital energy produced by mitochondria to meet the high energetic requirements of the immune response [57, 60].
Significant evidence that inflammatory cytokines can cause a stereotypical symptom complex dominated by severe fatigue derives from monitoring the effects of exogenous administration of IFNα. IFNα is a cytokine which is a key player in the innate response displaying both antiviral and antiproliferative capabilities and is thus often used to treat infectious diseases and cancer [61–63]. IFNα has often been detected in the CSF of patients suffering from a number of different infectious diseases such viral and bacterial meningitis [64]. The presence of elevated levels of this cytokine in the CSF of people with systemic lupus erythematosus and Aicardi-Goutieres syndrome is perhaps more surprising. It is thus worth noting that elevated concentrations of IFNα in the blood and CSF of patients diagnosed with these conditions is associated with diffuse chronic neuropathology [65, 66]. IFN α administration results in the development of severe fatigue together with psychomotor retardation [67–69]. It is worthy of note that these INFα-induced symptoms are refractory to therapy with selective serotonin reuptake inhibitors [70, 71]. This phenomenon is in agreement with observations in cancer patients, treated with inflammation and fatigue inducing chemotherapy, whose fatigue is also unresponsive to selective serotonin reuptake inhibitors [72, 73].
There is now considerable evidence implicating neuro-inflammation and the accompanying disruption of the blood-brain barrier (BBB) in the genesis of mental fatigue [74]. The relationship between the development of neuro-inflammation and the presence of elevated pro-inflammatory cytokines in the periphery is well documented [55, 56]. BBB disruption is observed quite early in neuro-inflammation, and parallels the release of pro-inflammatory cytokines [75–77]. Mechanisms underpinning BBB disruption in these conditions may involve effects of pro-inflammatory cytokines on endothelial cell tight junctions [56, 78, 79]. Cytokines, including TNFα and IL-1β, play a key role in the maintenance of neuronal functions and modulate neurotransmitter systems and neurocircuits in the brain, thereby resulting in behavior changes [80, 81]. Under normal physiologic conditions, these cytokines are involved in many indispensable brain processes such as long-term potentiation, synaptic plasticity, and neurogenesis [81].
Peripheral administration of IFN α leads to an increase in central IFN α levels, which consequently induce immune-inflammatory responses typified by elevations in IL-6 and monocyte chemoattractant protein-1 (MCP-1) [82–84], which are responsible for summoning monocytes into the CNS [85, 86]. The induction of MCP-1 is the main mechanism by which IFNα evokes inflammatory responses in the CNS [87]. MCP-1 elevations together with accompanied elevations of IL-6 have the capacity to prime microglia in the brain subsequent to administration of several different inflammatory stimuli to the CNS, including LPS [88]. It also seems worthy of note that loss of BBB integrity often noted during IFNα therapy may be facilitated by MCP-1, offering a potential mechanism by which a temporary but massive increase in IFNα levels following a prolonged viral infection may lead to chronic neuropathology. In fact, numerous studies have revealed that prolonged elevation of MCP-1 levels can markedly increase the permeability of the BBB via effects on the C-C motif chemokine receptor-2 receptor [85].
Microglia, astrocytes, and plasmacytoid dendritic cells [PDCs], which are normally resident within the meninges but are recruited to the parenchymal tissue of the brain following immune activation, have the capacity to produce IFNα [89, 90]. It has been demonstrated that IFNα-stimulated microglia in vitro generate IL-1, superoxide production and elevated levels of oxidative stress [91], suggesting that IFNα powers neuro-inflammation induced by microglia [92].
Although IFNα levels in the brain during exogenous administration are low in comparison to levels of MCP-1 and IL-6 [93], the amount detected is probably the result of the administered IFNα [82, 84]. The CNS responses in IFNα-treated patients indicate that IFNα likely enters the brain via leaky regions in the BBB and or provokes cells at the BBB to produce IFNα on the CNS side of the BBB. The latter explanation appears particularly attractive as IFNα has the capacity to upregulate numerous immune genes in endothelial cells such as those forming an integral part of the BBB. These include genes for Toll-Like Receptor (TLR) 3 and IFN regulatory factor 7 (IFR7), which play indispensable roles in IFNα production following numerous immunological stimuli such as a virus infection [94–96]. All in all, the weight of evidence demonstrates that IFNα has the capability of accessing and likely entering the brain [82, 97] despite the apparent lack of a saturable transport system in the BBB [98].
Oxidative and nitrosative stress pathways and incapacitating fatigue
A review involving patients with cancer or chemotherapy-induced fatigue referenced four studies where oxidative stress was held to be the cause of severe fatigue experienced by patients in those studies [99]. Elevated O&NS is also causatively implicated in the development of severe central or “systemic” fatigue [100–103]. A recent review highlighted seven studies where authors cited a significant positive association between oxidative stress and severity of fatigue in patients diagnosed with CFS [5]. Oxidative stress is dramatically elevated in patients with CFS compared to healthy controls during the conduct and aftermath of exercise [104, 105].
There is now ample evidence that rapid and massive increases in ROS production in the immediate aftermath of acute ischemic stroke overwhelm cellular antioxidant defenses, leading to further tissue damage in the penumbra [106]. These radicals can cause severe damage to lipids, proteins, DNA, and cell membranes leading to further neuronal loss by apoptosis or necrotic cell death [106, 107]. Furthermore, the rapid restoration of blood flow massively increases mitochondrial ROS production resulting in cellular damage and death characteristic of reperfusion injury [107, 27]. Wang et al. demonstrated a positive and significant correlation between elevated biomarkers of O&NS and objective measures of disease activity in systemic lupus erythematosus [108]. The existence of elevated O&NS also been repeatedly demonstrated in patients afforded a diagnosis of Sjögrens syndrome [24, 109], rheumatoid arthritis [110, 111, 112], Parkinson’s disease, and multiple sclerosis [5].
Numerous studies have demonstrated increased levels of ROS and RNS during exercise [113, 114] and the role of these radical species as essential signaling molecules in normal muscle function is well established [115, 116]. Skeletal muscles generate ROS and RNS at elevated rates during contraction [113, 114]. O&NS accelerates the development of muscle fatigue [117–119], weakness, and overall muscle dysfunction [120–122]. In similar vein, increased levels of RNS secondary to iNOS production cause muscle weakness and dysfunction in inflammatory pathways [120–122].
Administration of N-acetyl cysteine, an antioxidant, suppresses fatigue originating during activation of the tibialis anterior muscle, supporting the view that ROS scavenging plays a role in human muscle fatigue [123, 124]. Numerous studies have shown a role of ROS and ROS scavenging in muscle fatigue and force modulation [125, 126]. Interestingly, there is additional evidence that blockade of RNS can attenuate muscle fatigue [127, 128].
The adverse effects of increased O&NS on muscle cell function may be induced via a number of different mechanisms, which include dysregulation of the sarcoplasmic reticulum (SR), calcium release channel [129], depression of SR dependent ATPase activity [130], diminishing the use of oxygen by mitochondria overall and suppressing the activity of cytochrome c oxidase [21, 131, 132]. NO donors may cause a wide range of adverse reactions including decreasing the activity of cytochrome c oxidase [131], the disruption of calcium regulation [133], depression of contractile force [134] and diminished mitochondrial oxygen utilization [131], which conspire to accelerate the development of fatigue [117–119].
ROS that are generated by TNFα stimulation inhibit myogenesis via NF-κB-related mechanisms [135, 136]. NF-κB signaling appears to play an auxiliary role in atrophy of skeletal muscle induced by ROS [137]. NF-κB and ROS negatively impact skeletal muscle differentiation [138, 139]. Myoblast differentiation into myotubes is of paramount importance for muscle repair, regeneration, and function [140]. O&NS is known to at least partly underpin the pathology in a wide range of muscular pathologies characterized by abnormal differentiation [138, 141, 142].
The toll-like receptor radical cycle and incapacitating fatigue
Engagement of TLRs by pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) leads to the initiation of the innate immune response [143]. Activation of the TLR2/4 complex induces the expression of intracellular signaling networks, such as NF-κB and mitogen-activated protein kinases (MAPK) with subsequent upregulation of pro-inflammatory cytokines, ROS and RNS. [143–145].
Classical DAMPS capable of triggering pattern recognition receptors and the subsequent activation of the immune response include high-mobility group protein B1 (HMGB1), ATP, degraded hyaluronan, heat shock proteins, substance P, uric acid, the S100 proteins and damaged or degraded DNA [146]. HMGB1 and the S100 family of proteins are released following necrotic cell death [147, 148]. HMGB1 activates TLR2 and TLR4 [149, 150], while S100 DAMPS are capable of upregulating TLR4 receptor activity directly [151] or indirectly [147]. Increased levels of ROS and RNS produced by TLR activation can attack unsaturated membrane fatty acids and other cellular molecules generating a range of damaged molecules, including oxidized phospholipids, 4-hydroxynonenal (4-HNE), malondialdehyde (MDA), oxidized low density lipoprotein, and nitroso-protein adducts, protein carbonyls and oxidized and degraded DNA [152, 153]. Alpha beta ketones and aldehydes, protein carbonyls, and other products of lipid peroxidation and protein oxidation also act as DAMPs [152, 154].
These damaged molecules in turn function as redox-derived DAMPs exacerbating TLR2 and/or TLR4 activity, thereby driving increased synthesis of pro-inflammatory cytokines, ROS, and RNS [143]. 4-HNE and MDA are particularly aggressive entities, which attack proteins leading to the creation of reactive carbonyl adducts [155, 156], which, in turn, behave as potent redox-derived DAMPS [154]. Degraded mitochondrial DNA is also known to act as a DAMP and activate TLR-2 and TLR-4 receptors [157, 158]. The TLR radical cycle once further activated by redox-derived DAMPs may rapidly become self-sustaining and self-amplifying and may well underlie the excessive levels of nitro-oxidative stress and chronic immune activation seen in patients with neuro-inflammatory, neurodegenerative and autoimmune diseases [143].
Many patients with a diagnosis of CFS display increased IgM/IgA responses directed against LPS from gram-negative enterobacteria [45], which are known to act as PAMPS capable of activating TLR2/4. Numerous studies have also reported antibodies to an almost bewildering array of pathogens in people with CFS at levels significantly higher than in control subjects [5]. The pathogens that may mediate immune responses via engagement of TLR2/4 include Human Parvovirus 19 [159], Coxsackievirus B [160], HHV-6 [161], Coxiella burnetii [162], Borrelia burgdorferi [163], and Campylobacter species [164]. Activated TLR2/4 complexes are found in many inflammatory and neuro-immune disorders. Gow et al. detected activated TLR4 in patients they described as having “post infectious CFS” [165]. Elevated TLR4 signaling was also detected in a cohort of patients with CFS by Light et al. [166]. TLR4 receptors are also upregulated in the brain and peripheral immune system in patients with multiple sclerosis [167–169]. The same is true of patients with Parkinson’s disease [170] and depression [171]. Upregulated TLR4 signaling is also commonly observed in patients with autoimmune diseases and as a phenomenon observed in for example systemic lupus erythematosus [172], Sjögren’s disease [173], and rheumatoid arthritis [174]. Finally, TLR4 is upregulated in people with cerebrovascular disease and chronically following a major or minor infarct [175]. All in all, an activated TLR radical cycle stimulated by PAMPs and classical and redox-derived DAMPs is associated with chronic immune-inflammatory and O&NS processes which in turn play a pathogenetic role in chronic immune-inflammatory disease and in the disabling fatigue that accompanies these diseases and processes.
IO&NS pathways, neurotransmitters, and fatigue
IO&NS pathways, glutamate metabolism, and fatigue
Another pathway through which pro-inflammatory cytokines may induce chronic fatigue is through effects on glutamate neurotransmission. One major theory is that elevated levels of pro-inflammatory cytokines underpin the pathophysiology of mental fatigue as a result of their capacity to impair glutamate clearance by astrocytes and their widespread deleterious effects on astroglial cells and, consequently, the supply of metabolites to neurons leading to the attenuation of glutamate transmission [74].
Glutamate neurotransmission is vitally important in information processing within the CNS [76] and for long-term potential formation and memory formation [176]. In the brain, extracellular glutamate has to be maintained within narrow limits to ensure a high-precision signal during normal glutamate neurotransmission [177] and also, to prevent glutamate induced neuron excitotoxicity. Glutamate clearance is enabled by sodium (Na+)-dependent uptake transporters, e.g., the glutamate aspartate transporter (GLAST) and glutamate transporter 1 (GLT-1). Both are found at highest concentrations on astrocytes enwrapping synapses of neurons bearing glutamate [178]. Astrocytes play the lead role in maintaining extracellular glutamate levels within physiological norms via glutamate transporters on their surface and relaying calcium ions signals within local and remote neural networks [179, 180]. There is now overwhelming evidence that the optimal performance of these glial cells regulating extracellular glutamate levels underpins information processing, including memory formation and retrieval [181, 182].
Pro-inflammatory cytokines, such as TNFα, IL-1β, and IL-6 impair astroglial glutamate uptake leading to the excessive levels of extracellular glutamate thought to be the ultimate source of mental fatigue. A synthesis of the data from several studies indicates that TNFα inhibits astroglial reuptake of glutamate via the promotion of glutamate induced oxidative stress leading to reduced levels of astrocytic expression and function [183, 184]. Elevated levels of IL-1β and IL-6 appear to inhibit glutamate reuptake by astrocytes via a very similar if not identical mechanism [185, 186]. Lowered astroglial glutamate uptake capacity decreases neuronal uptake of astroglial glucose and metabolic substrates [187, 188].
Adverse consequences on information processing also flow from the disruption of the blood-brain barrier which is an early event in the development of neuro-inflammation [189]. While glutamate transporters on astrocytes and neurons (EAAT 1, EAAT 2, and EAAT 3) play a vital role in keeping glutamate levels within the brain, low glutamate receptors on the abluminal membranes of the BBB also make a major contribution to this endeavor by promoting the removal of glutamate from the brain while simultaneously preventing entry from the systemic circulation [190, 191]. Neuro-inflammation reduces astrocyte gap junction activity impairing the regulation of glutaminergic neurotransmission [192]. Neuro-inflammation also leads to the downregulation of astrocytic glutamate transporter expression via excess levels of glutamate secreted by activated microglia and hence leads to elevated levels of glutamate [193].
Another mechanism exists involving the effects of tryptophan catabolites (TRYCATs), such as kynurenine and quinolinic acid, which have multiple effects on glutamate neurotransmission in the brain. Indoleamine 2,3 dioxygenase (IDO) is activated by pro-inflammatory cytokines, including IFNγ, IL-1, IL-6, and TNFα, acting alone or together or in combination with NF-κB and p38 MAPK [194–196]. IDO is synthesized in several cell types which include microglia and macrophages and catalyses the breakdown of tryptophan to TRYCATs locally in the CNS or incorporated from the periphery [197, 198]. Kynurenine is further catabolized into quinolinic acid in microglia and kynurenic acid in astrocytes. These TRYCATs are both elevated in the CSF of IFNα-treated patients [199]. Elevated levels of quinolinic acid not only contribute to the generation of increased oxidative stress, but an also activate the N-methyl-d-aspartate (NMDA) receptor causing glutamate release [199, 200]. Glutamate release together with oxidative stress can cause excitotoxicity and therefore many authors have implicated excessive levels of quinolinic acid as a contributor to the development of several neurodegenerative illnesses including Huntington’s and Alzheimer’s disease [199, 201, 202]. Kynurenic acid, however, inhibits glutamate release, and acts as an a-amino-3-hydroxy-5-methyl-4-isoxazolepropionicacid (AMPA) and NMDA receptor antagonist [203].
Finally, cytokines may attenuate the expression of glutamate transporters and increase glutamate release [204, 205]. Once released, glutamate may target extrasynaptic NMDA receptors causing a lowered production of neurotrophic factors, e.g., brain-derived neurotrophic factor [206, 207]. There is now considerable evidence that elevated levels of glutamate and subsequent glutamate excitotoxicity is a source of neuropathy or neurodegeneration in Alzheimer’s disease [208], Parkinson’s disease [209], amyotrophic lateral sclerosis [210], Huntington’s disease [211], stroke [210], multiple sclerosis [212, 213], depression [214–216], and other neuropsychiatric disorders [217]. A grim illustration of the neurotoxic potential of TRYCATS can be seen in data appertaining to their production in Alzheimer’s disease and the aftermath of ischaemic stroke [218, 219]. As described above, TRYCATs cause increased glutamate levels and thus could be associated with the mental fatigue that accompanies TRYCAT-related diseases. Figure 1 shows the relationships between the TRYCAT pathway, glutamate, EATT and dopamine (see also next section).

Microglia and astrocytes become activated by signals relaying the presence of increased concentrations of pro-inflammatory cytokines (PICs) in the systemic circulation and the spleen which reach the brain by a variety of routes. Once activated, glial cells produce pro-inflammatory cytokines (PICs) and oxidative and nitrosative stress (O&Ns). Interferon (IFN) α and a range of other neurotoxic and neuromodulatory substances such as the tryptophan catabolites (TRYCATs), including quinolinic acid, kynurenine, and kynurenic acid (kyna). PICs and TRYCATs conspire to inhibit the reuptake of glutamate. An environment of glutamate excitotoxicity and a decrease in dopamine levels are crucial factors in the genesis of cognitive and sensory fatigue. IFNα and activated astrocytes result in impaired glucose metabolism and delivery of oxygen and energy to areas of elevated neural activity. These abnormalities associate with the development of severe intractable fatigue
IO&NS pathways, dopamine metabolism, and fatigue
The weight of evidence now strongly suggests that pro-inflammatory cytokines may target dopamine in the basal ganglia [80, 220]. These actions of pro-inflammatory cytokines on dopamine neurotransmission may be particularly important, relevant to the development of fatigue [92]. Dopamine plays a key role in motivation, drive, psychomotor function, and the promotion of goal-directed activity, and hence in energy and mood regulation [221, 222].
Cytokines have the capacity to decrease dopamine metabolism in the basal ganglia by several different mechanisms which include impairing its synthesis and packaging as well as stimulating its reuptake. Several enzymes which are indispensable in the synthesis of dopamine from tyrosine require the presence of an essential highly redox sensitive coenzyme called tetrahydrobiopterin (BH4). This enzyme may be irreversibly oxidized and hence inactivated in an environment of elevated oxidative stress generated directly or indirectly by pro-inflammatory cytokines [80, 223].
The optimal functioning of dopaminergic neurotransmission is dependent on the packaging of dopamine into vesicles before its release via the vesicular monoamine receptor-2 (VMAT-2). Without this process, dopamine is highly liable to auto-oxidation with the resultant production of a range of neurotoxins and ROS [224]. Decreased VMAT-2 activity can indeed lead to auto-oxidation and ROS production [224, 225]. There is some evidence that elevated levels of IL-1β and TNFα can reduce the activity of vesicular monoamine receptor [226]. The weight of evidence indicates that activation of the p38 MAPK pathway by pro-inflammatory cytokines influences the activity of dopamine transporters leading to an increased reuptake of dopamine [227–229]. Finally, the release of dopamine is partly mediated via glutamatergic systems and hence increased kynurenine acid may inhibit dopamine metabolism [199].
A further pathway whereby dopamine influences energy is its direct effects on mitochondrial function. Dopamine has direct effects on mitochondrial function, causing mitochondrial permeability pore transition, reducing oxygen consumption, and uncoupling [230]. In Parkinson’s, dysregulated dopamine transmission is linked to mitochondrial dysfunction. Dopamine has direct effects on mitochondrial complex I in particular [231]. Agents driving dopamine, prototypically amphetamines, cause mitochondrial dysfunction [232].
Decreased dopamine is one of the accepted causes of the severe incapacitating fatigue in people suffering from major depression as discussed previously. The relationship between impaired dopamine production and the production of the severe physical fatigue experienced by many people with Parkinson’s disease however was a matter of some considerable controversy until 2003. In that year, Lin and others presented the results of an elegant double blind study demonstrating conclusively that physical fatigue suffered by Parkinson’s disease patients was indeed mediated largely by impaired dopamine production [233].
IO&NS pathways, mitochondria dysfunction, and fatigue
ATP and the electron transport chain
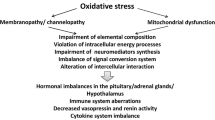
Central and peripheral fatigue often with concomitant exercise intolerance, are common symptoms of mitochondrial diseases [234, 235]. Mitochondrial diseases may be classified as syndromic or non syndromic [21] and the bewildering array of symptoms presented by people with mitochondrial disease is ultimately due to impaired oxidative phosphorylation and generation of ATP caused by mutations in mitochondrial or nuclear DNA [21, 234, 236]. While congenital and acquired mutations are both known to cause mitochondrial disease, there is a growing awareness that secondary mitochondrial dysfunction can occur as a result of chronic O&NS and elevation of pro-inflammatory cytokines [21, 237]. Such dysfunction likely impairs oxidative phosphorylation and ATP production via a number of different mechanisms [21]. Excessive levels of nitric oxide inhibit the activity of enzymes which form complex I and complex IV of the electron transport chain [238–240]. These enzymes are also inhibited, probably irreversibly, by another RNS namely peroxinitrite [21, 240]. Inhibition of these enzymes not only blocks the production of ATP but leads to a massive increase in ROS generation by mitochondria, due to incomplete oxygen reduction, and this in turn begets more RNS and more damage to the electron transport chain and other essential mitochondrial components leading to a spiral of increasing bioenergetic decline [21]. O&NS also drives a shift towards glycolysis rather than oxidative energy generation [237].
IO&NS, coenzyme Q10, and fatigue
Coenzyme Q10 deficiency together with elevated levels of oxidative stress and mitochondrial dysfunction has been detected in plasma or white blood cells of patients with CFS [241, 242] and fibromyalgia [243–246]. Interestingly, the level of coenzyme Q deficiency is associated with the severity of fatigue, pain, and neurocognitive deficits experienced by patients with CFS [241].
A number of treatment studies with coenzyme Q10, typically at doses of 300 mg/day, have demonstrated significant improvements in markers of O&NS, mitochondrial biogenesis and ATP generation following supplementation, which correlated significantly and positively with reductions in pain and fatigue in people with a diagnosis of fibromyalgia [246, 247]. These results are very much in line with other studies examining the benefits of coenzyme Q10 on ATP generation and improved mitochondrial function [248]. There is also good evidence for objective improvements in muscle function and muscle fatigue at relatively high doses and or long durations of treatment [249, 250]. Confidence in the effectiveness and safety of coenzyme Q10 supplementation has been further increased by a recent landmark study by Alehagen et al. [251] where 200 mg of coenzyme Q10 daily given for 4 years produced an absolute reduction of cardiovascular mortality of 6 % compared to control with minimal side effects in a population or elderly patients. The favorable safety profile of far higher doses (>2 g daily) was established in studies examining the effects of coenzyme Q10 supplementation in Parkinson’s disease [248].
IO&NS pathways, the master gene PGC-1α, and fatigue
The PGC-1 family of transcription factor orchestrates the regulation of energy metabolism. Their expression activates the transcription of regulatory genes involved in driving an increased capability for energy generation [252]. Animal experiments involving ablation of the PGC-1a gene demonstrated a diminished expression of genes governing fatty acid oxidation, the KREBS cycle, and oxidative phosphorylation [253]. There is growing evidence implicating impaired PGC-1 expression in the development of mitochondrial dysfunction and neurodegenerative processes [254–256]. Dysregulated expression of this transcription factor is involved in the etiopathogenesis of Parkinson’s disease [255]. Reduced expression of PGC-1a found in the brains of multiple sclerosis patients is associated with decreased mitochondrial membrane potential, reduced production of proteins associated with oxidative phosphorylation, and cellular antioxidant defenses. It is particularly noteworthy that decreases in all these parameters correlate significantly and positively with loss of cortical neurons [256].
PGC-1a also plays a crucial role in regulating muscle performance and long-term adaption to exercise and hence chronic downregulation of this transcription factor which can occur in an environment of chronic oxidative stress, as discussed below, can have serious consequences for exercise performance in an affected person. Elevated levels of this transcription factor, which takes place during exercise, drives a switch from fast type II striated muscle fibers, which rely on ATP produced by glycolysis as their energy source, to slow type I muscle fibers utilizing energy produced by mitochondrial respiration [257, 258]. PG1-a activity regulates glucose and fatty acid refueling of exercising muscle by promoting fatty acid oxidation [259, 260] and lactate homeostasis and hence its expression may be responsible for delaying exercise-induced accumulation of lactate in striated muscle [257]. The sum of these positive actions is responsible for conferring resistance to exercise-induced muscle fatigue [233]. The loss or under-expression of the PGC-1a gene makes individuals and animals prone to develop muscle fatigue at a much less intense or prolonged level of exercise than control subjects [260, 261]. For example, mitochondrial dysfunctions and lowered levels of PGC-1α, a trigger of mitochondrial biogenesis, may cause muscle weakness, fatigue and exercise intolerance [262]. Other data show that Korean mistletoe extract improves endurance capacity in mice by activating mitochondrial activity probably through enhancing effects on PGC-1α [263]. PGC-1α additionally promotes resistance to muscle fatigability by driving fiber type switching possibly via effects on intracellular calcium levels [259]. Muscle PGC-1α knockout animals show lowered endurance capacity and more fiber damage and increased inflammation following exercise [264].
Given that elevated levels of TNFα leads to the downregulation of PGC-1α [265, 266], it is tempting to speculate that insufficient levels of this transcription factor contribute to the severe muscle fatigability seen in many patients with a diagnosis of CFS. There is as yet no evidence of this but this possibility would seem to be worthy of further investigation. Clearly, however, decreased levels of PGC-1a expression lead to impairments in oxidative phosphorylation which lead to dramatically elevated ROS production by mitochondria within myocytes [21, 78]. To make matters worse, diminished PGC-1α expression also impairs the activation of the antioxidant defense systems needed to resist the damaging effects of increasing radical species in a toxic cellular environment [267, 268].
IO&NS pathways, brain dysfunctions, and fatigue
Several areas within the brain and in particular the anterior and/or posterior cingulate gyrus and the insular cortex have been implicated in the development of fatigue [166, 269]. PET scan studies using healthy volunteers examining regional blood flow during a fatiguing task have revealed that the medial orbitofrontal cortex exhibits a positive correlation in activity with fatigue (Brodmann’s area 10/11), providing good evidence that the medial orbitofrontal cortex is a region of the brain whose activity is closely associated with generating mental fatigue [270]. In neurological disorders, pathological changes within the structures of the frontal cortex and deep gray matter are neural correlates of the development of pathological fatigue [271]. Fatigue in different clinical populations has additionally been linked with changes in brain structures, such as the ascending reticular formation [272], the monoaminergic nuclei [273], and the frontostriatal network [271].
A study using positron emission tomography to examine the effects of IFNα on fatigue revealed the existence of an elevated metabolism of glucose in the basal ganglia [274]. The presence of elevated glucose metabolism in the putamen and nucleus accumbens correlates significantly with patient reports of fatigue [275]. This distribution of increased basal ganglia glucose metabolism is strikingly similar to that found in Parkinson’s disease [276]. Functional magnetic resonance imaging has also revealed denuded levels of neural activation in the basal ganglia in patients subjected to exogenously administrated IFNα [92]. Inoculation of healthy volunteers with LPS or typhoid vaccine (which are both inducers of pro-inflammatory cytokines) has been demonstrated to produce similar responses in the basal ganglia, indicating that administration of IFNα and other inflammatory inputs yield similar effects [277, 278].
Dysfunction in prefrontal cortex activity is thought to play a significant role in generating fatigue in patients with neuro-immune disorders, such as multiple sclerosis [279, 280] and CFS [281, 282]. Numerous studies have confirmed significant associations between abnormalities in the ventromedial prefrontal cortex and fatigue levels in multiple sclerosis [283, 284] and yet further support for this position stems from the fact that trauma based disruption of the ventromedial prefrontal cortex results in the production of severe chronic fatigue [285]. Pardini et al. [286] using fMRI investigated the brain areas associated with the generation of fatigue in patients with multiple sclerosis with minimal levels of disability who nevertheless suffered from severe fatigue. The authors actually detected a positive correlation between severe chronic fatigue and accuracy levels in timed tasks which they described as a “fatigue-motor performance paradox” mediated by oribitofrontal cortex and cerebellar activity [286]. Cortical atrophy involving the posterior parietal lobe (a region playing a major role in information integration and motor planning) also displays robust associations with the sensation of fatigue indicating that impairments in higher order parameters of motor control also play a large part in determining levels of fatigue in multiple sclerosis [287].
Many people with a diagnosis of CFS display significant lateral prefrontal cortex gray matter volume reductions [281, 282]. Cook and fellow workers examined the brain regions associated with reported fatigue during a task designed to generate cognitive fatigue in patients with CFS using functional MRI technology. The authors detected positive relationships between fatigue sensation and responsiveness in the cerebella, cingulate, frontal, temporal, and cerebellar regions [288]. Another study involving patients with CFS demonstrated increased fMRI activation in the posterior cingulate gyrus and in the occipitoparietal cortex, together with impaired activation in the dorsolateral dorsomedial and prefrontal cortices during fatigue provocation [289]. Significant reductions in gray matter volume located primarily in the prefrontal cortex have also been reported in many people afforded a diagnosis of CFS [281, 282] and decreased glucose metabolism in the prefrontal and orbitofrontal cortex is also seen in people with this illness. Hypoperfusion of the brain and brainstem also appears to be widespread in such patients [290, 291]. It is worthy of note that decreased cerebral blood flow in deep gray matter associated with fatigue development in some people with a CFS diagnosis is associated with the severity of fatigue in multiple sclerosis [292, 293]. Similar widespread reductions in blood flow are seen in depression and appear to be state related [294].
The consequences of such disruption are not immediately apparent until it is realized that the prefrontal territories are involved in effort evaluation and other cognitive tasks and hence dysfunction in these areas may well underpin the development of chronic fatigue [295, 296]. Rich connections exist between medial prefrontal structures and regions such as the hypothalamus, which play pivotal roles in regulating visceromotor and neuroendocrine responses [297] and the orbital prefrontal cortex which receives and integrates sensory information from virtually every system [298]. All in all, the weight of evidence demonstrates that the ventromedial prefrontal cortex plays an indispensable role in the selection and evaluation of motor and cognitive outputs and is the region of the brain responsible for a “cost benefit analysis” of and given activity [295, 296]. It is thus hardly surprising that disruption of this area produces sensations of severe mental fatigue [285].
IO&NS pathways, muscle dysfunctions, and fatigue
Muscle fatigue and impaired feedback from muscle afferents
Muscle fatigue is a common occurrence in a range of autoimmune diseases including systemic lupus erythematosus [299, 300], Sjögren’s disease [301, 302], and rheumatoid arthritis [303, 304], and is a diagnostic criterion for CFS. Severe muscle fatigue and weakness is one of the most disabling features in the overall motor impairment in Parkinson’s disease [305, 306]. de Haan and fellow workers reported that the disabling levels of muscle fatigue suffered by people with multiple sclerosis was not due to reduced muscle usage but rather to lower oxidative capacity in the muscle groups studied [307]. Steens et al. reported a central contribution to the muscle fatigue experienced by patients where there was an inadequate increase in cortical activation to compensate for changes in voluntary activation [308]. This built on an earlier study by the same team which noted impaired voluntary activation in multiple sclerosis patients and, for the first time, demonstrated a positive relationship between objective measures of muscle fatigue and perceived levels of general fatigue in patients suffering from this illness [309]. Many patients afforded a diagnosis of fibromyalgia also suffer from pathological levels of muscle fatigue linked to mitochondrial dysfunction within the affected muscle groups. However, other patients diagnosed with fibromyalgia seem to have muscle fatigue resulting from central motor control failure [310, 311] and yet others display muscle membrane abnormalities as evidenced by surface EMG readings [312]. Muscle fatigue can be defined as a reduction in maximal voluntary muscle force induced by exercise. It may result from a combination of factors which include muscle level peripheral changes and central nervous system failure to adequately drive motor neurons. All in all, the multiple lines of evidence demonstrate that muscle fatigue is not simply resident in striated muscle [313].
Maximal muscle voluntary activation is in general significantly lower than maximal muscle force and furthermore this level of voluntary activation decreases rapidly over time. Voluntary activation of elbow flexor muscles for example may be optimal when performing maximal voluntary muscle force of relatively short duration but central fatigue quickly develops when the muscle contractions are repetitive or sustained [314, 315] due at least in part to decreased rate of firing of motor neuron units [316]. The origin of fatigue during maximal voluntary muscle force is primarily located in muscles but some originate from progressive decrease in voluntary activation of muscle, which is termed central fatigue, and also as a result of impaired output signals from the motor cortex [317].
Central fatigue appears to originate in regions of the brain located upstream the motor cortex [317]. Central fatigue is defined as the inability of the CNS to drive motor neurons efficiently via the exertion of powerful inhibitory effects on central motor drive during the performance of intermittent or prolonged aerobic exercise [318]. Changes in cortical and spinal neuron activity take place during the development of muscle fatigue, but their effects are separate from supraspinal fatigue, which is generated as a result of firing of fatigue sensitive muscle afferent fibers which act to decrease the voluntary descending drive [317]. The weight of evidence indicates the existence of two different types of muscle receptors with different responses following mechanical stimulation [319]. The bulk of these sensory afferents are group III. It is however worth emphasizing that a minimum of 40 % of group IV afferents are likely non-nociceptive [319]. There is now evidence that feedback from opioid-mediated, otherwise described above as group III and IV, muscle afferents conspire to inhibit central motor drive, thereby inducing central fatigue and limiting the development of peripheral muscle fatigue during exercise [318]. However, feedback from group III/IV muscle afferents exerts a number of different effects in addition to limiting central motor drive, importantly such feedback during aerobic exercise improves ventilatory and cardiovascular responses [320] and hence positively affects VO2 measurements. All in all, copious amounts of data exists demonstrating that opioid-mediated muscle afferents play a key role as part of a homeostatic mechanism designed to inhibit central motor drive thereby limiting muscle fatigue and perhaps permanent muscle damage [321]. Mechanistically, sensory information from muscle afferents activate an inhibitory system which inhibits primary motor cortex output in a phenomenon which is often described as supraspinal fatigue. In response to such inhibition, motivational systems activate in an attempt to compensate for this level of inhibition and ultimately the motor output is a resultant of the balance of activity between these inhibitory and facilitatory systems [322]. It is also worth noting, if just for the sake of completeness, opioid receptor-sensitive muscle afferents in the spinal chord also play a facilitative role in the muscle fatigue-induced increase in intracortical inhibition seen in exercising healthy humans [323].
Unsurprisingly, voluntary muscle contraction has been the focus of the majority of studies into muscle fatigue, but many lines of evidence now indicate that impaired function ultimately provoking voluntary muscle contraction failure likely occurs at all levels of the neuromuscular system. There is much debate over which mechanisms if any should be given primacy. Some authors argue most common failure stems from reduced motor command signal emanating from the motor cortex [324]. Noakes and fellow workers [325] have proposed that failures in voluntary muscle contraction emanate from the activity of a central comparator region in the prefrontal cortex capable of integrating homeostatic inputs from numerous physiological systems and then acting to terminate motor commands, via the activation of compensatory homeostatic systems, following sensory input from muscle afferents indicating the existence of sub-optimal energy resources. In this model, sensory fatigue is regarded as the conscious awareness of these homeostatic mechanisms at work [325].
IO&NS pathways, the sodium and potassium ion pump, and fatigue
Another source of muscle fatigue is the deleterious effects of prolonged ROS elevation on the sodium-potassium pump in muscle fibers whose optimum function is essential in prolonging muscle activity by minimizing fatigue. Interestingly, dysfunctions in the sodium potassium pump have been demonstrated in patients diagnosed with CFS [326], as well as depression and bipolar disorder [327, 328]. The sodium potassium ion pump plays a major role in the function of skeletal muscle by regulating concentration gradients of sodium and potassium across cellular membranes. The contractions of skeletal muscle induce a profound loss of potassium ions (K+) and gain of sodium ions (Na+) within muscle cells [329], in spite of a marked compensatory increase in the Na+/K+-ATPase sodium potassium pump [330]. Therefore, this pump is a crucial player in minimizing muscle Na+ and K+ disturbances, and consequently in the preservation of membrane excitability and production of muscle force [329, 330].
Numerous studies indicate significant perturbations in muscle K+ homeostasis in exercising humans, which correlate positively with the development of fatigue [331, 332]. Several authors have proposed that elevated levels of ROS, normally produced during contractions of skeletal muscle during exercise could act to depress the optimal activity of the sodium potassium pump thereby providing an important connection between elevated ROS and disturbed Na+ and K+ homeostasis [333, 334]. This pump has an optimal redox range [335, 336] and hence the well-established role of elevated ROS in generating muscle fatigue [124, 334] stems, in a large part, from the inactivation of the sodium potassium ion pump [334].
The sodium potassium pump plays a crucial role in maximizing muscle performance and delaying and minimizing muscle fatigue during exercise [334, 337]. McKenna et al. found that the exercise-induced attenuation of sodium potassium pump activity was significantly improved by N-acetyl cysteine as evidenced by the lowering of the exercise-induced elevation of plasma [K+] thereby indication a strong association between ROS and inactivation of sodium further implicating ROS, and reduced sodium potassium pump and K+ activity in muscle fatigue [334]. This is clearly a likely mechanism by which prolonged oxidative stress could cause muscle pathology in people with CFS [326].
Increased muscle glutathione, cysteine, and increased muscle N-acetyl cysteine would all act to remove ROS from muscle cells and could thus protect the sodium potassium pump from the damaging effects of increased oxygen radicals. This proposition is consistent with other lines of evidence demonstrating that endogenous ROS scavengers such as catalase superoxide dismutase [338] and antioxidant agents such as dithiotreitol cysteine [339], α-tocopherol, carnosine [336], and histidine [340] display protective effects against ROS-induced attenuation of sodium potassium pump activity. The improvement in sodium potassium ion pump activity with increased muscle glutathione and cysteine availability was also demonstrated with N-acetyl cysteine infusions [124]. N-acetylcysteine administered orally, or via infusion, acts as an antioxidant, reduces muscle fatigability and enhances ergonomic performance by attenuating the decrease in Na+/K+-pump activity caused by the elevated ROS levels accompanying aerobic exercise [124, 334, 341]. The positive effects are dose- and duration-related and objective improvements in mitochondrial membrane potential, VO2, antioxidant capacity, markers of ROS, and RNS tissue damage and immune profiles have all been reported [124, 334, 341–343]. The most striking benefits in humans with a disease associated with chronically elevated O&NS, such as CFS, appear to occur following prolonged administration at between 2.8 and 4.2 g/day [342]. However, as discussed previously, numerous studies have demonstrated increased levels of ROS and RNS during exercise [113, 114] and at physiologically normal concentrations these radical species are indispensable signaling molecules modulating several essential physiological processes in muscle cells in response to increased levels of exercise [344, 345].
IO&NS pathways and muscle metabolite built up
The role of metabolite buildup in signaling between the muscles and brain has been the subject of intense interest. Ion-sensing channels in muscle (acid-sensing ion channels) with the capability of detecting pH and ATP changes and activating sensory nerves once a pH threshold is exceeded facilitates this direct muscle brain signaling [346, 347]. These “acid-sensing” ion channels are predominant in sensory nerve fibers innervating skeletal muscle [166] and have the capacity to detect pH changes in contracting muscle. The sensory nerve fibers containing acid-sensing ion channels are located on the exterior surfaces of venules and arterioles located in the fascia surrounding the muscles [348]. Although incapable of initiating the activity of sensory neurons on its own, ATP, at the trace concentrations located in muscle interstitium in contracting muscles, greatly increases the sensitivity of acid-sensing ion channels to pH and lactate thereby maintaining the gated current needed to activate sensory muscle afferents [346, 347]. The weight of evidence now indicates that the receptors mediating responses to these metabolites are a number of purigenic receptors a P2 acid-sensing ion channel receptor. Several combinations of metabolites produced in fatiguing muscles have the capacity of activating dorsal root ganglion neurons [346, 347] which are exquisitely sensitive to combinations of ATP, protons and lactate at the normal physiological range of concentrations [349].
There is some interesting research implicating abnormalities in the transcription of the above receptors in fatigue generation in a cohort of people diagnosed with CFS [350]. This is particularly noteworthy in the light of the fact that the cardinal hallmark of the illness in many people is dramatically increased muscle fatigue and pain following even trivial increases in exercise [351] and minor exercise dramatically increases their other symptoms [166]. Light et al. compared longitudinal adrenergic α2, β1, and β2 receptors, IL-6, IL-10, TNFα, TLR4, and CD14 expression in the muscles of patients with a diagnosis of CFS under the CDC 1994 criteria compared to healthy controls [166, 350]. While the latter displayed no statistically significant increase in mRNA for any of the receptor genes examined, CFS patients displayed significant elevations in the expression in virtually all of these genes (save for IL-10 and IL-6) as soon as 30 min after the commencement of moderate exercise and were of far greater magnitude than seen in controls at maximal exercise. The authors additionally noted strong positive correlations between elevations in mRNA of adrenergic β1, β2, adrenergic TLR4, IL-10, α2A, COMT, and CD14 in CFS patients and numerous measurements of mental fatigue in the aftermath of the experiments which persisted for 48 h. The abnormalities discussed above could be a major contribution to the profound pathological levels of fatigue suffered by these patients.
IO&NS pathways, heat shock proteins, and fatigue
Lower baseline levels of heat HSPs, including HSP70 and HSP27, are observed in some patients with CFS [105]. Levels of HSP27 in lymphocytes and monocytes are additionally significantly and positively correlated with fatigue resistance [352]. A number of vital cytoprotective effects have been assigned to HSPs, especially the HSP70 group. They essentially cover chaperone activities being involved in the management of protein disposal folding, correction of misfolding, prevention of protein accumulation, and escorting proteins across external and internal cellular membranes [353]. HSPs provide cellular protection in the face of various stressors such as increased oxidative stress, and activation of proteases and release of proteolytic enzymes [354]. Many stress signals are capable of activating HSP transcription and promoting post translational modification including energy depletion and generation of ROS [355]. HSPs serve to modulate signals initiated by immune-inflammatory responses, in particular those leading to the production of cytokines [356]. Increases in the levels of intracellular HSP levels improve cell resistance to pro-inflammatory cytokines such as IL-1 and TNFα [357] and decreases their production [358]. However, when HSPs located on or within cells escape into the extracellular environment following necrotic cell death or viral infection, these proteins activate and enhance the immune response. Hsp70 also facilitates antigen presentation and provoke cytokine production in antigen presenting cells (APGs) such as dendritic cells and macrophages [359, 360].
Several authors have demonstrated acute exercise results in the production of elevated levels of heat shock proteins in contracting skeletal muscle [361]. The situation in many patients diagnosed with CFS is however entirely atypical. Such patients demonstrate abnormal HSP responses during exercise and for a prolonged period thereafter [362]. Many patients with CFS additionally display low baseline levels of certain HSPs and an impoverished and delayed increase during exercise [104, 105]. This becomes even more intriguing in the light of evidence that HSP70 and HSP27 fail to rise during exercise in patients who report an infective origin of their symptoms but not in those who do not [105]. Intriguingly, many viruses implicated in the development of symptoms in at least some patients with CFS such as Epstein Barr and other herpes viruses suppress production of host HSPs [363]. Elevated levels of HSPs during exercise protect muscle lipids, proteins, and DNA against the corrosive effects of elevated oxygen and nitrogen species [364, 104] and hence the pathological consequences stemming from the failure of this cytoprotective system cannot be overstated.
IO&Ns pathways, mitochondria, and muscle fatigue
O&NS signals adversely affect mitochondrial morphology and function in skeletal muscle [344, 345]. ROS are produced at a multiplicity of sites within muscle fibers including the metabolism of arachadinonic acid, phospholipase A2, and the activity of the electron transport chain within mitochondria [365, 366, 21]. Mitochondrial dysfunction in particular is known to be a source of elevated cellular ROS levels [21]. Oxidative stress corrupts muscle components via lipid peroxidation [367], oxidation of mitochondrial and nuclear DNA [368], protein carbonylation [369], tyrosine nitration [369], and thiol group oxidation [367]. A number of elements crucial for the optimal performance of muscles contain thiol proteins and are hence highly vulnerable to oxidative stress induced dysfunction. They include the ryodine receptor calcium ion release channel [370], SR Ca2+ ATPase [371], troponin [372], tropomyosin [373], myosin [374], and actin [375]. All in all, it is clear that a number of thiol-regulated proteins mediate muscle fatigue induced by oxidative stress and resultant thiol oxidation but the proteins contained within the Na+/K+ ATPase pump as discussed above appear to be the most important [115, 376].
Discussion
We aimed to review the evidence indicating that a range of systemically elevated pro-inflammatory cytokines are intimately involved in the genesis and maintenance of the pathological levels of fatigue endured by people suffering from cancer and a range of neuro-psychiatric, neuro-inflammatory, and autoimmune diseases. Figure 2 shows the different pathways that may cause fatigue. Overall, the evidence implicating a causal role for pro-inflammatory cytokines, at least in part, in the onset of fatigue is persuasive. The weight of evidence indicates that elevated levels of pro-inflammatory are also intimately involved in the genesis and maintenance of neuro-inflammation and chronic disruption of the BBB. Neuro-inflammation, invariably accompanied by disruption of the BBB, is causatively implicated in the development of mental fatigue.

Prolonged tissue inflammation as a result of a protracted immune activation or other inflammatory insult leads to chronically elevated levels of pro-inflammatory cytokines (PICs) and oxidative and nitrosative stress (O&NS) leading to damage of proteins, lipids, and DNA. Some of these damaged molecules may consecutively function as redox-derived damage-associated molecular patterns (DAMPs) capable of activating toll-like receptors (TLR) which once activated lead to the production of more PICs, O&N species and thus redox-derived DAMPs. In this manner, once activated by PAMPs and DAMPS, an environment of chronic immune activation and inflammation in the periphery may become self-sustaining and self-amplifying in a TLR radical cycle leading to damage to mitochondria impaired OXPHOS and direct inhibitory effects on muscle function. Signals of peripheral inflammation can reach the brain leading to the activation of microglia and astrocytes. This signaling is mediated directly by PICs in the circulation which can enter the CNS via a number of routes or via the activation of the vagus nerve. Once activated, astrocytes and microglia produce neuromodulatory and neurotoxic substances including tryptophan catabolites (TRYCATs), PICs, IFNα, and O&NS. PICs and TRYCATs conspire to raise glutamate and decrease dopamine levels while activated astrocytes and IFNα can adversely affect glucose metabolism and perfusion levels throughout the brain. These abnormalities could explain the development of severe intractable fatigue in neuro-immune and autoimmune diseases. In normal circumstances, muscle fatigue during exercise is facilitated by group III/IV afferents activated by the presence of certain metabolites which relay information to the prefrontal cortex which in turn inhibits muscle function. The metabolites in muscles resulting from chronic inflammation could lead to chronic activation of these efferents and chronic inhibition of muscle function
The effects of elevated O&NS in generating muscle fatigue and chronic muscle pathology are well documented and the corruptive effects of ROS and RNS on proteins with an essential role in muscle function are equally well reported. The corrosive effects of O&NS extend to the inactivation of the sodium potassium pump which impacts muscle performance and fatigue. Activation of the TLR2/4 Radical cycle by DAMPs including heat shock proteins may drive chronic inflammation and O&NS processes thereby further activating the pathways to chronic fatigue. Limited adaptability of mitochondria to increase energy output and mitochondrial dysfunctions, potentially secondary to elevated O&NS is another plausible explanation for the existence of peripheral and central fatigue.
Excessive levels of cytokines are also known to disrupt dopaminergic neurotransmission particularly in the basal ganglia which is a region of the brain seemingly involved in the genesis of fatigue in many neurological illnesses. The capacity of elevated cytokines to impede reuptake of glutamate by astrocytes is another commonly reported phenomenon. The proposal that elevated levels of extracellular glutamate underpins, at least in part, the development of cognitive or mental fatigue, is intriguing. The evidence that neuro-inflammation and BBB disruption make independent contributions to increased levels of extracellular glutamate strengthens both the argument that abnormal glutamate levels are involved in the genesis of mental fatigue and that elevated levels of pro-inflammatory cytokines drive abnormal glutamate levels. Another indirect route by which elevated levels of pro-inflammatory cytokines in the periphery and or the CNS might cause cognitive fatigue is via the production of tryptophan catabolites once again adversely affecting glutaminergic and dopaminergic neurotransmission. IFNα disrupts glucose homeostasis in the basal ganglia and suppresses neural activity in the same region, both potential causes of cognitive fatigue.
While the abovementioned neuro-immune abnormalities may be important generators of chronic fatigue, other central and peripheral mechanisms could account for the presence of central and peripheral chronic fatigue. Functional and structural abnormalities in several brain areas are implicated in the genesis of chronic fatigue. The most common areas cited being the frontal and prefrontal cortex and the basal ganglia. It is however entirely possible that activation of microglia and the adverse effects of pro-inflammatory cytokines and IFNα could be the source of dysfunction within the basal ganglia so often implicated in the genesis of fatigue. Other studies place specific emphasis on structural and functional abnormalities within gray matter such as low oxygen perfusion levels and impaired glucose metabolism, both of which could result from reactive astrogliosis and hence once again these observations could ultimately flow from the results of activated immune and inflammatory pathways in the periphery.
Peripheral fatigue involves muscle weakness which can originate within muscles (e.g., muscle opioid receptors, muscle membranes, toxic levels of chemical entities within muscles) and central components originating from multiple levels of the neural axis (e.g., higher brain centers, motor cortex, and spinal cord). These elements are all involved in the homeostatic control of muscle function aimed at preserving muscle function and preventing muscle damage during exercise. Abnormalities in the transcription of receptors on muscle membranes which communicate information about levels of toxic metabolites to the brain, via the activation of dorsal root ganglion neurons, is another potential mechanism whereby muscle fatigue can occur at unusually low levels of effort. There may also be an intriguing connection between a state of chronic inflammation in the muscles and the development of sensory fatigue. It is noteworthy that the toxic milieu of metabolites which activates group III/IV efferents, resulting in inhibition of muscle performance, is chronically present in muscle in a state of chronic inflammation with elevated O&NS and TNFα. Much of this milieu is created as a result of impaired oxidative phosphorylation and cellular stress. The point however is that this is very much part of the danger signal which activates an inhibitory response from the prefrontal cortex and would at least theoretically result in chronic inhibition of muscle function and a severely compromised ability to exercise. Under the model proposed by some authors, the homeostatic mechanisms normally responsible for protecting muscles from exercise-based damage would also be chronically activated and chronic sensory fatigue would result from a conscious interpretation of these homeostatic mechanisms in action.
References
Weis J (2011) Cancer-related fatigue: prevalence, assessment and treatment strategies. Expert Rev Pharmacoecon Outcomes Res 11(4):441–6. doi:10.1586/erp.11.44
Segal B, Thomas W, Rogers T, Leon JM, Hughes P, Patel D, Patel K, Novitzke J, Rohrer M, Gopalakrishnan R, Myers S, Nazmul-Hossain A, Emamian E, Huang A, Rhodus N, Moser K (2008) Prevalence, severity, and predictors of fatigue in subjects with primary Sjögren's syndrome. Arthritis Rheum 59(12):1780–7. doi:10.1002/art.24311
Ahn GE, Ramsey-Goldman R (2012) Fatigue in systemic lupus erythematosus. Int J Clin Rheumatol 7(2):217–227
Hewlett S, Ambler N, Almeida C, Cliss A, Hammond A, Kitchen K, Knops B, Pope D, Spears M, Swinkels A, Pollock J (2011) Self-management of fatigue in rheumatoid arthritis: a randomised controlled trial of group cognitive-behavioural therapy. Ann Rheum Dis 70(6):1060–7. doi:10.1136/ard.2010.144691
Morris G, Maes M (2013) Myalgic encephalomyelitis/chronic fatigue syndrome and encephalomyelitis disseminata/multiple sclerosis show remarkable levels of similarity in phenomenology and neuroimmune characteristics. BMC Med 11:205. doi:10.1186/1741-7015-11-205
Beiske AG, Svensson E (2010) Fatigue in Parkinson's disease: a short update. Acta Neurol Scand Suppl 190:78–81. doi:10.1111/j.1600-0404.2010.01381.x
Winward C, Sackley C, Metha Z, Rothwell PM (2009) A population-based study of the prevalence of fatigue after transient ischemic attack and minor stroke. Stroke 40(3):757–61. doi:10.1161/STROKEAHA.108.527101
Alsén P, Brink E, Persson LO, Brändström Y, Karlson BW (2010) Illness perceptions after myocardial infarction: relations to fatigue, emotional distress, and health-related quality of life. J Cardiovasc Nurs 25(2):E1–E10. doi:10.1097/JCN.0b013e3181c6dcfd
Maes M, Kubera M, Obuchowiczwa E, Goehler L, Brzeszcz J (2011) Depression’s multiple comorbidities explained by (neuro) inflammatory and oxidative & nitrosative stress pathways. Neuro Endocrinol Lett 32(1):7–24
Thase ME (2009) Atypical depression: useful concept, but it’s time to revise the DSM-IV criteria. Neuropsychopharmacology 34(13):2633–41. doi:10.1038/npp.2009.100
Al-shair K, Muellerova H, Yorke J, Rennard SI, Wouters EF, Hanania NA, Sharafkhaneh A, Vestbo J; ECLIPSE investigators. Examining fatigue in COPD: development, validity and reliability of a modified version of FACIT-F scale. Health Qual Life Outcomes. 2012 23;10:100. doi: 10.1186/1477-7525-10-100. PubMed PMID: 22913289; PubMed Central PMCID: PMC3491053.
Norrie J, Heitger M, Leathem J, Anderson T, Jones R, Flett R (2010) Mild traumatic brain injury and fatigue: a prospective longitudinal study. Brain Inj 24(13–14):1528–38. doi:10.3109/02699052.2010.531687
Römkens TE, van Vugt-van Pinxteren MW, Nagengast FM, van Oijen MG, de Jong DJ (2011) High prevalence of fatigue in inflammatory bowel disease: a case control study. J Crohns Colitis 5(4):332–7. doi:10.1016/j.crohns.2011.02.008
Colosimo C, Millefiorini E, Grasso MG, Vinci F, Fiorelli M, Koudriavtseva T, Pozzilli C (1995) Fatigue in MS is associated with specific clinical features. Acta Neurol Scand 92(5):353–5
Krupp LB, Pollina DA (1996) Mechanisms and management of fatigue in progressive neurological disorders. Curr Opin Neurol 9(6):456–60
Ford H, Trigwell P, Johnson M (1998) The nature of fatigue in multiple sclerosis. J Psychosom Res 45(1):33–8
Schreurs KM, de Ridder DT, Bensing JM (2002) Fatigue in multiple sclerosis: reciprocal relationships with physical disabilities and depression. J Psychosom Res 53(3):775–81
Flachenecker P, Bihler I, Weber F, Gottschalk M, Toyka KV, Rieckmann P (2004) Cytokine mRNA expression in patients with multiple sclerosis and fatigue. Mult Scler 10(2):165–9
Chaudhuri A, Behan PO (2000) Fatigue and basal ganglia. J Neurol Sci 179(S 1–2):34–42
Lindqvist G, Malmgren H (1993) Organic mental disorders as hypothetical pathogenetic processes. Acta Psychiatr Scand Suppl 373:5–17
Morris G, Maes M (2014) Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Metab Brain Dis 29(1):19–36. doi:10.1007/s11011-013-9435-x
Blach-Olszewska Z, Leszek J (2007) Mechanisms of over-activated innate immune system regulation in autoimmune and neurodegenerative disorders. Neuropsychiatr Dis Treat 3(3):365–72
Perl A (2013) Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol 9(11):674–86. doi:10.1038/nrrheum.2013.147
Pagano G, Castello G, Pallardó FV (2013) Sjøgren’s syndrome-associated oxidative stress and mitochondrial dysfunction: prospects for chemoprevention trials. Free Radic Res 47(2):71–3. doi:10.3109/10715762.2012.748904
Szabó-Taylor KE, Nagy G, Eggleton P, Winyard PG. 2013. Oxidative stress in rheumatoid arthritis. In: Studies on arthritis and joint disorders. pp.145–167. DOI 10.1007/978-1-4614-6166-1_8. Springer New York
Exner N, Lutz AK, Haass C, Winklhofer KF (2012) Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J 31(14):3038–62. doi:10.1038/emboj.2012.170
Allen CL, Bayraktutan U (2009) Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke 4(6):461–70. doi:10.1111/j.1747-4949.2009.00387.x
Sosa V, Moliné T, Somoza R, Paciucci R, Kondoh H, LLeonart ME (2013) Oxidative stress and cancer: an overview. Ageing Res Rev 12(1):376–90. doi:10.1016/j.arr.2012.10.004
Moylan S, Berk M, Dean OM, Samuni Y, Williams LJ, O’Neil A, Hayley AC, Pasco JA, Anderson G, Jacka FN, Maes M (2014) Oxidative & nitrosative stress in depression: why so much stress? Neurosci Biobehav Rev 45C:46–62. doi:10.1016/j.neubiorev.2014.05.007
Pfaffenseller B, Fries GR, Wollenhaupt-Aguiar B, Colpo GD, Stertz L, Panizzutti B, Magalhães PV, Kapczinski F (2013) Neurotrophins, inflammation and oxidative stress as illness activity biomarkers in bipolar disorder. Expert Rev Neurother 13(7):827–42. doi:10.1586/14737175.2013.811981
Pieczenik SR, Neustadt J (2007) Mitochondrial dysfunction and molecular pathways of disease. Exp Mol Pathol 83(1):84–92
Hwang O (2013) Role of oxidative stress in Parkinson’s disease. Exp Neurobiol 22:11–17. doi:10.5607/en.2013.22.1.11
Scalzo P, Kümmer A, Cardoso F, Teixeira AL (2009) Increased serum levels of soluble tumor necrosis factor-alpha receptor-1 in patients with Parkinson’s disease. J Neuroimmunol 216(1–2):122–5. doi:10.1016/j.jneuroim.2009.08.001
Heesen C, Nawrath L, Reich C, Bauer N, Schulz KH, Gold SM (2006) Fatigue in multiple sclerosis: an example of cytokine mediated sickness behaviour? J Neurol Neurosurg Psychiatry 77(1):34–9
Menzies V, Lyon DE (2010) Integrated review of the association of cytokines with fibromyalgia and fibromyalgia core symptoms. Biol Res Nurs 11(4):387–94. doi:10.1177/1099800409348328
Seruga B, Zhang H, Bernstein LJ, Tannock IF (2008) Cytokines and their relationship to the symptoms and outcome of cancer. Nat Rev Cancer 8(11):887–99. doi:10.1038/nrc2507
Patarca R (2001) Cytokines and chronic fatigue syndrome. Ann N Y Acad Sci 933:185–200
Nakamura T, Schwander SK, Donnelly R, Ortega F, Togo F, Broderick G, Yamamoto Y, Cherniack NS, Rapoport D, Natelson BH (2010) Cytokines across the night in chronic fatigue syndrome with and without fibromyalgia. Clin Vaccine Immunol 17(4):582–7. doi:10.1128/CVI.00379-09
Maes M (1993) A review on the acute phase response in major depression. Rev Neurosci 4(4):407–16
Tyrrell PJ, Smithard DG (2006) Fatigue after stroke. Therapy 2:865–869. doi:10.2217/14750708.2.6.865
Emsley HC, Tyrrell PJ (2002) Inflammation and infection in clinical stroke. J Cereb Blood Flow Metab 22(12):1399–419
Ormstad H, Aass HC, Amthor KF, Lund-Sørensen N, Sandvik L. Serum cytokine and glucose levels as predictors of poststroke fatigue in acute ischemic stroke patients. J Neurol. 2011 Apr;258(4):670–6. doi: 10.1007/s00415-011-5962-8. Epub 2011 Mar 2. Erratum in: J Neurol. 2012 Feb;259(2):399. PubMed PMID: 21365457; PubMed Central PMCID: PMC3065647
Yirmiya R (2000) Depression in medical illness: the role of the immune system. West J Med 173(5):333–6
Yirmiya R, Weidenfeld J, Pollak Y, Morag M, Morag A, Avitsur R, Barak O, Reichenberg A, Cohen E, Shavit Y, Ovadia H (1999) Cytokines, “depression due to a general medical condition,” and antidepressant drugs. Adv Exp Med Biol 461:283–316
Maes M, Kubera M, Leunis JC, Berk M (2012) Increased IgA and IgM responses against gut commensals in chronic depression: further evidence for increased bacterial translocation or leaky gut. J Affect Disord 141(1):55–62. doi:10.1016/j.jad.2012.02.023
Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55(5):453–62
Burton MD, Sparkman NL, Johnson RW (2011) Inhibition of interleukin-6 trans-signaling in the brain facilitates recovery from lipopolysaccharide-induced sickness behavior. J Neuroinflammation 8:54. doi:10.1186/1742-2094-8-54
Kluger MJ (1980) Fever Pediatrics 66(5):720–4
Leonard BE, Song C (2002) Changes in the immune system in rodent models of depression. Int J Neuropsychopharmacol 5(4):345–56
Song C, Leonard BE (2005) The olfactory bulbectomised rat as a model of depression. Neurosci Biobehav Rev 29(4–5):627–47
Goehler LE, Lyte M, Gaykema RP (2007) Infection-induced viscerosensory signals from the gut enhance anxiety: implications for psychoneuroimmunology. Brain Behav Immun 21(6):721–6
Anisman H, Merali Z (1999) Anhedonic and anxiogenic effects of cytokine exposure. Adv Exp Med Biol 461:199–233
Lyte M, Li W, Opitz N, Gaykema RP, Goehler LE (2006) Induction of anxiety-like behavior in mice during the initial stages of infection with the agent of murine colonic hyperplasia Citrobacter rodentium. Physiol Behav 89(3):350–7
Gaykema RP, Goehler LE (2009) Lipopolysaccharide challenge-induced suppression of Fos in hypothalamic orexin neurons: their potential role in sickness behavior. Brain Behav Immun 23(7):926–30. doi:10.1016/j.bbi.2009.03.005
Morris G, Anderson G, Galecki P, Berk M, Maes M (2013) A narrative review on the similarities and dissimilarities between myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and sickness behavior. BMC Med 11:64. doi:10.1186/1741-7015-11-64
Morris G, Maes M (2013) A neuro-immune model of myalgic encephalomyelitis/chronic fatigue syndrome. Metab Brain Dis 28(4):523–40. doi:10.1007/s11011-012-9324-8
Peters A (2006) The energy request of inflammation. Endocrinology 147(10):4550–2
Schaffner A (2006) [Fever—useful or noxious symptom that should be treated?]. Ther Umsch 63(3):185–8
Rantala S, Vuopio-Varkila J, Vuento R, Huhtala H, Syrjänen J (2009) Predictors of mortality in beta-hemolytic streptococcal bacteremia: a population-based study. J Infect 58(4):266–72. doi:10.1016/j.jinf.2009.01.015
Charlton BG (2000) The malaise theory of depression: major depressive disorder is sickness behavior and antidepressants are analgesic. Med Hypotheses 54(1):126–30
Felger JC, Li L, Marvar PJ, Woolwine BJ, Harrison DG, Raison CL, Miller AH (2013) Tyrosine metabolism during interferon-alpha administration: association with fatigue and CSF dopamine concentrations. Brain Behav Immun 31:153–60. doi:10.1016/j.bbi.2012.10.010
Kirkwood J (2002) Cancer immunotherapy: the interferon-alpha experience. Semin Oncol 29(3 Suppl 7):18–26
Dorr RT (1993) Interferon-alpha in malignant and viral diseases. A review Drugs 45(2):177–211
Ohga S, Aoki T, Okada K, Akeda H, Fujioka K, Ohshima A, Mori T, Minamishima I, Ueda K (1994) Cerebrospinal fluid concentrations of interleukin-1 beta, tumour necrosis factor-alpha, and interferon gamma in bacterial meningitis. Arch Dis Child 70(2):123–5
Barth PG. The neuropathology of Aicardi-Goutières syndrome. Eur J Paediatr Neurol. 2002;6 Suppl A:A27-31; discussion A37-9, A77-86. PubMed PMID: 12365358.
Jönsen A, Bengtsson AA, Nived O, Ryberg B, Truedsson L, Rönnblom L, Alm GV, Sturfelt G (2003) The heterogeneity of neuropsychiatric systemic lupus erythematosus is reflected in lack of association with cerebrospinal fluid cytokine profiles. Lupus 12(11):846–50
Wichers MC, Koek GH, Robaeys G et al (2005) Early increase in vegetative symptoms predicts IFN-alpha-induced cognitive-depressive changes. Psy- chol Med 35:433–441
Majer M, Welberg LA, Capuron L, Pagnoni G, Raison CL, Miller AH (2008) IFN-alpha-induced motor slowing is associated with increased depression and fatigue in patients with chronic hepatitis C. Brain Behav Immun 22(6):870–80. doi:10.1016/j.bbi.2007.12.009
Sunami M, Nishikawa T, Yorogi A, Shimoda M (2000) Intravenous administration of levodopa ameliorated a refractory akathisia case induced by interferon-alpha. Clin Neuropharmacol 23(1):59–61
Morrow GR, Hickok JT, Roscoe JA, Raubertas RF, Andrews PL, Flynn PJ, Hynes HE, Banerjee TK, Kirshner JJ, King DK (2003) University of Rochester Cancer Center Community Clinical Oncology Program. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol 21(24):4635–41
Raison CL, Demetrashvili M, Capuron L, Miller AH (2005) Neuropsychiatric adverse effects of interferon-alpha: recognition and management. CNS Drugs 19(2):105–23
Ahles TA, Saykin AJ, Furstenberg CT, Cole B, Mott LA, Skalla K, Whedon MB, Bivens S, Mitchell T, Greenberg ER, Silberfarb PM (2002) Neuropsychologic impact of standard-dose systemic chemotherapy in long-term survivors of breast cancer and lymphoma. J Clin Oncol 20(2):485–93
Bower JE, Ganz PA, Aziz N, Fahey JL (2002) Fatigue and proinflammatory cytokine activity in breast cancer survivors. Psychosom Med 64(4):604–11
Rönnbäck L, Hansson E (2004) On the potential role of glutamate transport in mental fatigue. J Neuroinflammation 1(1):22
Minagar A, Alexander JS (2003) Blood-brain barrier disruption in multiple sclerosis. Mult Scler 9(6):540–9
Lynch G (2004) AMPA receptor modulators as cognitive enhancers. Curr Opin Pharmacol 4(1):4–11
Sibson NR, Blamire AM, Bernades-Silva M, Laurent S, Boutry S, Muller RN, Styles P, Anthony DC. MRI detection of early endothelial activation in brain inflammation. Magn Reson Med. 2004 Feb;51(2):248–52. PubMed PMID: 14755648. 2004 Feb;4(1):4–11. Review. PubMed PMID: 15018832.
Morris G, Berk M, Galecki P, Maes M (2014) The emerging role of autoimmunity in myalgic encephalomyelitis/chronic fatigue syndrome (ME/cfs). Mol Neurobiol 49(2):741–56. doi:10.1007/s12035-013-8553-0
Lopez-Ramirez MA, Male DK, Wang C, Sharrack B, Wu D, Romero IA (2013) Cytokine-induced changes in the gene expression profile of a human cerebral microvascular endothelial cell-line, hCMEC/D3. Fluids Barriers CNS 10(1):27. doi:10.1186/2045-8118-10-27
Haroon E, Anand, R.; Chen, X.; Ford, R.; Parekh, S.; Woolwine, B.; Spivey, J.R.; Hu, X.; Miller, A.H. 2012. 178. Interferon-alpha-induced fatigue is associated with alterations in CNS glutamate metabolism as measured by magnetic resonance spectroscopy. Brain Behavior and Immunity vol. 26 September, Supple 1 p. S49–S50.
Yirmiya R, Goshen I (2011) Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25(2):181–213. doi:10.1016/j.bbi.2010.10.015
Collins JM, Riccardi R, Trown P, O’Neill D, Poplack DG (1985) Plasma and cerebrospinal fluid pharmacokinetics of recombinant interferon alpha A in monkeys: comparison of intravenous, intramuscular, and intraventricular delivery. Cancer Drug Deliv 2(4):247–53
Felger JC, Alagbe O, Hu F, Mook D, Freeman AA, Sanchez MM, Kalin NH, Ratti E, Nemeroff CB, Miller AH (2007) Effects of interferon-alpha on rhesus monkeys: a nonhuman primate model of cytokine-induced depression. Biol Psychiatry 62(11):1324–33
Smith RA, Norris F, Palmer D, Bernhardt L, Wills RJ (1985) Distribution of alpha interferon in serum and cerebrospinal fluid after systemic administration. Clin Pharmacol Ther 37(1):85–8
Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, Andjelkovic AV (2005) Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab 25(5):593–606
Weiss JM, Downie SA, Lyman WD, Berman JW (1998) Astrocyte-derived monocyte-chemoattractant protein-1 directs the transmigration of leukocytes across a model of the human blood-brain barrier. J Immunol 161(12):6896–903
Hokeness KL, Kuziel WA, Biron CA, Salazar-Mather TP (2005) Monocyte chemoattractant protein-1 and CCR2 interactions are required for IFN-alpha/beta-induced inflammatory responses and antiviral defense in liver. J Immunol 174(3):1549–56
Rankine EL, Hughes PM, Botham MS, Perry VH, Felton LM (2006) Brain cytokine synthesis induced by an intraparenchymal injection of LPS is reduced in MCP-1-deficient mice prior to leucocyte recruitment. Eur J Neurosci 24(1):77–86
Akiyama H, Ikeda K, Katoh M, McGeer EG, McGeer PL (1994) Expression of MRP14, 27E10, interferon-alpha and leukocyte common antigen by reactive microglia in postmortem human brain tissue. J Neuroimmunol 50(2):195–201
Curtin JF, King GD, Barcia C, Liu C, Hubert FX, Guillonneau C, Josien R, Anegon I, Lowenstein PR, Castro MG (2006) Fms-like tyrosine kinase 3 ligand recruits plasmacytoid dendritic cells to the brain. J Immunol 176(6):3566–77
Colton CA, Yao J, Keri JE, Gilbert D (1992) Regulation of microglial function by interferons. J Neuroimmunol 40(1):89–98
Felger JC, Miller AH (2012) Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front Neuroendocrinol 33(3):315–27. doi:10.1016/j.yfrne.2012.09.003
Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G, Spivey JR, Saito K, Miller AH (2010) CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry 15(4):393–403. doi:10.1038/mp.2009.116
Indraccolo S, Pfeffer U, Minuzzo S, Esposito G, Roni V, Mandruzzato S, Ferrari N, Anfosso L, Dell’Eva R, Noonan DM, Chieco-Bianchi L, Albini A, Amadori A (2007) Identification of genes selectively regulated by IFNs in endothelial cells. J Immunol 178(2):1122–35
Tissari J, Sirén J, Meri S, Julkunen I, Matikainen S (2005) IFN-alpha enhances TLR3-mediated antiviral cytokine expression in human endothelial and epithelial cells by up-regulating TLR3 expression. J Immunol 174(7):4289–94
Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T (2005) IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434(7034):772–7
Wang J, Campbell IL, Zhang H (2008) Systemic interferon-alpha regulates interferon-stimulated genes in the central nervous system. Mol Psychiatry 13(3):293–301
Pan W, Banks WA, Kastin AJ (1997) Permeability of the blood-brain and blood-spinal cord barriers to interferons. J Neuroimmunol 76(1–2):105–11
Nicolson GL, Settineri R (2011) Lipid replacement therapy: a functional food approach with new. Formulations for reducing cellular oxidative damage. Cancer-Associated Functional foods in health and disease 1(4):135–160
Segal BM, Thomas W, Zhu X, Diebes A, McElvain G, Baechler E, Gross M (2012) Oxidative stress and fatigue in systemic lupus erythematosus. Lupus 21(9):984–92. doi:10.1177/0961203312444772
Cordero MD, Alcocer-Gómez E, de Miguel M, Cano-García FJ, Luque CM, Fernández-Riejo P, Fernández AM, Sánchez-Alcazar JA (2011) Coenzyme Q(10): a novel therapeutic approach for fibromyalgia? Case series with 5 patients. Mitochondrion 11(4):623–5. doi:10.1016/j.mito.2011.03.122
Avalos I, Chung CP, Oeser A, Milne GL, Morrow JD, Gebretsadik T, Shintani A, Yu C, Stein CM (2007) Oxidative stress in systemic lupus erythematosus: relationship to disease activity and symptoms. Lupus 16(3):195–200
Kennedy G, Spence VA, McLaren M, Hill A, Underwood C, Belch JJ (2005) Oxidative stress levels are raised in chronic fatigue syndrome and are associated with clinical symptoms. Free Radic Biol Med 39(5):584–9
Jammes Y, Steinberg JG, Delliaux S, Brégeon F (2009) Chronic fatigue syndrome combines increased exercise-induced oxidative stress and reduced cytokine and Hsp responses. J Intern Med 266(2):196–206. doi:10.1111/j.1365-2796.2009.02079.x
Jammes Y, Steinberg JG, Delliaux S (2012) Chronic fatigue syndrome: acute infection and history of physical activity affect resting levels and response to exercise of plasma oxidant/antioxidant status and heat shock proteins. J Intern Med 272(1):74–84. doi:10.1111/j.1365-2796.2011.02488.x
Rodrigo R, Fernandez-Gajardo R, Gutierrez R et al (2013) Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets 12(5):698–714
Chen XM, Chen HS, Xu MJ, Shen JG (2013) Targeting reactive nitrogen species: a promising therapeutic strategy for cerebral ischemia-reperfusion injury. Acta Pharmacol Sin 34(1):67–77. doi:10.1038/aps.2012.82
Wang G, Pierangeli SS, Papalardo E, Ansari GA, Khan MF (2010) Markers of oxidative and nitrosative stress in systemic lupus erythematosus: correlation with disease activity. Arthritis Rheum 62(7):2064–72. doi:10.1002/art.27442
Wakamatsu TH, Dogru M, Matsumoto Y, Kojima T, Kaido M, Ibrahim OM, Sato EA, Igarashi A, Ichihashi Y, Satake Y, Shimazaki J, Tsubota K (2013) Evaluation of lipid oxidative stress status in Sjögren syndrome patients. Invest Ophthalmol Vis Sci 54(1):201–10. doi:10.1167/iovs.12-10325
Karatas F, Ozates I, Canatan H, Halifeoglu I, Karatepe M, Colakt R (2003) Antioxidant status & lipid peroxidation in patients with rheumatoid arthritis. Indian J Med Res 118:178–81
Chandankhede MS, Gupta MM (2013) Oxidative stress and antioxidant status in patients with rheumatoid arthritis. Int J Biol Med Res 4(2):3088–3090
Farooqui T, Farooqui AA (2011) Lipid-mediated oxidative stress and inflammation in the pathogenesis of Parkinson’s disease. Parkinsons Dis 2011:247467
O’Neill CA, Stebbins CL, Bonigut S, Halliwell B, Longhurst JC (1996) Production of hydroxyl radicals in contracting skeletal muscle of cats. J Appl Physiol 81(3):1197–1206
Pattwell DM, McArdle A, Morgan JE, Patridge TA, Jackson MJ (2004) Release of reactive oxygen and nitrogen species from contracting skeletal muscle cells. Free Radic Biol Med 37(7):1064–1072
Ferreira LF, Reid MB (2008) Muscle-derived ROS and thiol regulation in muscle fatigue. J Appl Physiol 104(3):853–860
Jackson MJ, Pye D, Palomero J (2007) The production of reactive oxygen and nitrogen species by skeletal muscle. J Appl Physiol 102(4):1664–1670
Albertini M, Lafortuna C, Aguggini G (1997) Effects of nitric oxide on diaphragmatic muscle endurance and strength in pigs. Exp Physiol 82(1):99–106
Belia S, Pietrangelo T, Fulle S, Menchetti G, Cecchini E, Felaco M, Vecchiet J, Fanò G (1998) Sodium nitroprusside, a NO donor, modifies Ca2+ transport and mechanical properties in frog skeletal muscle. J Muscle Res Cell Motil 19(8):865–76
Zhu X, Heunks LM, Versteeg EM, van der Heijden HF, Ennen L, van Kuppevelt TH, Vina J, Dekhuijzen PN (2005) Hypoxia-induced dysfunction of rat diaphragm: role of peroxynitrite. Am J Physiol Lung Cell Mol Physiol 288(1):L16–26
Boczkowski J, Lanone S, Ungureanu-Longrois D, Danialou G, Fournier T, Aubier M (1996) Induction of diaphragmatic nitric oxide synthase after endotoxin administration in rats: role on diaphragmatic contractile dysfunction. J Clin Invest 98(7):1550–1559
Hambrecht R, Adams V, Gielen S, Linke A, Möbius-Winkler S, Yu J, Niebauer J, Jiang H, Fiehn E, Schuler G (1999) Exercise intolerance in patients with chronic heart failure and increased expression of inducible nitric oxide synthase in the skeletal muscle. J Am Coll Cardiol 33(1):174–179
Rubinstein I, Abassi Z, Coleman R, Milman F, Winaver J, Better OS (1998) Involvement of nitric oxide system in experimental muscle crush injury. J Clin Invest 101(6):1325–1333
Reid MB, Stokić DS, Koch SM, Khawli FA, Leis AA (1994) N-acetylcysteine inhibits muscle fatigue in humans. J Clin Invest 94(6):2468–2474
Medved I, Brown MJ, Bjorksten AR, Murphy KT, Petersen AC, Sostaric S, Gong X, McKenna MJ (2004) N-acetylcysteine enhances muscle cysteine and glutathione availability and attenuates fatigue during prolonged exercise in endurance-trained individuals. J Appl Physiol 97:1477–1485
Reid MB (2001) Invited review: redox modulation of skeletal muscle contraction: what we know and what we don’t. J Appl Physiol 90(2):724–731
Smith MA, Reid MB (2006) Redox modulation of contractile function in respiratory and limb skeletal muscle. Respir Physiol Neurobiol 151(2–3):229–241
Fujii Y, Takahashi S, Toyooka H (2013) Protection from diaphragmatic fatigue by nitric oxide synthase inhibitor in dogs. Anaesth Intensive Care. 1999 Feb;27(1):45–8. Retraction in: Gibbs N. Anaesth Intensive Care 41(2):275
Grassi B, Hogan MC, Kelley KM, Howlett RA, Gladden LB (2005) Effects of nitric oxide synthase inhibition by l-NAME on oxygen uptake kinetics in isolated canine muscle in situ. J Physiol 568(Pt 3):1021–33
Hart JD, Dulhunty AF (2000) Nitric oxide activates or inhibits skeletal muscle ryanodine receptors depending on its concentration, membrane potential and ligand binding. J Membr Biol 173(3):227–36
Ishii T, Sunami O, Saitoh N, Nishio H, Takeuchi T, Hata F (1998) Inhibition of skeletal muscle sarcoplasmic reticulum Ca2+-ATPase by nitric oxide. FEBS Lett 440(1–2):218–22
Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH (1994) Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases FEBS Lett 345(1):50–4
Davies KJ, Quintanilha AT, Brooks GA, Packer L (1982) Free radicals and tissue damage produced by exercise. Biochem Biophys Res Commun 107(4):1198–205
Pouvreau S, Allard B, Berthier C, Jacquemond V (2004) Control of intracellular calcium in the presence of nitric oxide donors in isolated skeletal muscle fibres from mouse. J Physiol 560(Pt 3):779–794
Kobzik L, Reid MB, Bredt DS, Stamler JS (1994) Nitric oxide in skeletal muscle. Nature 372(6506):546–548
Thaloor D, Miller KJ, Gephart J, Mitchell PO, Pavlath GK (1999) Systemic administration of the NF-kappaB inhibitor curcumin stimulates muscle regeneration after traumatic injury. Am J Physiol 277(2 Pt 1):C320–C329
Mourkioti F, Kratsios P, Luedde T, Song YH, Delafontaine P, Adami R, Parente V, Bottinelli R, Pasparakis M, Rosenthal N (2006) Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J Clin Invest 116(11):2945–2954
Bar-Shai M, Carmeli E, Reznick AZ (2005) The role of NF-kappaB in protein breakdown in immobilization, aging, and exercise: from basic processes to promotion of health. Ann N Y Acad Sci 1057:431–47
Buck M, Chojkier M (1996) Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. EMBO J 15(8):1753–1765
Barbieri E, Sestili P. Reactive oxygen species in skeletal muscle signaling. J Signal Transduct. 2012;2012:982794. doi: 10.1155/2012/982794. Epub 2011 Dec 5. PubMed PMID: 22175016; PubMed Central PMCID: PMC3235811
Forcales SV, Puri PL (2005) Signaling to the chromatin during skeletal myogenesis: novel targets for pharmacological modulation of gene expression. Semin Cell Dev Biol 16(4–5):596–611
Messina S, Altavilla D, Aguennouz M, Seminara P, Minutoli L, Monici MC, Bitto A, Mazzeo A, Marini H, Squadrito F, Vita G (2006) Lipid peroxidation inhibition blunts nuclear factor-kappaB activation, reduces skeletal muscle degeneration, and enhances muscle function in mdx mice. Am J Pathol 168(3):918–26
Fulle S, Protasi F, Di Tano G, Pietrangelo T, Beltramin A, Boncompagni S, Vecchiet L, Fanò G (2004) The contribution of reactive oxygen species to sarcopenia and muscle ageing. Exp Gerontol 39(1):17–24
Lucas K, Maes M (2013) Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Mol Neurobiol 48(1):190–204. doi:10.1007/s12035-013-8425-7
Guijarro-Muñoz I, Compte M, Álvarez-Cienfuegos A, Álvarez-Vallina L, Sanz L (2014) Lipopolysaccharide activates Toll-like receptor 4 (TLR4)-mediated NF-κB signaling pathway and proinflammatory response in human pericytes. J Biol Chem 289(4):2457–68. doi:10.1074/jbc.M113.521161
Yang Y, Kim SC, Yu T, Yi YS, Rhee MH, Sung GH, Yoo BC, Cho JY (2014) Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediators Inflamm 2014:352371. doi:10.1155/2014/352371
Weismann D, Binder CJ (2012) The innate immune response to products of phospholipid peroxidation. Biochim Biophys Acta 1818(10):2465–75. doi:10.1016/j.bbamem.2012.01.018
Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A, Lotze MT (2007) Masquerader: high mobility group box-1 and cancer. Clin Cancer Res 13(10):2836–48
Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A (2008) HMGB1: endogenous danger signaling. Mol Med 14(7–8):476–84
Srikrishna G, Freeze HH (2009) Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia 11(7):615–28
Sato Y, Goto Y, Narita N, Hoon DS (2009) Cancer cells expressing toll-like receptors and the tumor microenvironment. Cancer Microenviron 2(Suppl 1):205–14
Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, Roth J (2007) Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 13(9):1042–9
Uchida K (2013) Redox-derived damage-associated molecular patterns: ligand function of lipid peroxidation adducts. Redox Biol 1(1):94–6. doi:10.1016/j.redox.2012.12.005
Carta S, Castellani P, Delfino L, Tassi S, Venè R, Rubartelli A (2009) DAMPs and inflammatory processes: the role of redox in the different outcomes. J Leukoc Biol 86(3):549–55. doi:10.1189/jlb.1008598
Moghaddam AE, Gartlan KH, Kong L, Sattentau QJ (2011) Reactive carbonyls are a major Th2-inducing damage-associated molecular pattern generated by oxidative stress. J Immunol 187(4):1626–33
Bruenner BA, Jones AD, German JB (1995) Direct characterization of protein adducts of the lipid peroxidation product 4-hydroxy-2-nonenal using electrospray mass spectrometry. Chem Res Toxicol 8:552–559
Ishii T, Kumazawa S, Sakurai T, Nakayama T, Uchida K (2006) Mass spectroscopic characterization of protein modification by malondialdehyde. Chem Res Toxicol 19(1):122–9
Simmons JD, Lee YL, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, Gillespie MN, Richards WO (2013) Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg 258(4):591–6. doi:10.1097/SLA.0b013e3182a4ea46
Mathew A, Lindsley TA, Sheridan A, Bhoiwala DL, Hushmendy SF, Yager EJ, Ruggiero EA, Crawford DR (2012) Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J Alzheimers Dis 30(3):617–27
Hsu GJ, Tzang BS, Tsai CC, Chiu CC, Huang CY, Hsu TC (2011) Effects of human parvovirus B19 on expression of defensins and Toll-like receptors. Chin J Physiol 54(5):367–76
Frisancho-Kiss S, Davis SE, Nyland JF et al (2007) Cutting edge: cross-regulation by TLR4 and T cell Ig mucin-3 determines sex differences in inflammatory heart disease. J Immunol 178:6710–6714
Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, Finberg RW (2003) Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol 77(8):4588–96
Zamboni DS, Campos MA, Torrecilhas AC, Kiss K, Samuel JE, Golenbock DT, Lauw FN, Roy CR, Almeida IC, Gazzinelli RT (2004) Stimulation of toll-like receptor 2 by Coxiella burnetii is required for macrophage production of pro-inflammatory cytokines and resistance to infection. J Biol Chem 279(52):54405–15
Salazar JC, Duhnam-Ems S, La Vake C, Cruz AR, Moore MW, Caimano MJ, Velez-Climent L, Shupe J, Krueger W, Radolf JD (2009) Activation of human monocytes by live Borrelia burgdorferi generates TLR2-dependent and -independent responses which include induction of IFN-beta. PLoS Pathog 5(5):e1000444
Friis LM, Keelan M, Taylor DE (2009) Campylobacter jejuni drives MyD88-independent interleukin-6 secretion via Toll-like receptor 2. Infect Immun 77(4):1553–60
Gow JW, Hagan S, Herzyk P, Cannon C, Behan PO, Chaudhuri A (2009) A gene signature for post-infectious chronic fatigue syndrome. BMC Med Genomics 2:38. doi:10.1186/1755-8794-2-38
Light AR, Vierck CJ, Light KC (2010) Myalgia and Fatigue: Translation from Mouse Sensory Neurons to Fibromyalgia and Chronic Fatigue Syndromes. In: Kruger L, Light AR (eds) Translational pain research: from mouse to man. CRC, Boca Raton, FL
Nagyoszi P, Wilhelm I, Farkas AE, Fazakas C, Dung NT, Haskó J, Krizbai IA (2010) Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem Int 57(5):556–64
Andersson A, Covacu R, Sunnemark D, Danilov AI, Dal Bianco A, Khademi M, Wallström E, Lobell A, Brundin L, Lassmann H, Harris RA (2008) Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. J Leukoc Biol 84(5):1248–55
Bsibsi M, Ravid R, Gveric D, van Noort JM (2002) Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol 61(11):1013–21
Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ (2013) Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4:1562
Pandey GN, Rizavi HS, Ren X, Bhaumik R, Dwivedi Y (2014) Toll-like receptors in the depressed and suicide brain. J Psychiatr Res 53:62–8. doi:10.1016/j.jpsychires.2014.01.021
Tiffin N, Adeyemo A, Okpechi I (2013) A diverse array of genetic factors contribute to the pathogenesis of systemic lupus erythematosus. Orphanet J Rare Dis 8:2. doi:10.1186/1750-1172-8-2
Low HZ, Witte T (2011) Aspects of innate immunity in Sjögren’s syndrome. Arthritis Res Ther 13(3):218. doi:10.1186/ar3318
Goh FG, Midwood KS (2012) Intrinsic danger: activation of Toll-like receptors in rheumatoid arthritis. Rheumatology (Oxford) 51(1):7–23. doi:10.1093/rheumatology/ker257
Wang PF, Fang H, Chen J, Lin S, Liu Y, Xiong XY, Wang YC, Xiong RP, Lv FL, Wang J, Yang QW (2014) Polyinosinic-polycytidylic acid has therapeutic effects against cerebral ischemia/reperfusion injury through the downregulation of TLR4 signaling via TLR3. J Immunol 192(10):4783–94. doi:10.4049/jimmunol.1303108
Vereker E, O’Donnell E, Lynch MA (2000) The inhibitory effect of interleukin-1beta on long-term potentiation is coupled with increased activity of stress-activated protein kinases. J Neurosci 20(18):6811–9
Yudkoff M, Nissim I, Daikhin Y, Lin ZP, Nelson D, Pleasure D, Erecinska M (1993) Brain glutamate metabolism: neuronal-astroglial relationships. Dev Neurosci 15(3–5):343–50
Huang YH, Bergles DE (2004) Glutamate transporters bring competition to the synapse. Curr Opin Neurobiol 14(3):346–52
Pannasch U, Derangeon M, Chever O, Rouach N (2012) Astroglial gap junctions shape neuronal network activity. Communicative & integrative biology 5(3):248–254
Araque A (2008) Astrocytes process synaptic information. Neuron Glia Biol 4(01):3–10
Newman LA, Korol DL, Gold PE (2011) Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One 6(12):e28427. doi:10.1371/journal.pone.0028427
Harrington, M. 2011. Astrocytes ‘feed’ memory formation. Lab animal, 40 (4), p. 98.
Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, Rothstein JD, Volsky DJ (2003) Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology 312(1):60–73
Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, Gelbard HA (1996) Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J Biol Chem 271(26):15303–6
Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC (2000) Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 7(3):153–9
Chao CC, Hu S, Ehrlich L, Peterson PK (1995) Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-d-aspartate receptors. Brain Behav Immun 9(4):355–65
Loaiza A, Porras OH, Barros LF (2003) Glutamate triggers rapid glucose transport stimulation in astrocytes as evidenced by real-time confocal microscopy. J Neurosci 23(19):7337–42
Hertz L. Intercellular metabolic compartmentation in the brain: past, present and future. Neurochem Int. 2004 Jul-Aug;45(2–3):285–96. Review. PubMed PMID: 15145544.
Waubant E (2006) Biomarkers indicative of blood-brain barrier disruption in multiple sclerosis. Dis Markers 22(4):235–244
Helms G (2001) Volume correction for edema in single-volume proton MR spectroscopy of contrast-enhancing multiple sclerosis lesions. Magn Reson Med 46(2):256–263
O’kane, R. L., Martínez-López, I., Dejoseph, M. R., Viña, J. R. and Hawkins, R. A. 1999. Na+-dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) of the blood-brain Barrier A MECHANISM FOR GLUTAMATE REMOVAL. Journal of Biological Chemistry, 274 (45), pp. 31891–31895
Karpuk N, Burkovetskaya M, Fritz T, Angle A, Kielian T (2011) Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. J Neurosci 31(2):414–25. doi:10.1523/JNEUROSCI.5247-10.2011
Takaki J, Fujimori K, Miura M, Suzuki T, Sekino Y, Sato K (2012) l-glutamate released from activated microglia downregulates astrocytic l-glutamate transporter expression in neuroinflammation: the ‘collusion’ hypothesis for increased extracellular l-glutamate concentration in neuroinflammation. J Neuroinflammation 9:275. doi:10.1186/1742-2094-9-275
Fujigaki H, Saito K, Fujigaki S, Takemura M, Sudo K, Ishiguro H, Seishima M (2006) The signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways, and synergistic effect of several proinflammatory cytokines. J Biochem 139(4):655–62
Guillemin GJ, Smith DG, Smythe GA, Armati PJ, Brew BJ (2003) Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv Exp Med Biol 527:105–12
Pemberton LA, Kerr SJ, Smythe G, Brew BJ (1997) Quinolinic acid production by macrophages stimulated with IFN-gamma, TNF-alpha, and IFN-alpha. J Interferon Cytokine Res 17(10):589–95
Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith QR (1991) Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem 56(6):2007–17
Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ (2012) Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 13(7):465–77. doi:10.1038/nrn3257
Schwarcz R, Pellicciari R (2002) Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther 303(1):1–10
Tavares RG, Schmidt AP, Abud J, Tasca CI, Souza DO (2005) In vivo quinolinic acid increases synaptosomal glutamate release in rats: reversal by guanosine. Neurochem Res 30(4):439–44
Guidetti P, Schwarcz R (2003) 3-Hydroxykynurenine and quinolinate: pathogenic synergism in early grade Huntington’s disease? Adv Exp Med Biol 527:137–45
Guillemin GJ, Wang L, Brew BJ (2005) Quinolinic acid selectively induces apoptosis of human astrocytes: potential role in AIDS dementia complex. J Neuroinflammation 2:16
Stone TW (2000) Development and therapeutic potential of kynurenic acid and kynurenine derivatives for neuroprotection. Trends Pharmacol Sci 21(4):149–54
Ida T, Hara M, Nakamura Y, Kozaki S, Tsunoda S, Ihara H (2008) Cytokine-induced enhancement of calcium-dependent glutamate release from astrocytes mediated by nitric oxide. Neurosci Lett 432(3):232–6. doi:10.1016/j.neulet.2007.12.047
Tilleux S, Hermans E (2007) Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res 85(10):2059–70
Hardingham GE, Fukunaga Y, Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5(5):405–14
Haydon PG, Carmignoto G (2006) Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev 86(3):1009–31
Esposito Z, Belli L, Toniolo S, Sancesario G, Bianconi C, Martorana A (2013) Amyloid β, glutamate, excitotoxicity in Alzheimer’s disease: are we on the right track? CNS Neurosci Ther 19(8):549–55. doi:10.1111/cns.12095
Zinger A, Barcia C, Herrero MT, Guillemin GJ (2011) The involvement of neuroinflammation and kynurenine pathway in Parkinson’s disease. Parkinsons Dis 2011:716859. doi:10.4061/2011/716859
Vaarmann A, Kovac S, Holmström KM, Gandhi S, Abramov AY (2013) Dopamine protects neurons against glutamate-induced excitotoxicity. Cell Death Dis 4:e455. doi:10.1038/cddis.2012.194
Anitha A, Nakamura K, Thanseem I, Yamada K, Iwayama Y, Toyota T, Matsuzaki H, Miyachi T, Yamada S, Tsujii M, Tsuchiya KJ, Matsumoto K, Iwata Y, Suzuki K, Ichikawa H, Sugiyama T, Yoshikawa T, Mori N (2012) Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol Autism 3(1):12. doi:10.1186/2040-2392-3-12
Kostic MS, Rajkovic RJ, Potic Floranovic MS, Dimov ID, Pavlovic D (2013) Multiple sclerosis and oxidative stress—a clinical perspective. Neurochem J 7(1):77–86
Lim CK, Brew BJ, Sundaram G, Guillemin GJ (2010) Understanding the roles of the kynurenine pathway in multiple sclerosis progression. Int J Tryptophan Res 3:157–67
Maes M, Mihaylova I, Ruyter MD, Kubera M, Bosmans E (2007) The immune effects of TRYCATs (tryptophan catabolites along the IDO pathway): relevance for depression—and other conditions characterized by tryptophan depletion induced by inflammation. Neuro Endocrinol Lett 28(6):826–31
Maes M, Berk M, Goehler L, Song C, Anderson G, Gałecki P, Leonard B (2012) Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med 10:66. doi:10.1186/1741-7015-10-66
Maes M, Galecki P, Verkerk R, Rief W (2011) Somatization, but not depression, is characterized by disorders in the tryptophan catabolite (TRYCAT) pathway, indicating increased indoleamine 2,3-dioxygenase and lowered kynurenine aminotransferase activity. Neuro Endocrinol Lett 32(3):264–73
Szalardy L, Klivenyi P, Zadori D, Fulop F, Toldi J, Vecsei L (2012) Mitochondrial disturbances, tryptophan metabolites and neurodegeneration: medicinal chemistry aspects. Curr Med Chem 19(13):1899–920
Kincses ZT, Toldi J, Vécsei L (2010) Kynurenines, neurodegeneration and Alzheimer’s disease. J Cell Mol Med 14(8):2045–54. doi:10.1111/j.1582-4934.2010.01123.x
Brouns R, Verkerk R, Aerts T, De Surgeloose D, Wauters A, Scharpé S, De Deyn PP (2010) The role of tryptophan catabolism along the kynurenine pathway in acute ischemic stroke. Neurochem Res 35(9):1315–22. doi:10.1007/s11064-010-0187-2
Thorne RG, Hanson LR, Ross TM, Tung D, Frey WH 2nd (2008) Delivery of interferon-beta to the monkey nervous system following intranasal administration. Neuroscience 152(3):785–97. doi:10.1016/j.neuroscience.2008.01.013
Malhi GS, Berk M (2007) Does dopamine dysfunction drive depression? Acta PsychiatrScand Suppl 433:116–24
Berk M, Dodd S, Kauer-Sant’anna M, Malhi GS, Bourin M, Kapczinski F, Norman T (2007) Dopamine dysregulation syndrome: implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatr Scand Suppl 434:41–9
Cunnington C, Channon KM (2010) Tetrahydrobiopterin: pleiotropic roles in cardiovascular pathophysiology. Heart 96(23):1872–7. doi:10.1136/hrt.2009.180430
Guillot TS, Miller GW (2009) Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol Neurobiol 39(2):149–70. doi:10.1007/s12035-009-8059-y
Riddle EL, Fleckenstein AE, Hanson GR (2006) Mechanisms of methamphetamine-induced dopaminergic neurotoxicity. AAPS J 8(2):E413–8
Kazumori H, Ishihara S, Rumi MA, Ortega-Cava CF, Kadowaki Y, Kinoshita Y (2004) Transforming growth factor-alpha directly augments histidine decarboxylase and vesicular monoamine transporter 2 production in rat enterochromaffin-like cells. Am J Physiol Gastrointest Liver Physiol 286(3):G508–14
Zhu CB, Carneiro AM, Dostmann WR, Hewlett WA, Blakely RD (2005) p38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2A-dependent process. J Biol Chem 280(16):15649–58
Zhu CB, Blakely RD, Hewlett WA (2006) The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 31(10):2121–31
Zhu CB, Lindler KM, Owens AW, Daws LC, Blakely RD, Hewlett WA (2010) Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology 35(13):2510–20. doi:10.1038/npp.2010.116
Gautam AH, Zeevalk GD (2011) Characterization of reduced and oxidized dopamine and 3,4-dihydrophenylacetic acid, on brain mitochondrial electron transport chain activities. Biochim Biophys Acta 1807(7):819–28. doi:10.1016/j.bbabio.2011.03.013
Ben-Shachar D (2009) The interplay between mitochondrial complex I, dopamine and Sp1 in schizophrenia. J Neural Transm 116(11):1383–96. doi:10.1007/s00702-009-0319-5
Feier G, Valvassori SS, Lopes-Borges J, Varela RB, Bavaresco DV, Scaini G, Morais MO, Andersen ML, Streck EL, Quevedo J (2012) Behavioral changes and brain energy metabolism dysfunction in rats treated with methamphetamine or dextroamphetamine. Neurosci Lett 530(1):75–9. doi:10.1016/j.neulet.2012.09.039
Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM (2002) Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418(6899):797–801
Angelini C, Bello L, Spinazzi M, Ferrati C (2009) Mitochondrial disorders of the nuclear genome. Acta Myol 28(1):16–23
Jeppesen TD, Schwartz M, Olsen DB, Wibrand F, Krag T, Dunø M, Hauerslev S, Vissing J (2006) Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain 129(Pt 12):3402–12
McFarland R, Turnbull DM (2009) Batteries not included: diagnosis and management of mitochondrial disease. J Intern Med 265(2):210–28. doi:10.1111/j.1365-2796.2008.02066.x
Rose S, Frye RE, Slattery J, Wynne R, Tippett M, Pavliv O, Melnyk S, James SJ (2014) Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS One 9(1):e85436. doi:10.1371/journal.pone.0085436. eCollection 2014
Sarti P, Forte E, Mastronicola D, Giuffrè A, Arese M (2012) Cytochrome c oxidase and nitric oxide in action: molecular mechanisms and pathophysiological implications. Biochim Biophys Acta 1817(4):610–9. doi:10.1016/j.bbabio.2011.09.002
Leavesley HB, Li L, Prabhakaran K, Borowitz JL, Isom GE (2008) Interaction of cyanide and nitric oxide with cytochrome c oxidase: implications for acute cyanide toxicity. Toxicol Sci 101(1):101–11
Brown GC, Borutaite V (2004) Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim Biophys Acta 1658(1–2):44–9
Maes M (2009) Inflammatory and oxidative and nitrosative stress pathways underpinning chronic fatigue, somatization and psychosomatic symptoms. Curr Opin Psychiatry 22(1):75–83
Vermeulen RC, Eck IW V v (2014) Decreased oxygen extraction during cardiopulmonary exercise test in patients with chronic fatigue syndrome. J Transl Med 12:20. doi:10.1186/1479-5876-12-20
Castro-Marrero J, Cordero MD, Sáez-Francas N, Jimenez-Gutierrez C, Aguilar-Montilla FJ, Aliste L, Alegre-Martin J. Could mitochondrial dysfunction be a differentiating marker between chronic fatigue syndrome and fibromyalgia? Antioxid Redox Signal. 2013 Nov 20;19(15):1855–60. doi: 0.1089/ars.2013.5346. Epub 2013 May 29. PubMed PMID: 23600892.
Alcocer-Gómez E, Sánchez-Alcázar JA, Cordero MD (2014) Coenzyme q10 regulates serotonin levels and depressive symptoms in fibromyalgia patients: results of a small clinical trial. J Clin Psychopharmacol 34(2):277–8. doi:10.1097/JCP.0000000000000097
Cordero, M. D., Cano-García, F. J., Alcocer-Gómez, E., De Miguel, M. and Sánchez-Alcázar, J. A. 2012. Oxidative stress correlates with headache symptoms in fibromyalgia: coenzyme Q10 effect on clinical improvement. PloS one, 7 (4), p. 35677.
Cordero MD, Cotán D, del-Pozo-Martín Y, Carrión AM, de Miguel M, Bullón P, Sánchez-Alcazar JA. Oral coenzyme Q10 supplementation improves clinical symptoms and recovers pathologic alterations in blood mononuclear cells in a fibromyalgia patient. Nutrition. 2012 Nov-Dec;28(11–12):1200–3. doi: 10.1016/j.nut.2012.03.018. Epub 2012 Aug 14. PubMed PMID: 22898267.
Cordero MD, Alcocer-Gómez E, de Miguel M, Culic O, Carrión AM, Alvarez-Suarez JM, Bullón P, Battino M, Fernández-Rodríguez A, Sánchez-Alcazar JA. Can coenzyme q10 improve clinical and molecular parameters in fibromyalgia? Antioxid Redox Signal. 2013 Oct 20;19(12):1356–61. doi: 0.1089/ars.2013.5260. Epub 2013 Apr 6. PubMed PMID: 23458405.
Morris G, Anderson G, Berk M, Maes M (2013) Coenzyme Q10 depletion in medical and neuropsychiatric disorders: potential repercussions and therapeutic implications. Mol Neurobiol 48(3):883–903. doi:10.1007/s12035-013-8477-8
Gökbel H, Gül I, Belviranl M, Okudan N (2010) The effects of coenzyme Q10 supplementation on performance during repeated bouts of supramaximal exercise in sedentary men. J Strength Cond Res 24(1):97–102
Cooke M, Iosia M, Buford T, Shelmadine B, Hudson G, Kerksick C, Rasmussen C, Greenwood M, Leutholtz B, Willoughby D, Kreider R (2008) Effects of acute and 14-day coenzyme Q10 supplementation on exercise performance in both trained and untrained individuals. J Int Soc Sports Nutr 5:8
Alehagen U, Johansson P, Björnstedt M, Rosén A, Dahlström U (2013) Cardiovascular mortality and N-terminal-proBNP reduced after combined selenium and coenzyme Q10 supplementation: a 5-year prospective randomized double-blind placebo-controlled trial among elderly Swedish citizens. Int J Cardiol 167(5):1860–6. doi:10.1016/j.ijcard.2012.04.156
Finck BN, Kelly DP (2007) Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation 115(19):2540–8
Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin II, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM (2005) Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab 1(4):259–71
Eschbach J, Schwalenstöcker B, Soyal SM, Bayer H, Wiesner D, Akimoto C, Nilsson AC, Birve A, Meyer T, Dupuis L, Danzer KM, Andersen PM, Witting A, Ludolph AC, Patsch W, Weydt P (2013) PGC-1α is a male-specific disease modifier of human and experimental amyotrophic lateral sclerosis. Hum Mol Genet 22(17):3477–84. doi:10.1093/hmg/ddt202
Tsunemi T, La Spada AR (2012) PGC-1α at the intersection of bioenergetics regulation and neuron function: from Huntington’s disease to Parkinson’s disease and beyond. Prog Neurobiol 97(2):142–51. doi:10.1016/j.pneurobio.2011.10.004
Witte ME, Nijland PG, Drexhage JA, Gerritsen W, Geerts D, van Het Hof B, Reijerkerk A, de Vries HE, van der Valk P, van Horssen J (2013) Reduced expression of PGC-1α partly underlies mitochondrial changes and correlates with neuronal loss in multiple sclerosis cortex. Acta Neuropathol 125(2):231–43. doi:10.1007/s00401-012-1052-y
Summermatter S, Santos G, Pérez-Schindler J, Handschin C (2013) Skeletal muscle PGC-1α controls whole-body lactate homeostasis through estrogen-related receptor α-dependent activation of LDH B and repression of LDH A. Proc Natl Acad Sci U S A 110(21):8738–43. doi:10.1073/pnas.1212976110
Selsby JT, Morine KJ, Pendrak K, Barton ER, Sweeney HL (2012) Rescue of dystrophic skeletal muscle by PGC-1α involves a fast to slow fiber type shift in the mdx mouse. PLoS One 7(1):e30063. doi:10.1371/journal.pone.0030063
Summermatter S, Shui G, Maag D, Santos G, Wenk MR, Handschin C (2013) PGC-1α improves glucose homeostasis in skeletal muscle in an activity-dependent manner. Diabetes 62(1):85–95. doi:10.2337/db12-0291
Bonen A (2009) PGC-1alpha-induced improvements in skeletal muscle metabolism and insulin sensitivity. Appl Physiol Nutr Metab 34(3):307–14. doi:10.1139/H09-008
Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP (2005) PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 3(4):e101
Yamada T, Ivarsson N, Hernández A, Fahlström A, Cheng AJ, Zhang SJ, Bruton JD, Ulfhake B, Westerblad H (2012) Impaired mitochondrial respiration and decreased fatigue resistance followed by severe muscle weakness in skeletal muscle of mitochondrial DNA mutator mice. J Physiol 590(Pt 23):6187–97. doi:10.1113/jphysiol.2012.240077
Jung HY, Lee AN, Song TJ, An HS, Kim YH, Kim KD, Kim IB, Kim KS, Han BS, Kim CH, Kim KS, Kim JB (2012) Korean mistletoe (Viscum album coloratum) extract improves endurance capacity in mice by stimulating mitochondrial activity. J Med Food 15(7):621–8. doi:10.1089/jmf.2010.1469
Handschin C, Spiegelman BM (2008) The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 454(7203):463–9. doi:10.1038/nature07206
Tang K, Wagner PD, Breen EC (2010) TNF-alpha-mediated reduction in PGC-1alpha may impair skeletal muscle function after cigarette smoke exposure. J Cell Physiol 222(2):320–7. doi:10.1002/jcp.21955
Palomer X, Alvarez-Guardia D, Rodríguez-Calvo R, Coll T, Laguna JC, Davidson MM, Chan TO, Feldman AM, Vázquez-Carrera M (2009) TNF-alpha reduces PGC-1alpha expression through NF-kappaB and p38 MAPK leading to increased glucose oxidation in a human cardiac cell model. Cardiovasc Res 81(4):703–12. doi:10.1093/cvr/cvn327
Austin S, St-Pierre J (2012) PGC1α and mitochondrial metabolism—emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci 125(Pt 21):4963–71. doi:10.1242/jcs.113662
Tufekci KU, Civi Bayin E, Genc S, Genc K (2011) The Nrf2/ARE pathway: a promising target to counteract mitochondrial dysfunction in Parkinson’s disease. Parkinsons Dis 2011:314082. doi:10.4061/2011/314082
Cook DB, O’Connor PJ, Lange G, Steffener J (2007) Functional neuroimaging correlates of mental fatigue induced by cognition among chronic fatigue syndrome patients and controls. Neuroimage 36(1):108–22
Tajima S, Yamamoto S, Tanaka M, Kataoka Y, Iwase M, Yoshikawa E, Okada H, Onoe H, Tsukada H, Kuratsune H, Ouchi Y, Watanabe Y (2010) Medial orbitofrontal cortex is associated with fatigue sensation. Neurol Res Int 2010:671421. doi:10.1155/2010/671421
Chaudhuri A, Behan PO (2004) Fatigue in neurological disorders. Lancet 363(9413):978–88
Bruno RL, Cohen JM, Galski T, Frick NM (1994) The neuroanatomy of post-polio fatigue. Arch Phys Med Rehabil 75(5):498–504
Blomstrand E, Perrett D, Parry-Billings M, Newsholme EA (1989) Effect of sustained exercise on plasma amino acid concentrations and on 5-hydroxytryptamine metabolism in six different brain regions in the rat. Acta Physiol Scand 136(3):473–81
Juengling FD, Ebert D, Gut O, Engelbrecht MA, Rasenack J, Nitzsche EU, Bauer J, Lieb K (2000) Prefrontal cortical hypometabolism during low-dose interferon alpha treatment. Psychopharmaco (Berl) 152(4):383–9
Capuron L, Pagnoni G, Demetrashvili MF, Lawson DH, Fornwalt FB, Woolwine B, Berns GS, Nemeroff CB, Miller AH (2007) Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology 32(11):2384–92
Eidelberg D, Moeller JR, Dhawan V, Spetsieris P, Takikawa S, Ishikawa T, Chaly T, Robeson W, Margouleff D, Przedborski S et al (1994) The metabolic topography of parkinsonism. J Cereb Blood Flow Metab 14(5):783–801
Brydon L, Harrison NA, Walker C, Steptoe A, Critchley HD (2008) Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol Psychiatry 63(11):1022–9. doi:10.1016/j.biopsych.2007.12.007
Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR (2010) Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry 68(8):748–54. doi:10.1016/j.biopsych.2010.06.010
Rosen HJ, Allison SC, Schauer GF, Gorno-Tempini ML, Weiner MW, Miller BL (2005) Neuroanatomical correlates of behavioural disorders in dementia. Brain 128(Pt 11):2612–25
Sepulcre J, Masdeu JC, Goñi J, Arrondo G, Vélez de Mendizábal N, Bejarano B, Villoslada P (2009) Fatigue in multiple sclerosis is associated with the disruption of frontal and parietal pathways. Mult Scler 15(3):337–44. doi:10.1177/1352458508098373
Okada T, Tanaka M, Kuratsune H, Watanabe Y, Sadato N (2004) Mechanisms underlying fatigue: a voxel-based morphometric study of chronic fatigue syndrome. BMC Neurol 4(1):14
de Lange FP, Koers A, Kalkman JS, Bleijenberg G, Hagoort P, van der Meer JW, Toni I (2008) Increase in prefrontal cortical volume following cognitive behavioural therapy in patients with chronic fatigue syndrome. Brain 131(Pt 8):2172–80. doi:10.1093/brain/awn140
DeLuca J, Genova HM, Hillary FG, Wylie G (2008) Neural correlates of cognitive fatigue in multiple sclerosis using functional MRI. J Neurol Sci 270(1–2):28–39. doi:10.1016/j.jns.2008.01.018
Flachenecker P, Kümpfel T, Kallmann B, Gottschalk M, Grauer O, Rieckmann P, Trenkwalder C, Toyka KV (2002) Fatigue in multiple sclerosis: a comparison of different rating scales and correlation to clinical parameters. Mult Scler 8(6):523–6
Pardini M, Krueger F, Raymont V, Grafman J (2010) Ventromedial prefrontal cortex modulates fatigue after penetrating traumatic brain injury. Neurology 74(9):749–54. doi:10.1212/WNL.0b013e3181d25b6b
Pardini M, Bonzano L, Roccatagliata L, Mancardi GL, Bove M (2013) The fatigue-motor performance paradox in multiple sclerosis. Sci Rep 3:2001. doi:10.1038/srep02001
Pellicano C, Gallo A, Li X, Ikonomidou VN, Evangelou IE, Ohayon JM, Stern SK, Ehrmantraut M, Cantor F, McFarland HF, Bagnato F (2010) Relationship of cortical atrophy to fatigue in patients with multiple sclerosis. Arch Neurol 67(4):447–53. doi:10.1001/archneurol.2010.48
Cook DB, O’Connor PJ, Lange G, Steffener J (2007) Functional neuroimaging correlates of mental fatigue induced by cognition among chronic fatigue syndrome patients and controls. Neuroimage 36(1):108–22
Caseras X, Mataix-Cols D, Rimes KA, Giampietro V, Brammer M, Zelaya F, Chalder T, Godfrey E (2008) The neural correlates of fatigue: an exploratory imaginal fatigue provocation study in chronic fatigue syndrome. Psychol Med 38(7):941–51. doi:10.1017/S0033291708003450
Costa DC, Tannock C, Brostoff J (1995) Brainstem perfusion is impaired in chronic fatigue syndrome. QJM 88:767–773
Ichise M, Salit I, Abbey S, Chung DG, Gray B, Kirsh JC, Freedman M (1992) Assessment of regional cerebral perfusion by Tcm-HMPAO SPECT in chronic fatigue syndrome. Nucl Med Commun 13:767–772
Inglese M, Park SJ, Johnson G, Babb JS, Miles L, Jaggi H, Herbert J, Grossman RI (2007) Deep gray matter perfusion in multiple sclerosis: dynamic susceptibility contrast perfusion magnetic resonance imaging at 3 T. Arch Neurol 64(2):196–202
Niepel G, Tench CR, Morgan PS, Evangelou N, Auer DP, Constantinescu CS (2006) Deep gray matter and fatigue in MS: a T1 relaxation time study. J Neurol 253(7):896–902
Lajoie C, Levasseur MA, Paquet N (2013) Complete normalization of severe brain 18F-FDG hypometabolism following electroconvulsive therapy in a major depressive episode. Clin Nucl Med 38(9):735–6. doi:10.1097/RLU.0b013e31829b9bd9
Thorpe SJ, Rolls ET, Maddison S (1983) The orbitofrontal cortex: neuronal activity in the behaving monkey. Exp Brain Res 49(1):93–115
Walton ME, Bannerman DM, Rushworth MF (2002) The role of rat medial frontal cortex in effort-based decision making. J Neurosci 22(24):10996–1003
Ongür D, An X, Price JL (1998) Prefrontal cortical projections to the hypothalamus in macaque monkeys. J Comp Neurol 401(4):480–505
Carmichael ST, Price JL (1996) Connectional networks within the orbital and medial prefrontal cortex of macaque monkeys. J Comp Neurol 371(2):179–207
Stockton K, Iah K, Paratz DJ, Bennell K (2012) Fatigue, muscle strength and vitamin D status in women with systemic lupus erythematosus compared with healthy controls. Lupus 21(3):271–278
Tench C, Bentley D, Vleck V, Mccurdie I, White P, D’cruz D (2002) Aerobic fitness, fatigue, and physical disability in systemic lupus erythematosus. J rheumatology 29(3):474–481
Kanellopoulos P, Baltoyiannis C, Tzioufas A (2002) Primary Sjögren’s syndrome associated with inclusion body myositis. Rheumatology 41(4):440–444
Vrethem M, Lindvall B, Holmgren H, Henriksson K, Lindström F, Ernerudh J (1990) Neuropathy and myopathy in primary Sjögren’s syndrome: neurophysiological, immunological and muscle biopsy results. Acta Neurol Scand 82(2):126–131
Willer, B., Stucki, G., Hoppeler, H., Brühlmann, P. and Krähenbühl, S. 2000. Effects of creatine supplementation on muscle weakness in patients with rheumatoid arthritis. Rheumatology, 39 (3), pp. 293–298
Yates D (1963) Muscular changes in rheumatoid arthritis. Ann Rheum Dis 22(5):342–347
Friedman JH, Abrantes AM (2012) Self perceived weakness in Parkinson’s disease. Parkinsonism Relat Disord 18(7):887–889
Stevens-Lapsley J, Kluger BM, Schenkman M (2012) Quadriceps muscle weakness, activation deficits, and fatigue with Parkinson disease. Neurorehabil Neural Repair 26(5):533–541
De Haan A, De Ruiter CJ, Van Der Woude LH, Jongen PJ (2000) Contractile properties and fatigue of quadriceps muscles in multiple sclerosis. Muscle Nerve 23(10):1534–1541
Steens A, Heersema D, Maurits NM, Renken R, Zijdewind I (2012) Mechanisms underlying muscle fatigue differ between multiple sclerosis patients and controls: a combined electrophysiological and neuroimaging study. Neuroimage 59(4):3110–3118
Steens A, de Vries A, Hemmen J, Heersema T, Heerings M, Maurits N, Zijdewind I (2012) Fatigue perceived by multiple sclerosis patients is associated with muscle fatigue. Neurorehabil Neural Repair 26(1):48–57. doi:10.1177/1545968311416991
Bandak E, Amris K, Bliddal H, Danneskiold-SamsOe B, Henriksen M (2013) Muscle fatigue in fibromyalgia is in the brain, not in the muscles: a case–control study of perceived versus objective muscle fatigue. Ann Rheum Dis 72(6):963–966
Casale, R., Sarzi-Puttini, P., Atzeni, F., Gazzoni, M., Buskila, D. and Rainoldi, A. 2009. Central motor control failure in fibromyalgia: a surface electromyography study. BMC musculoskeletal disorders, 10 (1), p. 78.
Klaver-Kr’Ol E, Zwarts M, Ten Klooster P, Rasker J (2012) Abnormal muscle membrane function in fibromyalgia patients and its relationship to the number of tender points. Clin Exp Rheumatol 30(6 suppl 74):44–50
Gandevia SC (2001) Spinal and supraspinal factors in human muscle fatigue. Physiol Rev 81(4):1725–89
Søgaard K, Gandevia SC, Todd G, Petersen NT, Taylor JL (2006) The effect of sustained low-intensity contractions on supraspinal fatigue in human elbow flexor muscles. J Physiol 573(Pt 2):511–23
Gandevia SC, Allen GM, Butler JE, Taylor JL (1996) Supraspinal factors in human muscle fatigue: evidence for suboptimal output from the motor cortex. J Physiol 490(Pt 2):529–36
Ranieri F, Di Lazzaro V (2012) The role of motor neuron drive in muscle fatigue. Neuromuscul Disord 22(Suppl 3):S157–61. doi:10.1016/j.nmd.2012.10.006
Taylor JL, Todd G, Gandevia SC (2006) Evidence for a supraspinal contribution to human muscle fatigue. Clin Exp Pharmacol Physiol 33:400–405
Amann M (2012) Significance of group III and IV muscle afferents for the endurance exercising human. Proceedings of the Australian Physiological Society 43:1–7
Mense S, Meyer H (1985) Different types of slowly conducting afferent units in cat skeletal muscle and tendon. J Physiol 363:403–17
Amann M, Blain GM, Proctor LT, Sebranek JJ, Pegelow DF, Dempsey JA (2011) Implications of group III and IV muscle afferents for high-intensity endurance exercise performance in humans. J Physiol 589(Pt 21):5299–309. doi:10.1113/jphysiol.2011.213769
Amann M, Blain GM, Proctor LT, Sebranek JJ, Pegelow DF, Dempsey JA. Group III and IV muscle afferents contribute to ventilatory and cardiovascular response to rhythmic exercise in humans. J Appl Physiol (1985). 2010 Oct;109(4):966–76. doi: 10.1152/japplphysiol.00462.2010. Epub 2010 Jul 15. PubMed PMID: 20634355; PubMed Central PMCID: PMC2963332.
Tanaka M, Watanabe Y. Supraspinal regulation of physical fatigue. Neurosci Biobehav Rev. 2012 Jan;36(1):727–34. doi: 0.1016/j.neubiorev.2011.10.004. Epub 2011 Oct 25. Review. PubMed PMID: 22040772.
Hilty L, Jäncke L, Luechinger R, Boutellier U, Lutz K (2011) Limitation of physical performance in a muscle fatiguing handgrip exercise is mediated by thalamo-insular activity. Hum Brain Mapp 32(12):2151–60. doi:10.1002/hbm.21177
St Clair Gibson A, Noakes TD. Evidence for complex system integration and dynamic neural regulation of skeletal muscle recruitment during exercise in humans. Br J Sports Med. 2004 Dec;38(6):797–806. Review. PubMed PMID: 15562183; PubMed Central PMCID: PMC1724966.
Noakes TD (2007) The central governor model of exercise regulation applied to the marathon. Sports Med 37(4–5):374–7
Fulle S, Belia S, Vecchiet J, Morabito C, Vecchiet L, Fanò G (2003) Modification of the functional capacity of sarcoplasmic reticulum membranes in patients suffering from chronic fatigue syndrome. Neuromuscul Disord 13(6):479–484
Goldstein D, Bennett B, Friedlander M, Davenport T, Hickie I, Lloyd A (2006) Fatigue states after cancer treatment occur both in association with, and independent of, mood disorder: a longitudinal study. BMC Cancer 6:240
Banerjee M, Siddique S, Dutta A, Mukherjee B, Ranjan Ray M (2012) Cooking with biomass increases the risk of depression in pre-menopausal women in India. Soc Sci Med 75(3):565–72. doi:10.1016/j.socscimed.2012.03.021
Sejersted OM, Sjøgaard G (2000) Dynamics and consequences of potassium shifts in skeletal muscle and heart during exercise. Physiol Rev 80(4):1411–1481
Clausen T (2003) Na+-K+ pump regulation and skeletal muscle contractility. Physiol Rev 83(4):1269–1324
Nielsen JJ, Kristensen M, Hellsten Y, Bangsbo J, Juel C (2003) Localization and function of ATP-sensitive potassium channels in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol 284(2):R558–R563
Street D, Nielsen JJ, Bangsbo J, Juel C (2005) Metabolic alkalosis reduces exercise-induced acidosis and potassium accumulation in human skeletal muscle interstitium. J Physiol 566(Pt 2):481–489
Bailey DM, Davies B, Young IS, Jackson MJ, Davison GW, Isaacson R, Richardson RS (2003) EPR spectroscopic detection of free radical outflow from an isolated muscle bed in exercising humans. J Appl Physiol 94(5):1714–1718
McKenna MJ, Medved I, Goodman CA, Brown MJ, Bjorksten AR, Murphy KT, Petersen AC, Sostaric S, Gong X (2006) N-acetylcysteine attenuates the decline in muscle Na+, K + −pump activity and delays fatigue during prolonged exercise in humans. J Physiol 576(Pt 1):279–288
Petrushanko I, Bogdanov N, Bulygina E, Grenacher B, Leinsoo T, Boldyrev A, Gassmann M, Bogdanova A (2006) Na-K-ATPase in rat cerebellar granule cells is redox sensitive. Am J Physiol Regul Integr Comp Physiol 290(4):R916–25
Kurella EG, Tyulina OV, Boldyrev AA (1999) Oxidative resistance of Na/K-ATPase. Cell Mol Neurobiol 19(1):133–40
Aughey RJ, Gore CJ, Hahn AG, Garnham AP, Clark SA, Petersen AC, Roberts AD, McKenna MJ. Chronic intermittent hypoxia and incremental cycling exercise independently depress muscle in vitro maximal Na-K+-ATPase activity in well-trained athletes. J Appl Physiol (1985). 2005 Jan;98(1):186–92. Epub 2004 Mar 19. PubMed PMID: 15033968.
Kim MS, Akera T (1987) O2 free radicals: cause of ischemia-reperfusion injury to cardiac Na+-K+-ATPase. Am J Physiol 252(2 Pt 2):H252–H257
Boldyrev AA, Bulygina ER (1997) Na/K-ATPase and oxidative stress. Ann N Y Acad Sci 834:666–668
Vinnikova AK, Kukreja RC, Hess ML (1992) Singlet oxygen-induced inhibition of cardiac sarcolemmal Na+K(+)-ATPase. J Mol Cell Cardiol 24(5):465–470
Cobley JN, McGlory C, Morton JP, Close GL (2011) N-Acetylcysteine’s attenuation of fatigue after repeated bouts of intermittent exercise: practical implications for tournament situations. Int J Sport Nutr Exerc Metab 21(6):451–61
Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, Miklossy G, Jimah J, Doherty E, Tily H, Francis L, Garcia R, Dawood M, Yu J, Ramos I, Coman I, Faraone SV, Phillips PE, Perl A (2012) N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 64(9):2937–46. doi:10.1002/art.34502
Stanislaus R, Gilg AG, Singh AK, Singh I (2005) N-acetyl-l-cysteine ameliorates the inflammatory disease process in experimental autoimmune encephalomyelitis in Lewis rats. J Autoimmune Dis 2(1):4
Jackson WM, Aragon AB, Djouad F, Song Y, Koehler SM, Nesti LJ, Tuan RS (2009) Mesenchymal progenitor cells derived from traumatized human muscle. J Tissue Eng Regen Med 3(2):129–38. doi:10.1002/term.149
Musarò A, Fulle S, Fanò G (2010) Oxidative stress and muscle homeostasis. Curr Opin Clin Nutr Metab Care 13(3):236–42. doi:10.1097/MCO.0b013e3283368188
Naves LA, McCleskey EW (2005) An acid-sensing ion channel that detects ischemic pain. Braz J Med Biol Res 38(11):1561–9
Yagi J, Wenk HN, Naves LA, McCleskey EW (2006) Sustained currents through ASIC3 ion channels at the modest pH changes that occur during myocardial ischemia. Circ Res 99(5):501–9
Connor M, Naves LA, McCleskey EW (2005) Contrasting phenotypes of putative proprioceptive and nociceptive trigeminal neurons innervating jaw muscle in rat. Mol Pain 1:31
Light AR, Hughen RW, Zhang J, Rainier J, Liu Z, Lee J (2008) Dorsal root ganglion neurons innervating skeletal muscle respond to physiological combinations of protons, ATP, and lactate mediated by ASIC, P2X, and TRPV1. J Neurophysiol 100:1184–1201
Fukuda K, Straus SE, Hickie I, Sharpe MC, Dobbins JG, Komaroff A (1994) The chronic fatigue syndrome: a comprehensive approach to its definition and study International Chonic Fatigue Syndrome Study Group. Ann Intern Med 121(12):953–9
Holmes GP, Kaplan JE, Gantz NM, Komaroff AL, Schonberger LB, Straus SE, Jones JF, Dubois RE, Cunningham-Rundles C, Pahwa S et al (1988) Chronic fatigue syndrome: a working case definition. Ann Intern Med 108(3):387–9
Beyer I, Njemini R, Bautmans I, Demanet C, Bergmann P, Mets T (2012) Inflammation-related muscle weakness and fatigue in geriatric patients. Exp Gerontol 47(1):52–9. doi:10.1016/j.exger.2011.10.005
Deshaies RJ, Koch BD, Werner-Washburne M, Craig EA, Schekman R (1988) A subfamily of stress proteins facilitates translocation of secretory and mitochondrial precursor polypeptides. Nature 332(6167):800–5
Kregel KC. Heat shock proteins: modifying factors in physiological stress responses and acquired thermotolerance. J Appl Physiol (1985). 2002 May;92(5):2177–86. Review. PubMed PMID: 11960972.
Hall TJ (1994) Role of hsp70 in cytokine production. Experientia 50(11–12):1048–53
Moseley P (2000) Stress proteins and the immune response. Immunopharmacology 48(3):299–302
Müller E, Munker R, Issels R, Wilmanns W (1993) Interaction between tumor necrosis factor-alpha and HSP 70 in human leukemia cells. Leuk Res 17(6):523–6
Ensor JE, Wiener SM, McCrea KA, Viscardi RM, Crawford EK, Hasday JD (1994) Differential effects of hyperthermia on macrophage interleukin-6 and tumor necrosis factor-alpha expression. Am J Physiol 266(4 Pt 1):C967–74
Srivastava PK, Udono H, Blachere NE, Li Z (1994) Heat shock proteins transfer peptides during antigen processing and CTL priming. Immunogenetics 39(2):93–8
Todryk SM, Melcher AA, Dalgleish AG, Vile RG (2000) Heat shock proteins refine the danger theory. Immunology 99(3):334–7
Locke M (1997) The cellular stress response to exercise: role of stress proteins. Exerc Sport Sci Rev 25:105–36
Thambirajah AA, Sleigh K, Stiver HG, Chow AW (2008) Differential heat shock protein responses to strenuous standardized exercise in chronic fatigue syndrome patients and matched healthy controls. Clin Invest Med 31(6):E319–27
Hooper PL, Hightower LE, Hooper PL (2012) Loss of stress response as a consequence of viral infection: implications for disease and therapy. Cell Stress Chaperones 17(6):647–55. doi:10.1007/s12192-012-0352-4
Liu CC, Lin CH, Lin CY, Lee CC, Lin MT, Wen HC (2013) Transgenic overexpression of heat shock protein 72 in mouse muscle protects against exhaustive exercise-induced skeletal muscle damage. J Formos Med Assoc 112(1):24–30. doi:10.1016/j.jfma.2012.02.007
Kim C, Kim JY, Kim JH (2008) Cytosolic phospholipase A(2), lipoxygenase metabolites, and reactive oxygen species. BMB Rep 41(8):555–9
Vasilaki A, van der Meulen JH, Larkin L, Harrison DC, Pearson T, Van Remmen H, Richardson A, Brooks SV, Jackson MJ, McArdle A (2010) The age-related failure of adaptive responses to contractile activity in skeletal muscle is mimicked in young mice by deletion of Cu, Zn superoxide dismutase. Aging Cell 9(6):979–90. doi:10.1111/j.1474-9726.2010.00635.x
Sen CK, Atalay M, Hänninen O (1994) Exercise-induced oxidative stress: glutathione supplementation and deficiency. J Appl Physiol 77(5):2177–2187
Nakamoto H, Kaneko T, Tahara S, Hayashi E, Naito H, Radak Z, Goto S (2007) Regular exercise reduces 8-oxodG in the nuclear and mitochondrial DNA and modulates the DNA repair activity in the liver of old rats. Exp Gerontol 42(4):287–95
Barreiro E, Gáldiz JB, Mariñán M, Alvarez FJ, Hussain SN, Gea J (2006) Respiratory loading intensity and diaphragm oxidative stress: N-acetyl-cysteine effects. J Appl Physiol 100(2):555–63
Aghdasi B, Zhang JZ, Wu Y, Reid MB, Hamilton SL (1997) Multiple classes of sulfhydryls modulate the skeletal muscle Ca2+ release channel. J Biol Chem 272(6):3739–48
Xu KY, Zweier JL, Becker LC (1997) Hydroxyl radical inhibits sarcoplasmic reticulum Ca(2+)-ATPase function by direct attack on the ATP binding site. Circ Res 80(1):76–81
Putkey JA, Dotson DG, Mouawad P (1993) Formation of inter- and intramolecular disulfide bonds can activate cardiac troponin C. J Biol Chem 268(10):6827–30
Williams DL Jr, Swenson CA (1982) Disulfide bridges in tropomyosin. Effect on ATPase activity of actomyosin Eur J Biochem 127(3):495–9
Ajtai K, Burghardt TP (1989) Fluorescent modification and orientation of myosin sulfhydryl 2 in skeletal muscle fibers. Biochemistry 28(5):2204–10
DalleDonne I, Milzani A, Colombo R (1995) H2O2-treated actin: assembly and polymer interactions with cross-linking proteins. Biophys J 69(6):2710–9
Boldyrev A, Kurella E (1996) Mechanism of oxidative damage of dog kidney Na/K-ATPase. Biochem Biophys Res Commun 222(2):483–7
Funding and competing interests
No specific funding was obtained for this specific review.
GM, MB, PG, and MM declare that they have no competing interests.
Authors’ contributions
GM and MM participated in the design of this review, while PG and MB helped to draft the paper. All authors read and approved the final version.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Morris, G., Berk, M., Galecki, P. et al. The Neuro-Immune Pathophysiology of Central and Peripheral Fatigue in Systemic Immune-Inflammatory and Neuro-Immune Diseases. Mol Neurobiol 53, 1195–1219 (2016). https://doi.org/10.1007/s12035-015-9090-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9090-9