
Abstract
Autophagy is a constitutive lysosomal catabolic pathway that degrades damaged organelles and protein aggregates. Neuronal survival is highly dependent on autophagy due to its post-mitotic nature, polarized morphology, and active protein trafficking. Autophagic dysfunction has been linked to several neuronal diseases. Our understanding is still incomplete but may highlight up-to-date findings on how autophagy is executed and regulated at the molecular level and its role in neurodegenerative diseases (including Alzheimer’s disease (AD), Huntington’s disease (HD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS)), brain ischemia, and myelin diseases, hence providing attractive new avenues for the development of treatment strategies to combat neuronal diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The Autophagic/Lysosomal Pathway
The dynamic, tightly regulated balance between the formation and degradation of proteins and organelles maintains normal cell growth and development. Autophagy and the ubiquitin-proteasome system (UPS) are the two major degradative pathways in mammalian cells. The latter is responsible for degrading damaged proteins in the proteasome and is specific in its selection of proteins and protein aggregates that are tagged with ubiquitin. Autophagy, on the other hand, is a cell self-digestive, lysosomal degradation pathway. It is highly conserved from yeast to mammals, both morphologically and with regard to the protein constituents that make up the core autophagy machinery.
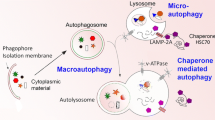
There are three basic forms of autophagy, namely, macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), which primarily differ in the way in which cytosolic components are delivered to lysosomes. Moreover, the different types of autophagy share a common endpoint, the lysosome, but differ in the substrates targeted, their regulation, and the conditions in which each of them is preferentially activated [1–3]. Both micro- and macroautophagy can be selective or nonselective and these processes have been best characterized in yeast. Microautophagy is a much simpler process and delivers the cytoplasmic contents by invagination of the lysosomal membrane into its lumen [4, 5]. Microautophagy is dependent on GTP hydrolysis and on calcium [6]. However, the molecular regulation of microautophagy remains to be unraveled. Bulk microautophagy does not seem to be dependent on Atg proteins, whereas selective forms of microautophagy require different sets of Atg proteins [7–9]. CMA incorporates cytosolic proteins that are brought by chaperones to the lysosome membrane [10, 11]. CMA involves the selective sequestration of proteins with a KFERQ-like motif into lysosomes via chaperones Hsc70 and Lysosomal-Associated Membrane Protein-2A (LAMP-2A) complex, which transfers protein substrates to the lysosomal membrane, and there, through binding to the receptor LAMP-2A, they are translocated into the lysosome. CMA performs several general functions, such as the elimination of oxidized proteins and the removal of misfolded proteins, and also provides amino acids during prolonged periods of starvation. Macroautophagy, usually identified simply as autophagy, is the well-characterized form of autophagy that involves the sequestration of cytosolic components into lysosomes in a nonselective manner. Macroautophagy occurs constitutively in cells and is markedly induced under stress conditions, such as starvation, and has two major purposes: as a source to generate essential macromolecules and energy in conditions of nutritional scarcity or as a mechanism for the removal of altered intracellular components [12]. Originally viewed as a nonselective, bulk process, it is now known that there are some selective forms of autophagy [13]. Selective autophagy of different cargoes is named after the organelle or the type of material ingested, so that mitophagy is used for mitochondria, reticulophagy for endoplasmic reticulum, pexophagy for peroxisomes, lipophagy for lipid droplets, and xenophagy for heterologous substrates (e.g., pathogens) [14]. It is interesting to note that cross-talk occurs between macroautophagy and CMA during starvation [15, 16]. When CMA is stimulated, macroautophagy is first induced and then declines. The molecular basis for this switch remains to be identified.
Cross-Talk Between the Ubiquitin-Proteasome System and Macroautophagy
Proteins are targeted for destruction by the UPS via a series of enzymatic reactions that tag them with homopolymers of a small, 76-amino acid residue protein called ubiquitin [17]. Polyubiquitylation marks the UPS clients for transportation by a poorly understood shuttling machinery to a specialized organelle called the proteasome, where proteins are degraded to oligopeptides, which are released into the cytoplasm or nucleoplasm, where they can be digested into amino acids by soluble peptidases. The narrow size of the proteasomal catalytic pore suggests that protein substrates need to be partially unfolded prior to entry into the 20S subunit. Thus, protein complexes and aggregates can only be digested if disassembled, which makes them poor proteasome substrates [18]. In contrast to the UPS, autophagy is restricted to the cytoplasm but is capable of degrading a much wider spectrum of substrates, which, on average, tend to be longer-lived and bulkier. These include functional or misfolded soluble proteins, protein complexes, oligomers, and aggregates. Although the two pathways differ significantly, there appears to be a certain overlap in function between the two degradative pathways; it is speculated that ubiquitin coordinates the catabolism of cellular targets by both the UPS and autophagy. Indeed, many proteins are known to be substrates of both degradative systems, and in certain conditions, ubiquitylated proteasomal substrates, which are normally degraded by the UPS, can also be digested by autophagy and vice versa [19, 20]. Moreover, misfolded protein aggregates, which are often polyubiquitinated, can contribute to the pathogenesis of many human diseases [21]. Notably, proteasome functions are often impaired in these cases and are likely not able to degrade the aggregate fully [22–24]. Autophagic vacuoles were frequently found in these cases [22, 25, 26] and, indeed, autophagy has been shown to be able to participate in the removal of misfolded/unfolded proteins or protein aggregates [27–31]. These studies indicate that impairment of proteasome activity was found to activate autophagy, which was thought to be a compensatory mechanism allowing the cell to reduce the levels of UPS substrates. Some potential overlap may result from incomplete specificity of the different adaptor molecules that have been proposed to retrieve ubiquitylated substrates for each degradative pathway. These proteins have emerged as important players in mediating this cross-talk, including histone deacetylase 6 (HDAC6) [32], neighbor of BRCA1 gene 1 (NBR1), p62/sequestosome 1 (p62) [33], and the FYVE-domain containing protein Alfy [34]. Notably, these proteins have all been found to regulate or be essential for aggresome formation.
Impaired autophagy also leads to the impaired degradation of specific UPS clients. The data suggested that the decreased UPS flux in autophagy-compromised cells was not due to impaired catalytic activity of proteasomes isolated from them. Instead, the block in the UPS function is mediated by accumulation of p62, as its knockdown rescued the levels of UPS substrates in autophagy-deficient cells [35]. In addition, overexpression of p62 alone was sufficient to inhibit the UPS, an effect partially dependent on its UBA domain. Since p62 competes with other ubiquitin-binding proteins involved in proteasomal degradation, like p97/valosin-containing protein (VCP), for binding to ubiquitylated proteins, we proposed that elevated levels of p62 may deny such shuttling proteins access to ubiquitylated UPS substrates [36]. Thus, p62 has been implicated in two different, but not mutually exclusive, mechanisms of cross-talk between the UPS and autophagy.
Unfortunately, there is little consensus on the exact mechanism(s) of this cross-talk, as several potential explanations have been suggested. One such proposed mechanism involves ER stress is critical for autophagy induction and is in turn mitigated by autophagy [37].
The Autophagy Flux
Genetic, biochemical, and morphological studies have demonstrated that macroautophagy is a multi-step process. Autophagy starts with the formation of double-membraned cisternae that subsequently engulf cytoplasmic organelles to form double-membrane vacuoles, known as autophagosomes (APs), which expand around portions of the cytoplasm containing proteins, aggregated proteins, and organelles, and then sequester cargo and fuse with acidic lysosomes to form autolysosomes. Hence, the appearance of APs under transmission electron microscopy (EM) is a morphological hallmark unique to autophagy. Most mature APs are approximately 0.5 to 1 μm in diameter; they can fuse with endocytic pathway vesicles including early endosomes, multivesicular bodies (MVBs), and late endosomes thus forming amphisomes. Eventually, the AP fuses with the lysosome, becoming an autolysosome, and the acidic hydrolases within break down the captured components to macromolecules and amino acids that are released back to the cytosol by transporters and permeases delivered with the lysosomal membrane. After degradation by autophagy, the recycled products can be used in biosynthetic pathways and to generate new proteins [38, 39].
Autophagy Machinery
An understanding of the molecular mechanism of mammalian macroautophagy was less well known, until a group of autophagy-related genes (Atgs) and their encoded proteins (ATGs) was recently discovered only within the last couple of decades [40, 41]. Currently, there are 35 known ATG proteins involved in both autophagy and cytoplasm-to-vacuole-targeting (Cvt) pathway (a biosynthetic autophagy-like pathway) in yeast and most, if not all, human counterparts of the autophagy-specific proteins that have been discovered [40]. The core machinery can be grouped into six functional units: (1) the ULK1 kinase complex (ULK:Atg13:FIP200) for the induction of autophagy, (2) Atg9 for recycling membrane, (3) class III phosphatidylinositol 3-kinase (PI3K) complex (Vps34-Beclin1-Vps15) for vesicle nucleation, (4) phosphatidylinositol 3-phosphate[PI(3)P]-binding Atg2-Atg18 complex (WIPI1/2 in mammals), (5) Atg12-Atg5-Atg16L conjugation system, and (6) LC3 conjugation system involving phosphatidylethanolamine (LC3-PE) for membrane expansion [42]. These six functional groups act at an early stage in the autophagy pathway and are recruited to the phagophore. We now know it requires these components as well as other cytosolic proteins and intracellular organelles such as the endoplasmic reticulum (ER), Golgi apparatus, and perhaps mitochondria, in addition to the endosome-lysosome system, to control autophagosome formation and thus the rate of degradation of sequestered material and cellular energy levels (Fig. 1).

Steps of autophagy and a series of autophagy-related (Atg) proteins involved in the specific processes of autophagy in mammal cells
Here, we present each step separately, accompanied by an indication of genes associated with each sub-process.
Autophagic Induction
Autophagosome formation appears to start at pre-autophagosomal structures (autophagosome precursor; also known as phagophore)-assembly sites (PAS). The source of the membrane is unclear, although recent data support a contribution from the ER [43]. Autophagosome initiation and formation in yeast is centrally controlled by the Atg1:Atg13:Atg17 complex [44–46]. This complex recruits multiple autophagy proteins to a site called the pre-autophagosomal structure (PAS) [47], where it is thought to play a critical role in autophagosome formation [48, 49]. Unfortunately, detailed structural information regarding the PAS is not currently available, and it is even unknown whether it is a membranous structure. Equivalent structures have not been observed in mammalian cells.
It has recently been shown that the mammalian counterpart of this complex is ULK:Atg13:FIP200 (200-kDa focal adhesion kinase family-interacting protein) [50–52]. The two mammalian homologues of serine/threonine protein kinase Atg1, unc-51-like kinase-1 (ULK1) and unc-51-like kinase-2 (ULK2), and the homologue of Atg17, FIP200, interact with mammalian Atg13 to form a stable complex, which is located at the phagophore during starvation. Recent evidence suggests that the structure and function of the ULK:Atg13:FIP200 complex in mammals differ from those of the equivalent Atg1 complex in yeast. The ULK:Atg13:FIP200 complex is a stable complex that is not regulated by nutrition conditions. Inactivation of mTOR by starvation or rapamycin activates ULKs and results in phosphorylation of Atg13 and FIP200. This differs from what is observed in yeast, where the inactivation of TOR increases the binding affinity of Atg1 to Atg13 and Atg17 [53]. Moreover, two teams identified a novel mammalian Atg13 binding protein, named Atg101. Atg101 shows no homology with other Atg proteins and is conserved in various eukaryotes, but not in Saccharomyces cerevisiae. Atg101 is essential for autophagy and contributes to Atg13 function by protecting Atg13 from proteasomal degradation [54, 55]. Corroborating the importance of ULK1 and its interactions with its adaptor proteins and mTOR itself for the initiation of autophagy, a recent study of the hierarchy of mammalian autophagy proteins places ULK1 and FIP200 as the most upstream at the site of autophagosome formation.
Nucleation
In mammals, the nucleation and assembly of the initial phagophore membrane is dependent on the class III phosphatidylinositol 3-kinase (PI3K) complex. The class III PI3K (Vps34) is associated with Beclin 1 (Atg6) and p150, the homologue of Vps15 (phosphoinositide-3-kinase, regulatory subunit 4), to form the Vps34/class III PI3K core complex [56, 57]. Active Vps34 will phosphorylate phosphatidylinositol (PtdIns) to form phosphatidylinositol-3-phosphate (PtdIns3P) in the membrane platform for the formation of autophagosomes, thus creating PtdIns3P-rich omegasomes [58, 59]. This PtdIns3P serves as a recruitment signal for other autophagy proteins, which then positively regulate the further elongation of the phagophore.
There are several studies, conducted in both yeast and mammalian cells, indicating that Beclin 1 is a molecular platform which interacts with many proteins regulating autophagy and phagocytosis as well as the endosome-to-Golgi vacuolar retrieval pathway [56, 60–62]. Beclin 1 is an adaptor protein, which via its interacting proteins, called the Beclin 1 interactome, can either stimulate or suppress the onset of autophagy. It has been increasingly recognized that mammalian Beclin 1 protein assembled two core complexes, Atg14L or UVRAG complexes. Atg14L (the probable mammalian homologue of yeast Atg14) exists primarily in a Beclin1-Atg14L-Vps34-Vps15 complex that is essential for the phagophore formation and thus stimulating autophagocytosis [56, 63], and UV radiation resistance-associated gene (UVRAG) is present in a Beclin1-UVRAG-Vps34-Vps15 complex that controls other Beclin 1-dependent functions, e.g., phagocytosis. Atg14L and UVRAG are located in the Beclin1-Vps34-Vps15 complex in a mutually exclusive manner [64]. These two proteins are cellular targeting proteins which direct the Beclin 1-Vps34-Vps15 complexes to their distinct cellular locations, i.e., Atg14L to specialized subdomain of the ER, called the omegasome and UVRAG mostly to the membranes of endosomes and cell surface [65]. Other additional proteins, including Beclin 1-regulated autophagy (Ambra) [66], Bax-interacting factor 1 (Bif1) [67], PTEN-induced putative kinase 1 (PINK1), neuronal isoform of protein interaction, specifically with TC10 (nPIST) [68], IP3 receptor (IP3R) [69], the pancreatitis-associated protein, vacuole membrane protein 1 (VMP1) [70], and high mobility group box 1 (HMGB1) [71], have also been identified as Beclin1-interacting proteins upregulating autophagy. Recently, further insights have been provided into the mechanisms behind starvation-induced autophagy. Starvation induces Jun N-terminal kinase 1 (JNK1) activity, which phosphorylates Bcl2, thereby disrupting the interaction between Beclin 1 and Bcl-2 and inducing autophagy [72]. This mechanism might also account for the upregulation of autophagy after proteasome inhibition or ER stress, as one study showed that ER stress-induced autophagy is JNK1-dependent and that proteasome inhibition can induce ER stress [37].
In contrast to these positive regulators, there are negative regulators among Beclin1-interacting partners. Anti-apoptotic proteins BCL2 (B cell Lymphoma) and other Bcl-2 family members: Bcl-XL, Bcl-w and Mcl-1 and viral Bcl-2 proteins, bind to Beclin1 through their BH3 domain and inhibit autophagy by disrupting the interaction between Beclin1 and Vps34/class III PI3K complex [60]. The Bcl-2/Beclin 1 interaction is clearly an important checkpoint in autophagy induction upon starvation or treatment with many different compounds. In normal conditions, Bcl-2 inhibits Beclin 1, while upon stress Beclin 1 dissociates from Bcl-2, allowing the activation of Vps34 and the subsequent stimulation of autophagy [73]. The ability of Beclin 1 to switch between the two complexes can be regulated by the phosphorylation of Bcl-2 by JNK1 [72] or phosphorylation of Beclin 1 by DAPK [73]. Moreover, RUN domain- and cysteine-rich domain-containing Beclin1-interacting protein (Rubicon) negatively regulates autophagosome maturation by interacting with Beclin 1 in contrast to Atg14L [74] (Fig. 2).

Two distinct Vps34 protein complex involved in autophagy in mammalian cells
Membrane Flow During Autophagosome Formation
Atg9 and its mammalian ortholog(mAtg9 or Atg9L1) are the only characterized integral membrane proteins required for both autophagosome and Cvt vesicle formation [75]. Recent publications have revealed that Atg9 is in fact a key regulator of autophagy induction [76]. Atg9 provides membranes for the formation of the PAS in yeast [77] and possibly also in mammalian cells during starvation [78] and selective types of autophagy [79, 80]. Under normal conditions, Atg9L1 traffics between the trans-Golgi network (TGN) and late endosomes [81] but, in response to starvation, several Atg9L1 vesicles assembled individually into the pre-autophagosomal structure, and eventually, they are incorporated into the autophagosomal outer membrane [82]. However, Atg9L1 is absent from the completed vesicles, suggesting that it is retrieved before the vesicle sealing/completion step. Recent studies reveal that Atg9 cycles between the mitochondria and the PAS vesicle assembly site [83]. These characteristics make Atg9L1 a potential membrane carrier for vesicle formation. The putative role of Atg9L1 is to carry lipids and/or serve as a platform for recruiting effectors to the phagophore. More recent studies have also implicated Atg9L1 to be located in the outer mitochondrial membrane [84] and even the plasma membrane [85], suggesting that multiple membrane sources may contribute towards autophagosome biogenesis.
Elongation
Although the initiation of autophagy is well described, the exact mechanisms that contribute to elongation and closure of the phagosomal membrane are less understood. Two systems, ATG12-ATG5-ATG16L1 and LC3-PE (Microtubule-associated Light Chain 3-Phosphatidyl Ethanolamine) complexes, are essential for the forming autophagosome to expand and act sequentially [86]. Atg5 and Atg12 are involved in the first of two ubiquitylation-like reactions that control autophagy. Atg12 is conjugated to Atg5 in a reaction that requires Atg7 [ubiquitin-activating-enzyme(E1)-like] and Atg10 [ubiquitin-conjugating-enzyme(E2)-like]. Atg5-Atg12 conjugates are localized onto the PAS and dissociate upon completion of autophagosome formation. The process of Atg5-Atg12 conjugation depends on Vps34 function, and Vps34 activity, along with autophagy, is positively regulated by the small GTPase Rab5. Atg5-Atg12 interacts non-covalently with Atg16L (Atg16-like) to form a complex of approximately 800 kDa [87]. The second ubiquitylation-like reaction involves the conjugation of microtubule-associated protein 1 light chain 3 (MAP1-LC3; also known as Atg8 and LC3) to the lipid phosphatidylethanolamine (PE). LC3 is cleaved at its C-terminus by Atg4 to form cytosolic LC3-I, which is covalently conjugated to PE to form membrane-associated LC3-II, a process that requires the activities of Atg7 (E1-like) and Atg3 (E2-like) [88]. The Atg5-Atg12 conjugate seems to modulate LC3-I conjugation to PE by acting in an E3-like fashion [89] and the location of the Atg5-Atg12:Atg16L complex at the phagophore dictates the location of the LC3 conjugation reaction [90]. In this way, LC3 is specifically targeted to Atg5-Atg12-associated membranes, which are expanded phagophores. The LC3-PE system also involves cargo recognition through interactions with p62/SQSTM1 (Sequestosome 1), NBR1 (Neighbor of BRCA1 gene), and NDP52 (Nuclear Dot Protein 52) [91].
The lipidated LC3 (LC3-II) then associates with newly forming autophagosome membranes. LC3-II remains on mature autophagosomes until its fusion with lysosomes [92]. The conversion of LC3-I to LC3-II is thus widely used as a marker for autophagosome formation. However, the increase of LC3-II alone is not enough to show autophagy activation because the inhibition of LC3-II degradation in the lysosome by the impaired autophagy flux can also cause its accumulation. When the autophagosome is formed, the Atg12-Atg5:Atg16L complex leaves the autophagosome, and the pool of LC3 associated with the autophagosomal cytosolic surfaces is cleaved from the PE by the protease Atg4 and recycled. Cross-talk between the two ubiquitylation-like systems has also been suggested. Atg10 can interact with LC3-I and facilitate LC3-I conjugation to PE [93]. Similarly, Atg3 co-immunoprecipitates with Atg12, and overexpression of Atg3 increases Atg5-Atg12 conjugation [94].
Despite the extensive involvement of these two ubiquitin-like conjugation systems in autophagy, the presence of Atg5-, Atg7-, and LC3-independent autophagy pathways has been reported [95]. This pathway of autophagy was not associated with LC3 processing but appeared to specifically involve autophagosome formation from late endosomes and the trans-Golgi [95]. Atg7-independent autophagy had been implicated in mitochondrial clearance from reticulocytes [96], and it has consistently been shown that Ulk-1 (a mammalian homologue of Atg1) is required for both reticulocyte clearance of mitochondria [97] and, along with Beclin-1, for Atg5/Atg7-independent autophagy [95]. The exact molecular basis of Atg5/Atg7-independent autophagy remains to be elucidated (Fig. 2).
Maturation (Fusion with Lysosomes)
The late stage of autophagy depends on molecules that regulate the maturation of autophagosomes, including their fusion with endosomes and lysosomes, as well as on the acidification of the autophagic compartments, and the recycling of metabolites from the lysosomal compartment. These steps are of a fundamental importance for the flux (defined here as extending from the cargo sequestration step to that of its lysosomal degradation) of material through the autophagic pathway [98]. Any blockade in the maturation of autophagosomes, fusion with the lysosomal compartment, or impairment of the lysosomal function or biogenesis would result in an accumulation of autophagosomes that would inevitably slow down or interrupt the autophagic flux [99, 100]. Jaeger et al. observed that the deficiency in Beclin 1 expression markedly reduced the level of Vps34 protein and subsequently impaired the degradation of autophagosomes in cultured cells. They also reported that the inhibition of autophagosomal-lysosomal fusion with bafilomycin A triggered the accumulation of large, APP-containing vesicles in the perinuclear space and downregulated the levels of Beclin 1 and Vps34 proteins [101]. This implies a negative feedback mechanism, which could block the initiation of autophagy if the maturation of autophagosomes is impaired.
Although it is unclear how exactly autophagosomes fuse with lysosomes, their fusion is known to require several proteins such as LAMP2, the Rubicon-UVRAG complex, soluble N-ethylmalemide sensitive factor attachment protein receptor (SNAREs), homotypic fusion and protein sorting (HOPS), Ras [rat sarcoma] like in rat brain (Rab), endosormal sorting complex required for transport (ESCRT), and LC3 [3, 102, 103]. Once the autophagosome fuses with lysosomes, cytosolic components are degraded by hydrolases and lipases.
Signals for Autophagosome Formation
Several signaling pathways seem to regulate autophagy in mammalian cells. Similar to yeast, the classical pathway involves the serine/threonine kinase, mammalian target of rapamycin (mTOR). Various pathways and small molecules regulating autophagy via mTOR and mTOR-independent mechanisms have been identified in recent years.
mTOR-Dependent Pathway
The mTOR pathway involves two functional complexes: a rapamycin-sensitive mTOR complex 1 (mTORC1), which inhibits autophagy, consisting of the mTOR catalytic subunit, regulatory associated protein of mTOR (raptor), G protein β-subunit-like protein (GβL; also known as mLST8), and proline-rich Akt substrate of 40 kDa (PRAS40); and mTOR complex 2 (mTORC2) consisting of mTOR, rapamycin-sensitive companion of mTOR (rictor), GβL, SAPK-interacting protein 1 (SIN1), and protein observed with rictor (PROTOR) that is not a direct autophagy regulator [104].
Key upstream regulators of mTORC1 include the class I phosphoinositide 3-kinase-Akt pathway, which keeps mTORC1 active in cells with sufficient growth factors, and the AMP-activated protein kinase (AMPK) pathway that inhibits mTORC1 upon starvation and calcium signals [105, 106]. Especially, mTORC1 requires Rag GTPase, Rheb, and Vps34 for its activation and subsequent inhibition of autophagy in response to amino acids [107, 108]. Energy levels are primarily sensed by AMPK, a key factor for cellular energy homeostasis. In low energy states, AMPK is activated and the activated AMPK then inactivates mTORC1 through TSC1/TSC2 and Rheb protein [109]. The tumor suppressor gene, p53, has been reported to play a dual role in autophagy via the mTOR pathway [110, 111], with some studies suggesting transcription-dependent and transcription-independent positive regulation of autophagy [112, 113] and other studies suggesting the negative regulation of autophagy by wild type and mutant forms of p53 in the cytoplasm [114, 115].
p70S6 kinase or S6K, homologue of Sch9, is a downstream target of mTORC1. In mammals, the S6K1 [116] is a positive regulator of autophagy, whereas yeast Sch9 is a negative regulator [117]. The mechanism by which S6K promotes autophagy may involve synthesis of proteins that participate in autophagosome formation and maturation [118, 119] or modulation of pathways that directly regulate autophagy. While autophagy is a beneficial process that recycles cellular contents for survival, excessive self-eating can cause cell death. Thus, placing a positive regulator of autophagy under the control of mTOR ensures that extended starvation leading to mTOR inhibition prevents unrestrained autophagy, which causes cell death [120]. The negative role of mTOR and the positive role of S6K in autophagy may provide a balance in the levels of autophagy over extended periods of nutrient deprivation.
Another downstream target of mTOR involved in regulating excessive autophagy is death-associated protein 1 (DAP1) [121]. Under amino acid deprivation, decrease in mTOR-mediated DAP1 phosphorylation restores the anti-autophagic function of DAP1. Thus, DAP1 reactivation in the absence of mTOR function prevents excessive autophagy during nutrient-deprived condition. mTORC1 can also regulate autophagy by unknown mechanisms that are rapamycin-insensitive [122]. Finally, mTORC2 can negatively regulate autophagy indirectly through Akt [123, 124].
Intriguingly, a recent study suggests that mTOR signaling is inhibited during autophagy initiation, but reactivated upon prolonged starvation. As a consequence, reactivated mTOR attenuates autophagy and generates proto-lysosomal tubules and vesicles that extrude from autolysosomes. These mature into functional lysosomes, thereby restoring lysosome homeostasis [125]. This negative feedback mechanism ensures the reversion of autophagy upon nutrient replenishment and as such prevents excess cytoplasmic vacuolization, which could lead to autophagic cell death [126–128].
mTOR-Independent Pathway
Besides TOR, Ras signaling also plays a role in autophagy regulation by growth factors. Two downstream effector cascades of Ras, the Ras-PtdIns3K and Ras-Raf-1-ERK1/2 pathways, are likely to oppose each other in autophagy regulation through signaling in response to growth factors versus the absence of amino acids [39]. Other mTOR-independent autophagy activation pathways have been reported including that induced by lithium chloride, which appears to act through modulation of myo-inositol-1,4,5 triphosphate (IP3) levels [129]. Although conflicting data does exist, one group reports that resveratrol also drives autophagy independent of mTOR by activating sirtuins that deacetylate Atg’s, which activates autophagy [115]. Mammalian PKA negatively regulates autophagy either by directly phosphorylating LC3 [130] or by activating TORC1, which inhibits autophagy [131]. Autophagy is also modulated by reactive oxygen species (ROS). Starvation (a potent inducer of autophagy) increases levels of ROS in a PI3K-dependent manner, and treatment with antioxidants ameliorates the ability of starvation to induce autophagy [132]. One way in which ROS may be acting to regulate autophagy is by the modulation of the action of Atg4 on Atg8/LC3 [133]. It is possible that under oxidative conditions Atg4 is oxidized and inactive, which allows Atg8 to lipidate and thus initiates autophagy, while reduced Atg4 is active favoring Atg8 delipidation. A recent study showed that small-molecule enhancers of the cytostatic effects of rapamycin (called SMERs) induce autophagy independently of mTOR [134]. c-Jun NH2-terminal kinase 1 (JNK1) may induce autophagy by phosphorylating Bcl-2 or Bim and abolishing their inhibitory effects on autophagy [72].
Autophagy in Neurons
Neurons are differentiated cells with a polarized cell body. Their viability and function is closely connected to the availability of trophic factors (including for example neurotrophins like nerve growth factor (NGF)) but also critically depends on active membrane transport connecting the distant cell body with dendrites and axons. Neurons, because of their extreme polarization, size, and post-mitotic nature may be particularly sensitive to the accumulation of aggregated or damaged cytosolic compounds, or membranes [135]. Thus, the beneficial roles of autophagy in nervous system are mainly associated with maintenance of the normal balance between the formation and degradation of cellular proteins.
Recent findings show that autophagy in neurons is indeed constitutively active [136, 137] and that autophagosomes accumulate rapidly when their clearance is blocked [138], indicating fast basal turnover. Reports using mutant mice lacking the autophagy-related genes Atg5 or Atg7 indicate an active role of basal autophagy in maintaining neuronal homeostasis [53, 137]. Another physiological role for neuronal autophagy was shown using mutant mice that display a specific loss of Atg7 in Purkinje neurons [139, 140]. This study reveals the indispensability of autophagy in the maintenance of axonal homeostasis and the prevention of axonal dystrophy and degeneration.
Multiple studies have suggested that the levels of lipidated LC3 and the numbers of autophagosomes detected in neurons are relatively low in normal as well as nutrient withdrawal conditions [141, 142]. One hypothesis is that autophagosomes are formed normally, but there is an extremely high rate of turnover of autophagosomes due to efficient autophagic/lysosomal degradation in neurons [143]. An alternate hypothesis, however, is that autophagosome synthesis is tightly controlled in neurons; the constantly low rate of formation of typical autophagosomes may be sufficient to maintain the homeostatic balance. Perhaps, neurons are prevented from undergoing extensive autophagy induction in response to starvation because this could lead to loss of mass and cells, as neurons cannot be regenerated. When an organism encounters a period of nutrient deprivation, the brain is typically shielded by peripheral production of nutrients through autophagy, ensuring that they can maintain essential normal function regardless of such conditions. Future studies of the unique regulation of autophagy in neurons should examine these possibilities.
An Influence of Neuronal Subtype
Acute stressors such as axotomy or nerve crush lead to a rapid accumulation of autophagosome-like vesicles in axon terminals as well as the soma [144, 145]. These data indicated that autophagosome-like structures could be formed at the cell body, but importantly, may also be created distally in the axon. A membrane source for creating autophagosomes distally is readily available: the neuronal ER is not limited to the soma, but also found in the axons and terminals [146]. In light of the length of neurons, the ability to locally create autophagosomes would increase the efficiency of macroautophagic clearance by sequestering the cargo. Studies suggest that not only could cargo be sequestered distally, but autophagosomes could mature and form amphisomes while being transported retrogradely back to the soma [147–149]. These findings suggest that neuronal autophagy may be adapted to different neuronal functions in different subcompartments, and the degree of functional subcompartmentalization (soma, axons, and dendrites) of the neuron distinguishes it from other cell types.
Historically, both a proregenerative and prodestructive role for macroautophagy had been proposed: in the first instance, autophagosomes clear and recycle the damaged material to promote an environment that permitted axonal regeneration. In the latter instance, the induction of macroautophagy would promote cell death to eliminate the damaged cell [150]. Recently, new genetic and molecular tools have begun to establish that both models are likely correct and the interplay between neurons and glia may underlie how macroautophagy can be toxic in one cell type and protective in another [151, 152]. It is also possible that the basal dependence on macroautophagy by a neuron may indicate how it responds to macroautophagic induction. For example, the Purkinje cells of the cerebellum have been shown to rely heavily on macroautophagy to survive [139]. Thus, under conditions of excitotoxic stress, the autophagosome accumulation observed in axonal swellings is thought largely to be protective [68, 153]. In contrast, dopaminergic neurons depend less on macroautophagy for everyday survival [154]. A strong induction of macroautophagy in these neurons due to stress may, therefore, lead to an end-stage response that is destructive. Adding to the complexity, a recent study shows that cultured neurons from male rats more readily undergo autophagy in response to 24 h of nutrient deprivation compared to female neurons [155], indicating gender differences in autophagic capacity of neurons.
Autophagy Signaling in Neurons
Although autophagy occurs in virtually all cell types, and likely involves highly conserved molecular machinery, emerging evidence suggests cell type/tissue-specific regulation of autophagy. The investigations as to whether autophagy signaling in neurons requires mTOR have yielded somewhat mixed results. Some evidence has been provided that mTOR inhibition by rapamycin does indeed induce autophagic flux in neurons [53, 156, 157]. Importantly, Young et al. showed that insulin was the key factor and that the absence of insulin induced autophagy in primary neurons, in an mTOR-dependent manner [158]. However, other evidence points to an mTOR-independent mechanism [159–161], and in fact, a small molecule screen for inducers of autophagy in neuroblastoma cells found compounds that induced autophagy through distinct mechanism from rapamycin [162]. Recently, a siRNA screen searching for inducers of basal autophagy in neuroblastoma cells suggested that the Vps34 kinase complex but not mTORC1 regulates autophagy [163]. Taken together, these data suggest that there are parallel signaling pathways that regulate neuronal autophagy with distinct mechanisms.
Despite the observations noted above regarding autophagy signaling in neurons, it remains unknown whether or not similar results occur in the intact brain in vivo. It has been shown that neuronal autophagy can be induced by hypoxic/ischemia, overexcited glutamate receptors (caused by excitotoxic stimuli such as NMDA and kainic acid), methamphetamine, proteasome inhibition, and secondary to alterations in lysosomal enzyme activity or lipid storage [141]. New research adds other signaling pathways including IGF-1 [164], Cdk5/endophilin B1(Bif-1) [165], JNK/FoxO [166], and mutant LRRK2 [167] to the list of autophagy inducers in pathophysiological conditions. Carra et al. demonstrated that HSPB8 in complex with BAG3 and Hsp70 can stimulates autophagy and thereby enhance the clearance of protein aggregates which arise during neurodegenerative diseases [168]. HspB8·Bag3 activity was independent of the ER stress kinase PERK, demonstrating that its action is unrelated to ER stress and suggesting that it activates stress-mediated translational arrest and autophagy through a novel pathway.
Neuroprotective Versus Neurotoxic Role of Autophagy
Growing evidence supports the view that autophagy is essential for maintaining neuronal homeostasis: (i) deficiency in this pathway leads to neurodegeneration and human genetic diseases [169, 170]; and (ii) conditional knockout of key autophagic genes, Atg5 or Atg7, leads to accumulation of intracellular protein aggregates and neuronal death [139]. A protective role of autophagy was also observed in neonatal hypoxic/ischemia-induced brain injury [171]. Autophagy is required for preconditioning-induced cell protection [172]. This protective effect is not simply a function of autophagy liberating fuels for cells but appears to be related to a decrease in the amount of the mitochondria (because of mitophagy). This, in turn, results in less release of toxic molecules like cytochrome c from the mitochondria in response to proapoptotic insults. Koike and colleagues reported in vivo evidence of neuronal cell death requiring autophagy in the mammalian brain [173]. While most evidence points to autophagy as a protective process in neurons, other studies also provide genetic and cellular evidence that otherwise argues for a role of autophagy in promoting neuronal death. Increased autophagosome number has been observed during hippocampal neuron death and, in fact, Atg7 is critical for neuronal cell death after hypoxic/ischemia brain injury [174]. In another model of neuronal cell loss, FTD, inhibition of autophagy actually delays the neurodegeneration progression [175]. Also, loss of the critical autophagy protein Atg7 increases resistance to retrograde degeneration in axons after toxin injury [152]. Perhaps basal autophagy in the brain is essential for neuronal homeostasis, but in the case of acute neural injuries when autophagy is excessively induced, it can result in cell death, termed “programmed cell death type II” (PCD II) or “autophagic cell death” (ACD). This dichotomous role of autophagy is the result of a complex relationship between the autophagy and apoptosis pathways.
More recent studies have begun to dissect the autophagic process in neurons under various stress or pathological conditions. These studies suggest that, while accumulation of autophagosomes can arise from increased production in some cases [131], it can be caused by a mechanism that blocks fusion and degradation of autophagosomes through lysosomes in other scenarios [53]. Under circumstances in which lysosomes are destabilized or their function is compromised, it is more likely that autophagy inhibitors attenuate autophagic stress on compromised lysosomes by decreasing delivery of autophagic cargo rather than by attenuating an overaggressive auto-cannibalistic process. Indeed, healthy neurons in culture seem to tolerate robust autophagy induction [176] except when lysosomal function is also impaired. In situation where the prevention of autophagy is neuroprotective, counteraction of lysosome destabilization could be more suitable to the mechanism of cytoprotection than is the blockade of authentic ACD.
Impaired Autophagy in Neurodegenerative Diseases
Initial studies have revealed the accumulation of autophagic vacuoles in the brains of Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) patients [177, 178]. In addition, autophagy deregulation has also been linked to amyotrophic lateral sclerosis, frontotemporal dementia, and inclusion body myopathy associated with Paget’s disease of the bone and frontotemporal dementia [111, 179, 180]. More importantly, as the bulk degradative nature of autophagy would enable the removal of intracellular protein aggregates, extensive efforts have been devoted to understanding whether autophagy may be activated to eliminate these aggregated proteins, and whether the presence of aggregates may be attributed to malfunctioning of the autophagic pathway. Although increasing studies suggest that aggregation of the toxic species may actually be neuroprotective [177], autophagy may still be important for removing the oligomeric form of the toxic proteins. Nonetheless, despite the observation that elevated autophagy would facilitate protein aggregate degradation in HD, AD, PD, and amyotrophic lateral sclerosis (ALS) [181], recent advances reveal that each neurodegenerative disease may harbor different extent of autophagy deregulation, involving different steps in the autophagic pathway (Table 1) (Fig. 3).

Abnormal autophagic processes associated with specific neurodegenerative disease
Alzheimer’s Disease
AD is a neurodegenerative disorder characterized by progressive dementia and brain morphological changes such as atrophy, senile plaques with fibrillogenic beta amyloid (Aβ), and intraneuronal neurofibrillary tangles (NFT) with hyperphosphorylated tau. In AD brain, both APP proteins and cleaved amyloid-β peptides, along with a high level of PS1 protein, accumulate within the large pool of autophagic vacuoles in swollen dystrophic neurites, suggesting that the autophagy system is involved in AD pathogenesis [142]. Autophagy activation was found to be elevated after Aβ stimulation and in APP/PS1 mice, an AD mouse model [182, 183]. Importantly, compelling evidence indicates that Aβ is generated in autophagic vacuoles during autophagy, suggesting that autophagy activation in AD brains may exacerbate AD pathogenesis by increasing Aβ levels [142]. In contrast to these early observations, later articles have indicated that the APP processing is not directly dependent on autophagosome formation but it is driven by the APP sorting through the cell surface to the early endosomes and subsequently either to the TGN or to the late endosomes, also called multivesicular bodies [184–186].
Interestingly, elevated autophagy induction does not appear to be the only abnormality in the autophagic pathway in AD, the autophagic/lysosomal pathway is also implicated in the degradation of APP and Aβ. Emerging data indicates that the expression of Beclin 1 is reduced in AD brain, and thus provokes the characteristic pathological changes observed in AD brains [187, 188]. Moreover, the deficiency of Beclin 1 in cultured neurons and transgenic mice provokes the deposition of Aβ peptides whereas its overexpression reduces the accumulation of Aβ [189]. These data raise the possibility that upregulation of autophagy may be beneficial in AD by decreasing the levels of Aβ that characterize its pathology.
One possibility to reconcile this apparent discrepancy may be that autophagosome synthesis is partially defective very early on, possibly before overt pathology appears, and that the Beclin 1 deficiency may play an important role in the genesis of disease. Then, as disease progresses, there may be additional events that impair autophagosome clearance, leading to a situation where, despite reduced autophagosome synthesis, there is a build up of autophagosomes due to a “traffic jam” in their removal processes. This may compound the autophagy problem and further reduce autophagic flux. Furthermore, the abnormal pool of slowly turned over autophagic vesicles may additionally impact on AD pathology by serving as a site for Aβ generation [142]. Recent studies indicated that, although autophagic vacuoles are normally efficiently disposed of in healthy neurons, clearance of autophagic vacuoles is impaired in AD brains [53]. Furthermore, defective lysosomal proteolysis may underlie further pathogenic protein accumulations and neuronal cell death in AD. Knock down of PS1, a gene mutated in familial AD, leads to defects in autophagic vacuole clearance, lysosomal acidification, and decreased lysosomal proteolytic activity. Importantly, expression of PS1 mutants associated with early-onset AD also results in similar abnormality in the autophagic/lysosomal pathway [190].
In addition to regulating Aβ pathology, new data suggested possible role of autophagy on modulation of tau level and fragmentation. Both soluble and aggregated forms of tau are degraded by autophagy, and autophagy inhibition elevates tau aggregation and toxicity [191, 192]. In addition, rapamycin treatment decreases tau pathology [191, 193]. Many recent studies have revealed that the activation of autophagy can inhibit the formation of tau pathology in transgenic mouse models and subsequently ameliorate cognitive deficits [194–196].
Hence, in AD, the maturation of autophagolysosomes and their retrograde transport are impaired, which results in a massive accumulation of AVs within degenerating neurites. This combination of increased autophagy induction and defective clearance of Aβ-generating AVs results in Aβ accumulation [184]. Autophagy is now recognized as an arbiter of neuronal survival and death decisions in Alzheimer’s disease, and in full-blown Alzheimer’ disease, investigation of the regulation of autophagosome maturation and movement especially to neuronal cell body/soma is a promising area [142, 197]. Thus, autophagy regulation and stimulation might serve as a future therapy target for preventing and/or stopping Alzheimer’s disease neuropathogenesis.
Huntington’s Disease
HD is an autosomal dominant neurodegenerative disorder caused by a CAG trinucleotide repeat expansion in the huntingtin gene. This expansion produces a mutant form of the huntingtin (Htt) protein, which confers one or more toxic functions to mutant Htt leading to neurodegeneration [198]. Individuals harboring alleles with between 36 and 40 CAGs may or may not develop HD symptoms; those that do tend to become symptomatic late in life. However, those with alleles containing expansions greater than 40 CAGs repeats will eventually develop symptoms of HD if they live long enough [199]. The characteristic features of altered autophagy were first observed in postmortem brains of HD patients [200]. Activation of autophagy by treatment with mTOR inhibitor rapamycin attenuates htt toxicity in the fly [27]. This is further supported by the observation that knock down of TOR expression also attenuates neuronal loss [201]. Indeed, autophagy has been implicated in the degradation of both soluble and aggregated forms of cytosolic mutant htt [29, 202]. Despite the observed neuroprotective effect of autophagy activation in HD models, the precise mechanism that underlies autophagic dysfunction in HD is poorly understood. The inefficient engulfment of cytosolic components by autophagosomes has been proposed to be responsible for their slower turnover and the accumulation of Htt in HD cells. Recently, a defect in cargo recognition mediated by p62 has been described in different cell models of HD. The mutant Htt can interact with p62, impairing its ability to recognize cargo aggregates and organelles [203]. For this reason, though the synthesis of autophagosomes and their fusion to lysosomes is normal, their cargo content is decreased. This leads to an accumulation of aggregates and organelles like lipid droplets and mitochondria, which may contribute to neurodegeneration.
Another mechanism underlying the intracellular accumulation of mutant Htt is that the accumulated mutant Htt recruits Beclin 1 and impairs the Beclin 1-mediated long-lived protein turnover. Thus, the sequestration of beclin 1 in the vulnerable neuronal population of HD patients might further reduce Beclin 1 function and autophagic degradation of mutant Htt [29].
Interestingly, expression of mutant htt itself also stimulates autophagy [203, 204]. In addition, a recent study demonstrated that overexpression of a full-length htt that lacks the polyglutamine stretch stimulates autophagy activation, in turn facilitating clearance of mutant htt [205]. Together with the observation that htt is structurally similar to autophagy regulator mTOR and partially localizes with autophagosomes [206], it is tempting to speculate that wild-type htt may play a role in autophagy regulation and that the autophagy abnormality in HD may also involve a loss of function scenario.
The initial increase in AVs and autophagy observed in HD models may represent an attempt to remove mutant Htt protein and over time the autophagy machinery becomes dysfunctional, leading to neurodegeneration. Thus, the therapeutic induction and recovery of autophagy might may enhance the clearance of mutant Htt protein and reduce its toxic effect in HD neurons. Upregulating autophagy can ameliorate symptoms and pathology in many HD models. Inducing mammalian target of rapamycin (mTOR)-dependent autophagy reduced neurodegeneration in a fly HD model and improved behavior and motor performance in mouse HD models [27, 202, 207]. Inducing autophagy independently of mTOR also reduced mHtt aggregation and toxicity in various models [204, 208–210]. Rilmenidine, an imidazoline receptor 1 agonist, induces autophagy, enhances mutant huntingtin clearance, and reduces toxicity in an HD mouse model [210]. Since this drug is a centrally acting hypertensive agent with minimal side effects, it is currently being tested in a safety trial in HD patients. While autophagy is a promising therapeutic target, the degree of autophagy induction must be optimized if overactive autophagy is detrimental, as seen in some circumstances [211].
Parkinson’s Disease
PD is one of the most common neurodegenerative disorders and one of the first to be linked to autophagy. PD is caused by the selective cell death of dopamine neurons in the substantia nigra (SN) and is characterized by the presence of Lewy bodies, which are the intracytoplasmic inclusions containing α-synuclein and ubiquitin. The vast majority of PD cases are sporadic with unknown etiology. However, approximately 5 % of cases are inherited. Early ultrastructural examinations in PD patients revealed characteristics of apoptosis and autophagy in the degenerating melanized neurons of the SN [212]. Subsequently, the pathogenic role of autophagy in PD was demonstrated by the finding that α-synuclein, a major constituent of Lewy body, is degraded by macroautophagy and CMA [213]. Misfolded α-synuclein oligomers can be degraded by different catabolic pathways including CMA and macroautophagy, and they accumulate in different pathological situations underlying PD pathology [213, 214].
Emerging evidence has suggested that aberrant autophagy is one of the underlying mechanisms for hereditary forms of PD. Mutations of at least six genes have been linked with hereditary PD: α-synuclein (SNCA or PARK1), Parkin (PARK2), ubiquitin carboxyhydroxylase L1 (UCH-L1 or PARK5), PTEN-induced putative kinase 1 (PINK1 or PARK6), DJ-1 (PARK7), and leucine-rich repeat kinase 2 (LRRK2 or PARK8) [208]. Vast majority of these studies showed that these genetic defects lead to autophagy impairment [215]. For instance, abnormal expression of α-synuclein can interfere with different types of autophagy. In fact, upregulation of wild-type (wt) α-synuclein leads to significant inhibition of autophagy, and mutant forms of α-synuclein A30P and A53T have been shown to inhibit CMA [216–218]. Parkin and PINK1 are specifically involved in the target (damaged mitochondria) recognition phase of mitophagy [219, 220]. Hence, neurodegeneration in PINK1 and Parkin-positive familial forms of PD may result from a defect in mitophagy, leading to the accumulation of damaged mitochondria and excessive ROS production. In addition, PINK1 significantly enhances basal and starvation-induced autophagy, which is reduced by knocking down Beclin 1 expression or by inhibiting the Beclin partner Vps34 [221], suggesting that beclin 1 plays an important role in the intracellular degradation of α-synuclein either directly or indirectly through the autophagic pathway. A Beclin-independent autophagic clearance of defective mitochondria and ubiquitinated β-amyloid is also mediated by Parkin in Alzheimer’s disease models [222], implying that the protective role of Parkin and PINK1 is not restricted to PD.
Although neuronal autophagy appears primarily to be a protective process in the nervous system, it can also play a paradoxical role in neuronal death. Activation of autophagy by various stressors, including starvation, MPP + treatment, or overexpression of A53T-alpha-synuclein, results in neuronal loss that are attributable to autophagy [223–225]. Treatment with autophagy inhibitor 3-methylamphetamine (3-MA) or knock down of Atg5 or Atg12 expression attenuates the neuronal loss [165]. Oxidative stress, a major pathogenic mechanism of PD, has been shown to increase autophagic cell death in dopaminergic neurons by suppressing the expression of Tnfaip8/oxidative stress-regulated gene-α (Oxi-α), which encodes a novel mTOR activator [226]. These studies suggest that pathogenic autophagy associated with neuronal death does occur and may be distinct from basal neuronal autophagy. In fact, the autophagic cell death, proposed to occur in SN in response to excessive ROS and environmental toxins in animal models and also in PD patients, results from the interference in the last step of autophagy: lysosomal function, rather than excessively enhancing autophagosome formation. A recent review suggested lysosomal dysfunction as a unifying feature of PD [227]. Hence, different forms of autophagy play a critical role in homeostasis and mitochondrial clearance of dopaminergic neurons in SN while genetic defects in autophagy lead to the disease. Environmental parkinsonian toxins also perturb autophagy, leading to autophagic stress in dopaminergic neurons, but there is no agreement on the role of autophagy in the cellular response to these toxins [228]. Future studies should clarify the mechanistic aspects of autophagy dysregulation by parkinsonian neurotoxins and their interplay with genetic defects implicated in PD. Hopefully, understanding of these mechanisms will lead to the identification of targets for pharmacological intervention and specific drugs that correct dopaminergic dysfunction and prevent PD progression.
Amyotrophic Lateral Sclerosis
ALS is a heterogeneous group of inexorable neurodegenerative disorders characterized by a selective loss of upper and lower motor neurons in the brain and spinal cord [229, 230]. Enormous efforts have continuously been made towards defining the molecular pathogenesis of these devastating diseases. The autophagic/lysosomal system is among the underlying mechanisms, whose dysfunction is tightly associated with a variety of neurodegenerative diseases.
Indeed, the accumulation of autophagosomes was observed in the spinal cord of sporadic ALS patients [231], indicating autophagic dysfunction in ALS. Progressive enhancement of autophagy and/or decrease of autophagic flux are detected in a mutant SOD1 (SOD1G93A)-expressing ALS mouse model [232]. Another study also demonstrated that autophagosome formation was increased at the symptomatic stage and the percentage of motor neurons that contained phosphorylated mTOR/Ser2448 was decreased in the SOD1G93A transgenic mice [233], which suggests the possibility that increased and/or impaired autophagy underlies pathological phenomena in this model. Two studies suggest that autophagic clearance of mutant superoxide dismutase 1 (SOD1) is beneficial for motor neuron loss in ALS. The heat-shock protein HspB8 increases mutant SOD1 clearance by promoting autophagy in an ALS mouse mode. Furthermore, the unfolded protein response transcription factor X-box binding protein 1 (XBP-1) deficiency in mice has been shown to protect against ALS by autophagic induction and the subsequent degradation of mutant SOD1 [234]. Among putative therapeutic agents for their potential role in neuroprotection, agents promoting autophagy, such as lithium and rapamycin, have been suggested to be effective for the survival of motor neurons in ALS by autophagic clearance of SOD1 [235].
Additional familial ALS genes have been found recently. A mutation in an ESCRT protein causes ALS. Cells depleted of ESCRT have inhibited autophagic degradation, due to impaired autophagosome-lysosome fusion, causing accumulation of ubiquitinated aggregates [236]. Another protein mutated in ALS is VCP , which also appears to regulate autophagosome removal [237]. It is known that VCP is essential for maturation of ubiquitin-containing autophagosomes and mutant VCP toxicity is partially mediated through its effect on TDP-43 protein, a major constituent of ubiquitin inclusions that neuropathologically characterize ALS [238]. It is likely that imbalance between initiation (formation of autophagosomes) and/or maturation stages of autophagic processes results in aberrant accumulation of misfolded and/or aggregated proteins within cells. Such pathological conditions will contribute to the progression of this disease by enhancing the accumulation of aggregate-prone proteins, dysfunctional mitochondria, and increasing susceptibility to cell death.
Autophagy in Brain Ischemia
The effect of autophagy activation in ischemic neuronal death is still controversial. Koike et al. suggest a detrimental role, reporting that Atg7-deficient mice have significant lower cell death after hypoxic/ischemia [174]. Some recent studies have suggested a possible role of autophagy and lysosome in mediating neuronal cell death in animal models of cerebral ischemia. Uchiyama et al. reported that in a mouse hypoxic/ischemia model, many damaged neurons showed features of autophagic cell death [239]. More recently, they further found that hypoxic/ischemic brain injury induced autophagy in both neonatal and adult hippocampal pyramidal neurons, and the death of pyramidal neurons was prevented by Atg7 deficiency [239]. Moreover, Wen et al. showed that autophagy inhibitor 3-MA significantly reduced infarct volume after permanent occlusion of the rat’s middle cerebral artery (pMCAO) [240]. However, it was repeatedly demonstrated that autophagy plays an important role in protecting neurons from ischemia-induced death. 3-MA and wortmannin, two autophagy inhibitors, significantly reduced Beclin 1 expression and switched the mechanism of the cell death mode from apoptosis to necrosis [171, 241], thus attenuating the ischemic insults. Accumulation of p62 under hypoxic stress promotes neuronal cell death, which was partly blocked by autophagy inducer lithium chloride [242], supporting that autophagy promotes neuronal cell survival under hypoxic stress.
Wang et al. demonstrated that induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase (Nampt) in the early stage of cerebral ischemia [243]. A protective role of autophagy was also observed in neonatal hypoxic/ischemia-induced brain injury [171]. Autophagy is required for preconditioning-induced cell protection [172]. Currently, it seems that elimination of damaged mitochondria (mitophagy) and subsequent apoptosis interruption could represent the most important mechanism responsible for neuroprotective effects of autophagy in cerebral ischemia. The present study showed that the number of damaged mitochondria was not reduced, rather increased after brain ischemia. Accumulation of damaged mitochondria is likely a result of imperfect autophagy [244]. Liu et al. suggest that the autophagy pathway is upregulated moderately by ischemia but fails to operate at the full capacity, probably because of its impairment [245].
Cerebral ischemia results in damages to proteins, lipids, and all intracellular components. As a repair mechanism, autophagy is activated to eliminate damaged proteins that accumulate within the neuronal cells. At this stage, autophagy is prosurvival. If the ischemic stress persists for a long time, the autophagy intensity is strengthened consistently. In this case, not only the autophagy is further increased (“supply”) but also the cellular burden of damaged and/or dysfunctional macromolecules and organelles (“demand”) is increased. Hence, it may be inadequate autophagy, rather than excessive autophagy, that contributes to neuronal death after brain ischemia. Carloni et al. demonstrates that in neonatal hypoxic/ischemia-induced brain injury, autophagy is induced in neurons, but there is no change in the expression of Beclin 1 in GFAP-positive cells [171]. Qin et al. observed the activation of autophagy in astrocytes in both in vivo and in vitro cerebral hypoxic/ischemia models. Although the molecular mechanisms underlying the autophagic/lysosoal role in mediating cerebral ischemic astrocyte injury remain to be determined, the protective effects afforded by blocking autophagy suggest that an autophagic mechanism may at least partly contribute to astrocyte injury induced by cerebral ischemia. Therefore, therapeutic strategies to inhibit autophagy-induced astroglial cell death might be helpful in seeking effective new target treatments for cerebral ischemia [246]. Cui et al. proved that inhibiting autophagy activation and maturation can reduce I/R injury and improve cell survival in vivo and in vitro [247]. More recently, in the study of Gao et al., they suggested that the neuroprotective effects elicited by ischemic postconditioning (IPOC) are partially reversed by the autophagy inducer, rapamycin [248]. The autophagic inhibitor 3-MA can be used to reduce damage of fatal cerebral ischemia, which may provide novel strategies for clinical treatment of ischemic stroke [248]. However, several studies have reached the opposite conclusion that autophagy activation is associated with protective effects conferred by ischemic preconditioning (IPC) and that inhibition of autophagy may aggregate cerebral ischemic damage [171, 249]. Sheng et al. have demonstrated that autophagy activation was involved in the neuroprotective mechanism of IPC and that rapamycin pretreatment produced beneficial preconditioning-like effects, while pretreatment with 3-MA suppressed the neuroprotective effects of IPC [250]. Causes for the inconsistency are unclear. The different models of ischemia, the durations of the ischemic insults, and the different ages of animals are offered as possible explanations. It is reported that autophagy is more pronounced in adult mice than in neonatal mice subjected to hypoxic/ischemia injury and autophagic cell death may be more significant in mature animals [251]. Furthermore, although IPC and IPOC are both beneficial to neuronal survival, IPOC is conducted after reperfusion thus alters events after ischemia but IPC is administered prior to ischemia. The possible mechanisms and the conducted timing are different between the two maneuvers [252]. In addition, the timing of rapamycin and 3-MA administration might be crucial to the discrepancies.
Autophagy in Myelin Disease
Using explant cultures of dorsal root ganglion neurons, Rangaraju et al. have found that autophagy promotes membrane expansion in Schwann cells [253]. Lentivirus-mediated shRNA shutdown of Atg12 abrogates the improvements in myelin production, further demonstrating that autophagy is critical for the observed benefits. Enhancement of protein chaperones and upregulation of autophagy induced by dietary restriction by intermittent fasting (IF) may decrease PMP22 aggregates and attenuate neuropathic behavioral and morphological phenotypes in a spontaneous mouse model of CMT1A [254].
Upregulating autophagy in les oligodendrocytes in vitro increased the number of cells forming membrane extensions. Furthermore, by upregulating autophagy in vivo by intermittent fasting, both les and control animals had an increase in the number of myelinated axons as well as an increase in myelin sheath thickness [255]. Interestingly, disruptions in oligodendrocyte protein homeostasis have been described in several CNS myelin disorders, including vanishing white matter disease [256], multiple sclerosis [257], and Pelizeaus-Merzbacher disease [258]. Therefore, further investigation of this pathway may have broader implications for therapies to treat myelin disease.
Conclusion
The past several years have witnessed an extraordinary advance in our understanding of the molecular signaling involved in mammalian autophagy. In spite of those outcomes, there remain specific challenges to our understanding of autophagy in mammalian cells. From our knowledge, autophagy is a major contributor to maintain cellular homeostasis and metabolism. Mounting evidence has firmly established the importance of neuronal autophagy in both the normal functioning and pathophysiological conditions of the brain. Moreover, under neuronal disease conditions, a balanced level of autophagic flux can be induced and activated for neuronal cell survival and for the removal of toxic components in several neuronal diseases. In contrast, an insufficient level of autophagy can cause an abnormal level of flux and lead to neuropathy. We anticipate that we will make significant progress in this field and provide valuable knowledge to the design of drugs targeted at autophagy in the treatment of neuronal diseases.
References
Rubinsztein DC, Codogno P, Levine B (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11:709–730. doi:10.1038/nrd3802, Review
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147(4):728–741. doi:10.1016/j.cell.2011.10.026
Wirawan E, Vanden Berghe T, Lippens S et al (2012) Autophagy: for better or for worse. Cell Res 22:43–61. doi:10.1038/cr.2011.152
Mijaljica D, Prescott M, Devenish RJ (2011) V-ATPase engagement in autophagic processes. Autophagy 7(6):666–668
Sahu R, Kaushik S, Clement CC et al (2011) Microautophagy of cytosolic proteins by late endosomes. Dev Cell 20(1):131–139. doi:10.1016/j.devcel.2010.12.003
Uttenweiler A, Mayer A (2008) Microautophagy in the yeast Saccharomyces cerevisiae. Methods Mol Biol 445:245–259. doi:10.1007/978-1-59745-157-4-16
van der Vaart A, Mari M, Reggiori F (2008) A picky eater: exploring the mechanisms of selective autophagy in human pathologies. Traffic 9(3):281–289
Kraft C, Reggiori F, Peter M (2009) Selective types of autophagy in yeast. Biochim Biophys Acta 1793(9):1404–12. doi:10.1038/cdd.2012.73, Review
Beau I, Esclatine A, Codogno P (2008) Lost to translation: when autophagy targets mature ribosomes. Trends Cell Biol 18(7):311–314. doi:10.1016/j.tcb.2008.05.001, Review
Li W, Yang Q, Mao Z (2011) Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci 68(5):749–763. doi:10.1007/s00018-010-0565-6
Dice JF (1990) Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 15(8):305–309
Mizushima N (2005) The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ 12(Suppl 2):1535–41
Klionsky DJ, Cuervo AM, Dunn WA Jr et al (2007) How shall I eat thee? Autophagy 3(5):413–416
Kirkin V, McEwan DG, Novak I et al (2009) A role for ubiquitin in selective autophagy. Mol Cell 34(3):259–69. doi:10.1016/j.molcel.2009.04.026, Review
Massey AC, Kaushik S, Sovak G et al (2006) Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A 103(15):5805–5810
Kaushik S, Massey AC, Mizushima N et al (2008) Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell 19(5):2179–2192. doi:10.1091/mbc.E07-11-1155
Ciechanover A, Heller H, Elias S et al (1980) ATP-dependent conjugation of reticulocyte proteins with the polypeptide required for protein degradation. Proc Natl Acad Sci U S A 77(3):1365–1368
Nandi D, Tahiliani P, Kumar A (2006) The ubiquitin-proteasome system. J Biosci 31(1):137–155, Review
Fuertes G, Villarroya A, Knecht E (2003) Role of proteasomes in the degradation of short-lived proteins in human fibroblasts under various growth conditions. Int J Biochem Cell Biol 35(5):651–64
Fuertes G, Martín De Llano JJ, Villarroya A et al (2003) Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem J 375(Pt 1):75–86
Perlmutter DH (2002) The cellular response to aggregated proteins associated with human disease. J Clin Invest 110:1219–1220
Keller JN, Dimayuga E, Chen Q et al (2004) Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int J Biochem Cell Biol 36:2376–2391
Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292:1552–1555
Teckman JH, Burrows J, Hidvegi T et al (2001) The proteasome participates in degradation of mutant α1-antitrypsin Z in the endoplasmic reticulum of hepatoma-derived hepatocytes. J Biol Chem 276:44865–44872
Ravikumar B, Rubinsztein DC (2004) Can autophagy protect against neurodegeneration caused by aggregate-prone proteins? Neuroreport 15:2443–2445
Teckman JH, Perlmutter DH (2000) Retention of mutant α(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physio 279:G961–G974
Ravikumar B, Vacher C, Berger Z et al (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36:585–595
Kamimoto T, Shoji S, Hidvegi T et al (2006) Intracellular inclusions containing mutant α1-antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem 281:4467–4476
Shibata M, Lu T, Furuya T et al (2006) Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem 281:14474–14485
Kruse KB, Brodsky JL, McCracken AA (2006) Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human α-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell 17:203–212
Kruse KB, Dear A, Kaltenbrun ER et al (2006) Mutant fibrinogen cleared from the endoplasmic reticulum via endoplasmic reticulum-associated protein degradation and autophagy: an explanation for liver disease. Am J Pathol 168:1299–1308
Pandey UB, Batlevi Y, Baehrecke EH et al (2007) HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy 3(6):643–645
Bjørkøy G, Lamark T, Brech A et al (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171(4):603–614
Simonsen A, Birkeland HC, Gillooly DJ et al (2004) Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J Cell Sci 117(Pt 18):4239–4251
Korolchuk VI, Mansilla A et al (2009) Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 33(4):517–527. doi:10.1016/j.molcel.2009.01.021
Korolchuk VI, Menzies FM, Rubinsztein DC (2010) Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584(7):1393–1398. doi:10.1016/j.febslet.2009.12.047
Ding WX, Ni HM, Gao W et al (2007) Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 171(2):513–524
Rubinsztein DC, Shpilka T, Elazar Z (2012) Mechanisms of autophagosome biogenesis. Curr Biol 22:R229–R234. doi:10.1016/j.cub.2011.11.034, Review
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93. doi:10.1146/annurev-genet-102808-114910
Klionsky DJ, Cregg JM, Dunn WA Jr et al (2003) A unified nomenclature for yeast autophagy-related genes. Dev Cell 5(4):539–545
Thumm M, Egner R, Koch B et al (1994) Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett 349(2):275–280
Pyo JO, Nah J, Jung YK (2012) Molecules and their functions in autophagy. Exp Mol Med 44(2):73–80. doi:10.3858/emm.2012.44.2.029, Review
Axe EL, Walker SA, Manifava M et al (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182:685–701. doi:10.1083/jcb.200803137
Cheong H, Nair U, Geng J (2008) The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Mol Biol Cell 19:668–681
Kabeya Y, Kamada Y, Baba M et al (2005) Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell 16:2544–2553
Kamada Y, Funakoshi T, Shintani T et al (2000) Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 150:1507–1513
Cheong H, Klionsky DJ (2008) Dual role of Atg1 in regulation of autophagy-specific PAS assembly in Saccharomyces cerevisiae. Autophagy 4:724–726
Kawamata T, Kamada Y, Kabeya Y (2008) Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol Biol Cell 19:2039–2050. doi:10.1091/mbc.E07-10-1048
Suzuki K, Kubota Y, Sekito T, Ohsumi Y (2007) Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 12:209–218
Ganley IG, du Lam H, Wang J et al (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284:12297–12305. doi:10.1074/jbc.M900573200
Hosokawa N, Hara T, Kaizuka T et al (2009) Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 20:1981–1991. doi:10.1091/mbc.E08-12-1248
Jung CH, Jun CB, Ro SH et al (2009) ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20:1992–2003. doi:10.1091/mbc.E08-12-1249
Nakahara Y, Suzuki-Migishima R, Yokoyama M et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889
Mercer CA, Kaliappan A, Dennis PB (2009) A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 5(7):973–979
Hosokawa N, Sasaki T, Iemura S et al (2009) Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5(7):973–979
Itakura E, Kishi C, Inoue K et al (2008) Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19:5360–5372
Sun Q, Fan W, Chen K et al (2008) Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A 105:19211–19216. doi:10.1073/pnas.0810452105
Decuypere JP, Parys JB, Bultynck G (2012) Regulation of the autophagic bcl-2/beclin 1 interaction. Cells 1(3):284–312. doi:10.3390/cells1030284
Burda P, Padilla SM, Sarkar S (2002) Retromer function in endosome-to-Golgi retrograde transport is regulated by the yeast Vps34 PtdIns 3-kinase. J Cell Sci 115:3889–390060
He C, Levine B (2010) The Beclin 1 interactome. Curr Opin Cell Biol 22:140–149. doi:10.1016/j.ceb.2010.01.001
Gordy C, He YW (2012) The crosstalk between autophagy and apoptosis: where does this lead? Protein Cell 3:17–27. doi:10.1007/s13238-011-1127-x
Konishi A, Arakawa S, Yue Z et al (2012) Involvement of Beclin 1 in engulfment of apoptotic cells. J Biol Chem 287:13919–13929. doi:10.1074/jbc.M112.348375
Funderburk SF, Wang QJ, Yue Z (2010) The Beclin 1-VPS34 complex—at the crossroads of autophagy and beyond. Trends Cell Biol 20:355–362. doi:10.1016/j.tcb.2010.03.002
Liang C, Lee JS, Inn KS et al (2008) Beclin 1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 10:776–787. doi:10.1038/ncb1740
Matsunaga K, Saitoh T, Tabata K et al (2009) Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11:385–396. doi:10.1038/ncb1846
Fimia GM, Stoykova A, Romagnoli A et al (2007) Ambra1 regulates autophagy and development of the nervous system. Nature 447:1121–1125
Takahashi Y, Coppola D, Matsushita N et al (2007) Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 9:1142–1151
Yue Z, Horton A, Bravin M et al (2002) A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron 35:921–33
Vicencio JM, Ortiz C, Criollo A et al (2009) The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 16:1006–1017. doi:10.1038/cdd.2009.34
Ropolo A, Grasso D, Pardo R et al (2007) The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J Biol Chem 282:37124–37133
Kang R, Livesey KM, Zeh HJ et al (2010) HMGB1: a novel Beclin 1-binding protein active in autophagy. Autophagy 6:1209–1211
Wei Y, Pattingre S, Sinha S et al (2008) JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 30(6):678–88. doi:10.1016/j.molcel.2008.06.001
Pattingre S, Tassa A, Qu X et al (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939
Zalckvar E, Berissi H, Mizrachy L (2009) DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 10(3):285–292. doi:10.1038/embor.2008.246
Noda T, Kim J, Huang WP et al (2000) Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J Cell Biol 148:465–480
Yamamoto H, Kakuta S, Watanabe TM et al (2012) Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol 198(2):219–33. doi:10.1083/jcb.201202061
Mari MI, Griffith J, Rieter E (2010) An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol 190:1005–1022. doi:10.1083/jcb.200912089
Orsi AI, Razi M, Dooley HC, Robinson D (2012) Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 23:1860–1873. doi:10.1091/mbc.E11-09-0746
Kageyama S, Omori H, Saitoh T et al (2011) The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol Biol Cell 22(13):2290–2300. doi:10.1091/mbc.E10-11-0893
Itakura E, Mizushima N (2010) Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6:764–776. doi:10.4161/auto.6.6.12709
Young AR, Chan EY, Hu XW et al (2006) Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 119(Pt 18):3888–900
Longatti AI, Lamb CA, Razi M (2012) TBC1D14 regulates autophagosome formation via Rab11-and ULK1-positive recycling endosomes. J Cell Biol 197(5):659–675. doi:10.1083/jcb.201111079
Reggiori F, Shintani T, Nair U et al (2005) Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 1:101–109
Hailey DW, Rambold AS, Satpute-Krishnan P et al (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141(4):656–667. doi:10.1016/j.cell.2010.04.009
Moreau K, Ravikumar B, Renna M et al (2011) Autophagosome precursor maturation requires homotypic fusion. Cell 146(2):303–317. doi:10.1016/j.cell.2011.06.023
Esclatine A, Chaumorcel M, Codogno P (2009) Macroautophagy signaling and regulation. Curr Top Microbiol Immunol 335:33–70. doi:10.1007/978-3-642-00302-82, Review
Ravikumar B, Imarisio S, Sarkar S et al (2008) Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J Cell Sci 121:1649–1660. doi:10.1242/jcs.025726
Ohsumi Y, Mizushima N (2004) Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol 15:231–236
Hanada T, Noda NN, Satomi Y et al (2007) The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 282:37298–37302
Fujita N, Itoh T, Omori H et al (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19:2092–2100. doi:10.1091/mbc. E07-12-1257
Lee KM, Hwang SK, Lee JA (2013) Neuronal autophagy and neurodevelopmental disorders. Neurobiol 22(3):133–142. doi:10.5607/en.2013.22.3.133
Burman C, Ktistakis NT (2010) Autophagosome formation in mammalian cells. Semin Immunopathol 32:397–413. doi:10.1007/s00281-010-0222-z
Nemoto T, Tanida I, Tanida-Miyake E et al (2003) The mouse APG10 homologue, an E2-like enzyme for Apg12p conjugation, facilitates MAP-LC3 modification. J Biol Chem 278:39517–39526
Tanida I, Tanida-Miyake E, Komatsu M et al (2002) Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, facilitates the conjugation of hApg12p to hApg5p. J Biol Chem 277:13739–13744
Nishida Y, Arakawa S, Fujitani K et al (2009) Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461:654–658. doi:10.1038/nature08455
Zhang J, Randall MS, Loyd MR et al (2009) Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood 114(1):157–64. doi:10.1182/blood-2008-04-151639
Kundu M, Lindsten T, Yang CY et al (2008) Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112(4):1493–1502
Codogno P, Meijer AJ (2005) Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ Suppl 2:1509–1518
Eskelinen EL (2005) Maturation of autophagic vacuoles in mammalian cells. Autophagy 1:1–10
Rubinsztein DC, Cuervo AM, Ravikumar B et al (2009) In search of an “autophagomometer”. Autophagy 5:585–589
Jaeger PA, Wyss-Coray T (2010) Beclin 1 complex in autophagy and Alzheimer disease. Arch Neurol 67(10):1181–1184. doi:10.1001/archneurol.2010.258
Lee JA, Gao FB (2012) Neuronal functions of ESCRTs. Exp Neurobiol 21:9–15. doi:10.5607/en.2012.21.1.9
Lee JA, Beigneux A, Ahmad ST (2007) ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol 17:1561–1567
Guertin DA, Sabatini DM (2009) The pharmacology of mTOR inhibition. Sci Signal 2(67):pe24. doi:10.1126/scisignal.267pe24
Høyer-Hansen MI, Jäättelä M (2007) AMP-activated protein kinase: a universal regulator of autophagy. Autophagy 3:381–338
Zheng MI, Wang YH, Wu XN (2011) Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat Cell Biol 13:263–272. doi:10.1038/ncb2168
Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124:471–484, Review
Sancak YI, Bar-Peled L, Zoncu R (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303. doi:10.1016/j.cell.2010.02.024
Gwinn DMI, Shackelford DB, Egan DF (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226. doi:10.1016/j.molcel.2008.03.003
Levine B, Abrams J (2008) p53: the Janus of autophagy? Nat Cell Biol 10:637–639. doi:10.1038/ncb0608-637
Tasdemir E, Chiara Maiuri M, Morselli E et al (2008) A dual role of p53 in the control of autophagy. Autophagy 4:810–814
Feng Z, Zhang H, Levine AJ et al (2005) The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A 102:8204–8209
Crighton D, Wilkinson S, O’Prey J et al (2006) DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126:121–124
Morselli EI, Maiuri MC, Markaki M (2010) The life span-prolonging effect of sirtuin-1 is mediated by autophagy. Autophagy 6:186–188
Tasdemir E, Maiuri MC, Galluzzi L et al (2008) Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10:676–687. doi:10.1038/ncb1730
Armour SM, Baur JA, Hsieh SN et al (2009) Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging (Albany NY) 1:515–528
Yorimitsu T, Zaman S, Broach JR et al (2007) Protein kinase A and Sch9 cooperatively regulate induction of autophagy in Saccharomyces cerevisiae. Mol Biol Cell 18:4180–4189
Abeliovich H, Dunn WA Jr, Kim J et al (2000) Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol 151:1025–1034
Lawrence BP, Brown WJ (1993) Inhibition of protein synthesis separates autophagic sequestration from the delivery of lysosomal enzymes. J Cell Sci 105(Pt 2):473–480
Chang YY, Juhasz G, Goraksha-Hicks P et al (2009) Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem Soc Trans 37:232–236. doi:10.1042/BST0370232
Koren I, Reem E, Kimchi A (2010) DAP1, a novel substrate of mTOR, negatively regulates autophagy. Curr Biol 20:1093–1098. doi:10.1016/j.cub.2010.04.041
Thoreen CC, Kang SA, Chang JW et al (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 284:8023–8032. doi:10.1074/jbc.M900301200
Mammucari C, Milan G, Romanello V et al (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6:458–471
Brunet A, Bonni A, Zigmond MJ et al (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868
Yu L, McPhee CK, Zheng L et al (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465:942–946. doi:10.1038/nature09076
Yu L, Alva A, Su H et al (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304:1500–1502
Yu L, Lenardo MJ, Baehrecke EH (2004) Autophagy and caspases: a new cell death program. Cell Cycle 3:1124–1126, Review
Yu L, Strandberg L, Lenardo MJ (2008) The selectivity of autophagy and its role in cell death and survival. Autophagy 4(5):567–573
Berridge MJ (1993) Inositol trisphosphate and calcium signalling. Nature 361(6410):315–325, Review
Cherra SJ III, Kulich SM, Uechi G et al (2010) Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol 190:533–539. doi:10.1083/jcb.201002108
Mavrakis M, Lippincott-Schwartz J, Stratakis CA et al (2006) Depletion of type IA regulatory subunit (RI) of protein kinase A (PKA) in mammalian cells and tissues activates mTOR and causes autophagic deficiency. Hum Mol Genet 15:2962–2971
Sarkar S, Davies JE, Huang Z et al (2007) a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 282:5641–5652
Scherz-Shouval R, Shvets E, Fass E et al (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26:1749–1760
Sarkar S, Perlstein EO, Imarisio S et al (2007) Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol 3:331–338
Tooze SA, Schiavo G (2008) Liaisons dangereuses: autophagy, neuronal survival and neurodegeneration. Curr Opin Neurobiol 18:504–515. doi:10.1016/j.conb.2008.09.015
Lee JA (2009) Autophagy in neurodegeneration: two sides of the same coin. BMB Rep 42(6):324–330
Komatsu M, Waguri S, Chiba T et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441(7095):880–884
Boland B, Kumar A, Lee S et al (2008) Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci 28(27):6926–6937. doi:10.1523/JNEUROSCI.0800-08.2008
Komatsu M, Wang QJ, Holstein GR et al (2007) Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A 104:14489–14494
Yue Z, Friedman L, Komatsu M et al (2009) The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim Biophys Acta 1793:1496–1507. doi:10.1016/j.bbamcr.2009.01.016
Yu WH, Cuervo AM, Kumar A et al (2005) Macroautophagy-a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol 171(1):87–98
Nixon RA, Wegiel J, Kumar A et al (2005) Extensive involvement of autophagy in Alzheimer disease: an immunoelectronmicroscopy study. J Neuropathol Exp Neurol 64:113–122
Mizushima N (2004) Methods for monitoring autophagy. Int J Biochem Cell Biol 36(12):2491–2502 Review.
Dixon JS (1967) ‘Phagocytic’ lysosomes in chromatolytic neurones. Nature 215(5101):657–658
Matthews MR, Raisman G (1972) A light and electron microscopic study of the cellular response to axonal injury in the superior cervical ganglion of the rat. Proc R Soc Lond B Biol Sci 181(62):43–79
Broadwell RD, Cataldo AM (1984) The neuronal endoplasmic reticulum: its cytochemistry and contribution to the endomembrane system. II. Axons and terminals. J Comp Neurol 230(2):231–248
Hollenbeck PJ (1993) Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol 121(2):305–315
Maday S, Wallace KE, Holzbaur EL (2012) Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 196(4):407–417
Katsumata K, Nishiyama J, Inoue T et al (2010) Dynein- and activity-dependent retrograde transport of autophagosomes in neuronal axons. Autophagy 6(3):378–385. doi:10.1083/jcb.201106120
Rubinsztein DC, DiFiglia M, Heintz N, et al. (2005) Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 1(1):11–22 Review
Cheng HC, Kim SR, Oo TF et al (2011) Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J Neurosci 31(6):2125–2135. doi:10.1523/JNEUROSCI.5519-10.2011
Rodriguez-Muela N, Germain F, Marino G et al (2012) Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ 19(1):162–169. doi:10.1038/cdd.2011.88
Wang QJ, Ding Y, Kohtz DS et al (2006) Induction of autophagy in axonal dystrophy and degeneration. J Neurosci 26(31):8057–8068
Hara T, Nakamura K, Matsui M et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441(7095):885–889
Du L, Hickey RW, Bayir H et al (2009) Starving neurons show sex difference in autophagy. J Biol Chem 284(4):2383–2396. doi:10.1074/jbc.M804396200
Rose C, Menzies FM, Renna M et al (2010) Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum Mol Genet 19:2144–2153. doi:10.1093/hmg/ddq093
Crews L, Spencer B, Desplats P et al (2010) Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS ONE 5:e9313. doi:10.1371/journal.pone.0009313
Young JE, Martinez RA, La Spada AR (2009) Nutrient deprivation induces neuronal autophagy and implicates reduced insulin signaling in neuroprotective autophagy activation. J Biol Chem 284:2363–2373. doi:10.1074/jbc.M806088200
Yamamoto A, Cremona ML, Rothman JE (2006) Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol 172:719–731
Sarkar S, Ravikumar B, Floto RA et al (2009) Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine expanded huntingtin and related proteinopathies. Cell Death Differ 16:46–56. doi:10.1038/cdd.2008.110
Tsvetkov AS, Miller J, Arrasate M et al (2010) A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A 107:16982–16987. doi:10.1073/pnas.1004498107
Zhang L, Yu J, Pan H et al (2007) Small molecule regulators of autophagy identified by an image-based highthroughput screen. Proc Natl Acad Sci U S A 104:19023–19028
Lipinski MM, Hoffman G, Ng A et al (2010) A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell 18:1041–1052. doi:10.1016/j.devcel.2010.05.005
Bains M, Zaegel V, Mize-Berge J et al (2011) IGF-I stimulates Rab7-RILP interaction during neuronal autophagy. Neurosci Lett 488:112–117. doi:10.1016/j.neulet.2010.09.018
Wong AS, Lee RH, Cheung AY et al (2011) Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson’s disease. Nat Cell Biol 13:568–579. doi:10.1038/ncb2217
Xu P, Das M, Reilly J et al (2011) JNK regulates FoxO-dependent autophagy in neurons. Genes Dev 25:310–322. doi:10.1101/gad.1984311
Cherra SJ, Chu CT (2008) Autophagy in neuroprotection and neurodegeneration: a question of balance. Future Neurol 3:309–323
Carra S, Brunsting JF, Lambert H et al (2009) HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. J Biol Chem 284(9):5523–32
Eskelinen EL (2006) Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Asp Med 27:495–502
Nishino I (2006) Autophagic vacuolar myopathy. Semin Pediatr Neurol 13:90–95, Review
Carloni S, Buonocore G, Balduini W (2008) Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis Dec 32(3):329–39. doi:10.1016/j.nbd.2008.07.022
Yitzhaki S, Huang C, Liu W et al (2009) Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Basic Res Cardiol 104:157–67. doi:10.1007/s00395-009-0006-6
Ghavami S, Shojaei S, Yeganeh B et al (2014) Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol 112:24–49
Koike M, Shibata M, Tadakoshi M et al (2008) Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol 172:454–469. doi:10.2353/ajpath.2008.070876
Lee JA, Gao FB (2009) Inhibition of autophagy induction delays neuronal cell loss caused by dysfunctional ESCRT-III in frontotemporal dementia. J Neurosci 29:8506–8511. doi:10.1523/JNEUROSCI.0924-09.2009
Lee S, Sato Y, Nixon RA (2011) Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci 31:7817–7830. doi:10.1523/JNEUROSCI.6412-10.2011
Rubinsztein DC (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443:780–786
Ju JS, Fuentealba RA, Miller SE et al (2009) Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 187:875–878. doi:10.1083/jcb.200908115
Lee JA, Gao FB (2008) ESCRT, autophagy, and frontotemporal dementia. BMB Rep 41:827–832, Review
Pasquali L, Ruffoli R, Fulceri F et al (2010) The role of autophagy: what can be learned from the genetic forms of amyotrophic lateral sclerosis. CNS Neurol Disord Drug Targets 9:268–278
Mizushima N, Levine B, Cuervo AM et al (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075. doi:10.1038/nature06639
Hung SY, Huang WP, Liou HC et al (2009) Autophagy protects neuron from Abeta-induced cytotoxicity. Autophagy 5:502–510
Wang H, Ma J, Tan Y et al (2010) Amyloid-beta1-42 induces reactive oxygen species-mediated autophagic cell death in U87 and SH-SY5Y cells. J Alzheimers Dis 21:597–610. doi:10.3233/JAD-2010-091207
Boland B, Smith DA, Mooney D et al (2010) Macroautophagy is not directly involved in the metabolism of amyloid precursor protein. J Biol Chem 285:37415–37426. doi:10.1074/jbc.M110.186411
O’Brien RJ, Wong PC (2011) Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci 34:185–204. doi:10.1146/annurev-neuro-061010-113613, Review
Rajendran L, Annaert W (2012) Membrane trafficking pathways in Alzheimer’s disease. Traffic 13:759–770. doi:10.1111/j.1600-0854.2012.01332.x
Pickford F, Masliah E, Britschgi M et al (2008) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118:2190–2199. doi:10.1172/JCI33585
Jaeger PA, Pickford F, Sun CH et al (2010) Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS ONE 5:e11102. doi:10.1371/journal.pone.0011102
Spencer B, Potkar R, Trejo M et al (2009) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci 29:13578–13588. doi:10.1523/JNEUROSCI.4390-09.2009
Lee JH, Yu WH, Kumar A et al (2010) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer related PS1 mutations. Cell 141:1146–1158. doi:10.1016/j.cell.2010.05.008
Berger Z, Ravikumar B, Menzies FM et al (2006) Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet 15:433–442
Wang Y, Martinez-Vicente M, Kruger U et al (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18:4153–4170
Caccamo A, Majumder S, Richardson A et al (2010) Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem 285:13107–13120. doi:10.1093/hmg/ddp367
Rodriguez-Navarro JA, Rodriguez L, Casarejos MJ et al (2010) Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis 39:423–438. doi:10.1016/j.nbd.2010.05.014
Majumder S, Richardson A, Strong R et al (2011) Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 6:e25416. doi:10.1371/journal.pone.0025416
Schaeffer V, Lavenir I, Ozcelik S et al (2012) Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 135:2169–2177. doi:10.1093/brain/aws143
Lee JY, Koga H, Kawaguchi Y et al (2010) HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 29:969–980. doi:10.1038/emboj.2009.405
Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57:369–384, Review
Finkbeiner S (2011) Huntington’s disease. Cold Spring Harb Perspect Biol 3(6):a007476. doi:10.1101/cshperspect.a007476
Tellez-Nagel I, Johnson AB, Terry RD (1974) Studies on brain biopsies of patients with Huntington’s chorea. J Neuropathol Exp Neurol 33:308–332
Wang T, Lao U, Edgar BA (2009) TOR-mediated autophagy regulates cell death in Drosophila neurodegenerative disease. J Cell Biol 186:703–711. doi:10.1083/jcb.200904090
Sarkar S, Rubinsztein DC (2008) Huntington’s disease: degradation of mutant huntingtin by autophagy. Febs J 275:4263–4270
Martinez-Vicente M, Talloczy Z, Wong E et al (2010) Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci 13:567–576. doi:10.1038/nn.2528
Li X, Wang CE, Huang S et al (2010) Inhibiting the ubiquitin-proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments. Hum Mol Genet 19:2445–2455. doi:10.1093/hmg/ddq127
Zheng S, Clabough EB, Sarkar S et al (2010) Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet 6:e1000838. doi:10.1371/journal.pgen.1000838
Atwal RS, Truant R (2008) A stress sensitive ER membrane association domain in Huntingtin protein defines a potential role for Huntingtin in the regulation of autophagy. Autophagy 4:91–93
Berger Z, Ravikumar B, Menzies FM et al (2006) Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet 15:433–442. doi:10.1093/hmg/ddi458
Williams A, Sarkar S, Cuddon P et al (2008) Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol 4:295–305
Tsvetkov AS, Miller J, Arrasate M et al (2010) A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A 107:16982–16987
Rose C, Menzies FM, Renna M et al (2010) Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum Mol Genet 19:2144–2153
Chakrabarti L, Eng J, Ivanov N et al (2009) Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol Brain 2:2410
Anglade P, Vyas S, Javoy-Agid F et al (1997) Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol 12:25–31
Webb JL, Ravikumar B, Atkins J et al (2003) Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278:25009–25013
Cuervo AM, Stefanis L, Fredenburg R et al (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305:1292–1295
Nuytemans K, Theuns J, Cruts M et al (2010) Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31:763–780
Isidoro C, Biagioni F, Giorgi FS et al (2009) The role of autophagy on the survival of dopamine neurons. Curr Top Med Chem 9:869–879
Winslow AR, Chen CW, Corrochano S et al (2011) Alpha-synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol 190:1023–1037. doi:10.1083/jcb.201003122
Xilouri M, Vogiatzi T, Vekrellis K et al (2009) Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 4:e5515. doi:10.1371/journal.pone.0005515
Narendra D, Tanaka A, Suen DF et al (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803. doi:10.1083/jcb.200809125
Vives-Bauza C, Zhou C, Huang Y et al (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 107:378–383. doi:10.1073/pnas.0911187107
Michiorri S, Gelmetti V, Giarda E et al (2010) The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ 17:962–974. doi:10.1038/cdd.2009.200
Khandelwal PJ, Herman AM, Hoe HS et al (2011) Parkin mediates Beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum Mol Genet 20:2091–2102
Stefanis L, Larsen KE, Rideout HJ et al (2001) Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 21:9549–9560
Choubey V, Safiulina D, Vaarmann A et al (2011) Mutant A53T (alpha)-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 286:10814–10824. doi:10.1074/jbc.M110.132514
Zhu JH, Horbinski C, Guo F et al (2007) Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol 170:75–86
Choi KC, Kim SH, Ha JY et al (2010) A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death. J Neurochem 112:366–376. doi:10.1111/j.1471-4159.2009.06463.x
Tofaris GK (2012) Lysosome-dependent pathways as a unifying theme in Parkinson’s disease. Mov Disord 27:1364–1369. doi:10.1002/mds.25136
Janda E, Isidoro C, Carresi C et al (2012) Defective autophagy in Parkinson’s disease: role of oxidative stress. Mol Neurobiol 46:639–661. doi:10.1007/s12035-012-8318-1
Andersen PM, Al-Chalabi A (2011) Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol 7(11):603–615. doi:10.1038/nrneurol.2011.150
Hardiman O, van den Berg LH, Kiernan MC (2011) Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol 7(11):639–649
Sasaki S (2011) Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 70(5):349–359. doi:10.1038/nrneurol.2011.153
Li L, Zhang X, Le W (2008) Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 4:290–293
Morimoto N, Nagai M, Ohta Y et al (2007) Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res 1167:112–117
Crippa V et al (2010) The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum Mol Genet 19:3440–3456
Hetz C et al (2009) XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 23:2294–2306
Filimonenko M, Stuffers S, Raiborg C et al (2007) Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179:485–500
Johnson JO, Mandrioli J, Benatar M et al (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68:857–64. doi:10.1016/j.neuron.2010.11.036
Uchiyama Y (2001) Autophagic cell death and its execution by lysosomal cathepsins. Arch Histol Cytol 64:233–246
Uchiyama Y, Koike M, Shibata M (2008) Autophagic neuron death in neonatal brain ischemia/hypoxia. Autophagy 4:404–408
Wen YD, Sheng R, Zhang LS et al (2008) Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 4:762–769
Wang JY, Xia Q, Chu KT et al (2011) Severe global cerebral ischemia-induced programmed necrosis of hippocampal CA1 neurons in rat is prevented by 3-methyladenine: a widely used inhibitor of autophagy. J Neuropathol Exp Neurol 70:314–322. doi:10.1097/NEN.0b013e31821352bd
Tanabe F, Yone K, Kawabata N et al (2011) Accumulation of p62 in degenerated spinal cord under chronic mechanical compression: functional analysis of p62 and autophagy in hypoxic neuronal cells. Autophagy 7:1462–1471
Wang P, Guan YF, Du H et al (2012) Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy 8:77–87. doi:10.4161/auto.8.1.18274
Brunk UT, Terman A (2002) The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem 269:1996–2002
Liu C, Gao Y, Barrett J et al (2010) Autophagy and protein aggregation after brain ischemia. J Neurochem 115(1):68–78. doi:10.1111/j.1471-4159.2010.06905.x
Qin AP, Liu CF, Qin YY et al (2010) Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy 6(6):738–753
Cui D, Wang L, Qi A et al (2012) Propofol prevents autophagic cell death following oxygen and glucose deprivation in PC12 cells and cerebral ischemia-reperfusion injury in rats. PLoS ONE 7(4):e35324
Gao L, Jiang T, Guo J et al (2012) Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS One 7(9):e46092. doi:10.1371/journal.pone.0046092
Wang JY, Xia Q, Chu KT et al (2011) Severe global cerebral ischemia-induced programmed necrosis of hippocampal CA1 neurons in rat is prevented by 3-methyladenine: a widely used inhibitor of autophagy. J Neuropathol Exp Neurol 70:314–322
Sheng R, Zhang LS, Han R et al (2010) Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 6:482–494
Zhu C, Wang X, Xu F et al (2004) The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ 12:162–176
Dirnagl U, Simon RP, Hallenbeck JM (2003) Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 26:248–254
Rangaraju S, Notterpek L (2011) Autophagy aids membrane expansion by neuropathic Schwann cells. Autophagy 7:238–239
Madorsky I, Opalach K, Waber A et al (2009) Intermittent fasting alleviates the neuropathic phenotype in a mouse model of Charcot-Marie-Tooth disease. Neurobiol Dis 34:146–154
Smith CM, Mayer JA, Duncan ID (2013) Autophagy promotes oligodendrocyte survival and function following dysmyelination in a long-lived myelin mutant. J Neurosci 33nn:8088–8100. doi:10.1523/JNEUROSCI.0233-13.2013
van Kollenburg B, van Dijk J et al (2006) Glia-specific activation of all pathways of the unfolded protein response in vanishing white matter disease. J Neuropathol Exp Neurol 65:707–715
Getts MT, Getts DR, Kohm AP et al (2008) Endoplasmic reticulum stress response as a potential therapeutic target in multiple sclerosis. Therapy 5:631–640
Dhaunchak AS, Nave KA (2007) A common mechanism of PLP/DM20 misfolding causes cysteine-mediated endoplasmic reticulum retention in oligodendrocytes and Pelizaeus-Merzbacher disease. Proc Natl Acad Sci U S A 104:17813–17818
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hu, Z., Yang, B., Mo, X. et al. Mechanism and Regulation of Autophagy and Its Role in Neuronal Diseases. Mol Neurobiol 52, 1190–1209 (2015). https://doi.org/10.1007/s12035-014-8921-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8921-4