
Abstract
Ca-doped barium hexaferrite (Ba1−xCaxFe12O19; x = 0.1–0.6) samples were synthesized successfully using solid-state reaction route. Further, the limit of the Ca solubility in BaFe12O19 has been investigated in the 900–1200°C temperature range. X-ray analysis reveals maximum Ca solubility in BFO at 1200°C (≈50%). The emergence of a hematite secondary phase at Ca concentrations ≈50 and 60 at% was observed. The XRD analysis also confirmed a gradual reduction in the c-parameter from 23.195 Å (at x = 0.1) to 23.147 Å (at x = 0.5). The structural data further suggested an enhancement of sample density with Ca substitution. Field-emission transmission electron microscopy micrographs reveal (a) distinct grain morphology at lower Ca concentration (x = 0.1) and (b) enhanced secondary grain growth and grain amalgamation at higher Ca concentration (x = 0.5). The M–H studies reveal that Mr and Ms almost stay constant up to x = 0.2, beyond which they start rising rapidly. Mr ≈ 42.65 emu g−1 and Ms ≈ 92.19 emu g−1, exhibit a maximum at x = 0.4. The coercivity first decreases rapidly from 4168 Oe (at x = 0.0) to 2326 Oe (at x = 0.1) followed by a marginal decrease to 2045 Oe (at x = 0.4). An Hc value ≥1200 Oe in a majority of our samples makes them potentially suitable for perpendicular recording media and permanent magnet applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Ferrites have emerged as very effective electrical and magnetic materials due to their appealing physical characteristics and for being applicable in manifold fields of ferrofluids, magnetic recording media, catalysis, diagnostic imaging and biomedical fields, and electrical devices, especially in microwave/GHz frequencies [1]. The characteristics that make hexagonal technologically promising candidates are (a) low dielectric constant, (b) high resistivity, (c) high saturation magnetization, (d) high coercivity and (e) their chemically stable nature [2]. The excellence of their electric and magnetic characteristics owes to the stacking variation of various metallic ions present in their crystal structure [3]. The properties of hexaferrites can hence be manipulated by partially substituting these cations using foreign dopants. Among M, X, U, W, Z and Y hexaferrites, M-type hexaferrites (with MFe12O19 as general formula, M denotes any metal of the second group of the periodic table) are widely chosen since they are easier to synthesize [4]. For this class of hexaferrites, resistivity and coercivity are superior, magneto anisotropy is quite high, and electrical conductivity is low [5]. They are highly chemical stable and most importantly, they are affordable and thus commercially viable [6,7]. As a consequence, they are among the best contenders to be utilized in domestic appliances also. They are majorly applicable in the areas of loudspeakers, shielding devices, capacitors, refrigerator door magnet, magnetic resonance imaging, memory cells, radar systems, transformers, multichip inductors, etc. [8,9,10,11,12]. As far as the production of the materials for permanent magnets is concerned, an estimated 90% (out of the total) weight is accounted for M-type hexaferrites [13].
A single BFO unit cell consists of 38 oxygen ions (O2−), 2 barium ions (Ba2+), 24 iron ions (Fe3+) in total [5]. Their magnetic properties stem from Fe3+ ions which occupy trigonal bipyramidal, tetrahedral and octahedral sites (typically named 2b; 4f1; 12k, 2a and 4f2) [14,15,16]. These five spin sites are further grouped into two categories (a) spin-up (2a, 2b, 12k) and (b) spin-down (4f1 and 4f2) sites [17]. The distribution of Fe3+ ions into spin-up and spin-down sites results in a reduction of magnetic moment because of the cancellation of spin moments [18,19,20]. The magnetic characteristics of BFO can be modified by altering relative concentration of Fe3+ cations in these spin-up and spin-down sites [17,21,22,23,24,25,26,27,28,29,30,31,32]. However, A-site doping has also been increasingly employed to modify various hexaferrite characteristics. Ca (either by itself and also in combination) has also been used to modify the magnetic properties of a number of M-type ferrites [22,23,31,32]. Popa et al [33] studied grain size variation in 5 to 20 min thermal-treated (between 800 and 1000°C) pure and 1 mol Ca-doped BFO and SrFO systems and their magnetic properties. An enhancement of coercivity in the SrFO system was reported [33]. Ali et al [34] used sol–gel autocombustion method to synthesize Ba1−xCaxFe11.5Cr0.5O19 (x = 0.0–0.5) samples and investigated their structural, microstructural and magnetic characteristics. Hematite impurity was observed in all the samples. The highest value of Hc ≈ 4306 Oe was reported at x = 0.5 [34]. Khan et al [2] prepared Ba0.5−xCa0.5PrxNiyFe12−yO19 (x = 0.00–0.10 and y = 0.00–1.00) using sol–gel autocombustion technique. Ms ≈ 37.08 emu g−1 and Hc ≈ 1645 Oe were reported for x = 0.5 [2]. Ashima et al [35,36] reported the characterization of Ba1−xCaxFe12O19 (x = 0.0 and 0.5) synthesized using solid-state reaction method. Hematite was observed as an impurity phase for x = 0.5. The Ms decreased from 53.03 (x = 0.0) to 33.17 emu g−1 (x = 0.5) due to the effect of Ca doping into BFO. On the other hand, coercivity (Hc) increased with increasing Ca content from 2750 (x = 0.0) to 3200 Oe (x = 0.5). Kumar et al [18] investigated structural, elemental and magnetic properties for the Ba1−xCaxFe12O19 for (x = 0.00–0.20) system synthesized using sol–gel method. The maximum value of Ms (≈59.48 emu g−1) has been observed for x = 0.05. Godara and group [8] synthesized Ba1−xCaxFe12O19 (x = 0.00–0.50) samples using sol–gel autocombustion method. Samples calcined at 1200°C were claimed to be phase pure till x = 0.50. Ms was reported to increase from 63.85 (at x = 0.00) to 71.82 emu g−1 (at x = 0.50). A decreasing trend in the value of coercivity from ≈3765 Oe (for x = 0.00) to 1735 Oe (for x = 0.50) was reported [37].
In summary, some of these results in Ba1−xCaxFe12O19 (Ca-substituted BFO) samples are in contrast to each other. For example, an increase in coercivity (with x) was observed by Ashima et al [35,36] to decrease in coercivity was observed by Godara and group [8]. The trend of their Mr and Ms values are also in contrast to each other. Further, the coercivity values reported by Ali et al [34] ≈4306 Oe at x = 0.5 to Khan et al [2] reported value of ≈1645 Oe (at x = 0.5). Therefore, it is worth studying the modification of magnetic properties of BFO upon Ca substitution in a wide composition range. We hereby present the modification of structural, microstructural and magnetic properties by substituting Ca in BFO (Ba1−xCaxFe12O19) samples calcined at 1200°C for 6 h synthesized via solid-state reaction method.
2 Experimental
M-type barium hexaferrite Ba1−xCaxFe12O19 (x = 0.1–0.6) powders were synthesized by means of a solid-state reaction route technique. The chemical reaction involved in the synthesis of Ba1−xCaxFe12O19 sample is as follows:
The starting materials (BaCO3, CaCO3 and Fe2O3) chosen for the synthesis of the required material were weighed according to their stoichiometric amount. Then these weighed materials were poured into a bottle suspended in an acetone medium having 20 zirconium balls. A 24-h period was spent combining these materials in a ball mill. The resulting blended powders were then allowed to air dry for around 12 h. Then a sintering procedure was conducted in a muffle furnace between 900 and 1200°C for 6 h. At last, the calcined powders were converted into fine powder using grinding using a mortar pestle.
The structural analysis of the prepared samples was examined by collecting their respective X-ray diffraction patterns/diffractograms. The X-ray patterns were collected using MAXIMA XRD-7000 from SHIMADZU within the scanned 2θ range (20°–80°; scan step size of 0.02°) having a scanning rate of 1° min−1. FULLPROF SUITE software was employed to perform the Rietveld refinement. Field-emission transmission electron microscopy (FESEM) data was recorded (in pellet form) using Gemin SEM500 (from Carl Zeiss). The scale for the scanning images of these samples is 1 μm and the magnification is 25 Kx. The room temperature M–H data was recorded using EZ9 from Microsense.
3 Results and discussions
3.1 Crystal structure determination
As stated in the introductory text, the issue of Ca solubility in BaFe12O19 has been investigated in the 900–1200°C temperature range. In this regard, Ba1−xCaxFe12O19 (x = 0.1–0.6) samples were synthesized using standard ceramic technique. However, the x = 0.5 sample was only calcined at 1100 and 1200°C. The issue of phase purity of these samples was addressed using Rietveld refinement of the XRD data. A nice match between the experimental and the refined data (low value of GoF, and random distribution of the difference data about mean) using a single-phase (P63/mmc structural symmetry, (ICSD 98-010-5657)) model has been used as a criterion for affirming the phase purity of the sample under investigation. Any discrepancy between the experimental and refined data was seen as an indication of limitation of the single-phase/multiple-phase model, thereby implying the sample to be phase-impure.
The Rietveld refined (employing a single-phase structural model) data of 900°C calcined x = 0.2 and x = 0.6 samples are shown in figure 1a and c. The presence of unexplained peaks (only black data) in the main plot and sharp positive peaks in the difference plot is suggestive of the failure of the single-phase model to reasonably explain the diffraction data. It is to be noted that the above-stated discrepancies are relatively much more pronounced in the x = 0.6 sample as compared to x = 0.2 sample. The single-phase \(\chi\)2-values ≈ 2.50 and 6.49 have been observed for x = 0.2 and 0.6, respectively. These discrepancies are indicative of the fact that the Ca solubility in BaFe12O19 is below 20 at% at 900°C. In order to further improve the refinement, multiple-phase Rietveld refinement was initiated. By comparing the mismatched peaks with the ICSD database, (a) Fe2O3 phase (ICSD 98-002-2616) was found to exist as an impurity in x = 0.2 and (b) Fe2O3 along with Ca2Fe2O5 (ICSD 98-000-5960) phases were found to be present as impurity phases in x = 0.4 and 0.6 samples. The powder diffraction data for x = 0.2 was hence modeled by employing (Ca-doped BFO (P63/mmc) + Fe2O3 (R-3c)) phases and the refined data is shown in figure 1b. The two-phase model leads to an improvement in the \(\chi\) 2-values from 2.50 to 1.50. Rietveld refined data using (Ca-doped BFO (P63/mmc) + Fe2O3 (R-3c) + Ca2Fe2O5 (Pnma)) phases for x = 0.6 sample is shown in figure 1d. The multiple-phase fitting for this sample leads to a reduction in \(\chi\)2-values from 6.49 to 1.62. A similar approach was used to analyse the phase purity of x = 0.2–0.6 samples calcined at 900, 1000 and 1100°C. However, the Rietveld refined data has not been shown for all these samples. The parameters extracted from Rietveld refinement (phase fraction, space group and GoF) for all the samples (up to x = 0.6) calcined at ≤1100°C have been summarized in table 1. It is quite evident from table 1 that up to 40 at% Ca is soluble in BFO at 1100°C. Further in the x = 0.6 sample, the Ca2Fe4O5 phase disappears at 1100°C and Fe2O3 phase (19.93%) persists. This implies that Ca solubility in BFO at 1100°C could be ≈40 at%.

The Rietveld refined data of (a) x = 0.2 single phase, (b) x = 0.2 two-phase and (c) x = 0.6 single phase, (d) x = 0.6 three-phase Ba1−xCaxFe12−2xO19 samples calcined at 900°C. The red/black curves represent experimental/Rietveld refined data. # represents the unidentified peaks. The (hkl) corresponding to the main phase has been marked in black colour and that of secondary phases Fe2O3 and Ca2Fe2O5 in purple and green colour, respectively.
The Rietveld refined data of Ca-doped BFO samples (x = 0.1–0.6) calcined at 1200°C is shown in figure 2a–f. However, the single-phase structural model was only found to be suitable up to x = 0.4. Beyond x = 0.4, Fe2O3 was observed as a phase impurity. Therefore, the structural data of the samples beyond x = 0.4 was refined using a two-phase (Ca-doped BFO and Fe2O3) model. The \(\chi\)2 (GoF) values ≈1.76, 1.43, 1.84, 1.45, 1.77 and 1.50 were obtained for x = 0.1, 0.2, 0.3, 0.4, 0.5 and 0.6 samples, respectively. This value of GoF is indicative of a good level of refinement [38]. Surprisingly, the fraction of Fe2O3 only increased from 5.90% (in x = 0.5 sample) to 6.57% (in x = 0.6 sample). This is because Ca-solubility ≈44 at% would have resulted in a segregation of around 16% Fe2O3 in the x = 0.6 sample. The observed discrepancy could either be due to chemical inhomogeneity or thermal fluctuations. In a nutshell, the XRD studies point to Ca solubility ≈50 a% in BFO.

The Rietveld refined data of (a) x = 0.1, (b) x = 0.2, (c) x = 0.3, (d) x = 0.4, (e) x = 0.5 and (f) x = 0.6 Ba1−xCaxFe12−2xO19 samples calcined at 1200°C. The red/black curves represent experimental/Rietveld refined data.
The impact of Ca2+ substitution on the X-ray density (Dx), bulk density (Db) and sample porosity (P) of these samples was also examined. Their values have been calculated using the formulae Dx = \(\frac{ZM}{{N}_{a}{V}_{\mathrm{cell}}}\), Db \(= \frac{m}{{t \pi r}^{2}}\) and P \(=\) \(\left(\frac{{D}_{\mathrm{x}}- {D}_{\mathrm{b}}}{{D}_{\mathrm{x}}}\right)\times 100\) [8]. The lattice parameters (a), (c), c/a ratio cell volume (Vcell), Dx, Db and P of these samples are given in table 2. The incorporation of foreign ion/ions having different ionic radii into the BFO lattice might potentially affect the lattice parameters a and c. In this study, we observe (as given in table 2), lattice parameter (a) remains almost the same for all the compositions, whereas the lattice parameter (c) decreases marginally from 23.195 to 23.093 Å with an increase in the Ca concentration. This observed trend could be explained on the basis of the relative ionic radii of the host Ba2+ (1.34 Å) and doped Ca2+ (0.99 Å) ions. The substitution of a cation having lower ionic radii as compared to host ions will drive the remaining host ions closer to each other as the substituted ion occupies lesser space. As the host ions move close to each other, a net reduction in lattice parameters takes place. The lattice contraction may also be explained on the basis of empirical Vegard’s law for any solid solutions A–B as [39].
where X = XB is the mole fraction of component B and \({a}_{\mathrm{A}}^{0} \,\, \mathrm{ and } \,\, {a}_{\mathrm{B}}^{0}\) are the lattice parameters of the pure components A and B, respectively. As per this law, if we are making solid solutions of two materials with one having higher ionic radii (and hence higher lattice parameters) and the other having lower ionic radii (and hence lower lattice parameters), the lattice parameters of the resulting solid solution will be average of the two extremes. The dopant-induced lattice expansion (where dopant atom has higher ionic radii) has been reported by a number of research groups [40,41,42,43,44]. In contrast, we are observed lattice contraction as we are substituting a dopant with lower ionic radii as compared to the host. This observed trend is better depicted in figure 3. The lattice parameter ratio (c/a) is a marker for determining the kind of hexaferrite. A ratio of less than 3.98 is an indicator of the M-type hexaferrite [45]. For our samples, the c/a ratio is less than 3.98 confirming that they are M-type hexaferrite. The X-ray density decreases from 5.24 to 5.05 g cm−3 with the increase in calcium concentration. Since, the X-ray density is directly proportional to the atomic mass of the sample, the decrease in X-ray density could be due to the difference in the atomic masses of the doped calcium (Ca2+) (40.078 amu) ion and host Ba2+ (137.33 amu) ion [34]. Conversely, bulk density increases with an increase in calcium concentration from 3.15 g cm−3 (at x = 0.1) to 3.69 (at x = 0.5) g cm−3. This implies that enhanced Ca substitution results in a net reduction of the concentration of voids, which ultimately leads to enhanced bulk density [46]. The enhancement of sample densification is also apparent from the calculated porosity (P) values, which decrease from 37% at x = 0.1 to 23% at x = 0.5. Thus, in summary, XRD measurements confirm (a) Ca solubility in BFO is ≈50%, (b) the emergence of secondary phases at Ca concentrations ≈50 and 60 at%, (c) gradual reduction in the value of c-parameter and (d) the enhancement of sample density.

Variation of lattice parameters (a and c) with composition in Ba1−xCaxFe12−2xO19 samples calcined at 1200°C.
In the literature reports, only Ashima et al [35] reported Fe2O3 as a secondary phase impurity in the 50 at% Ca-substituted BFO samples. Most of the other authors [2,18,47] reported phase purity up to 50 at% Ca substitution in BFO. The differences could be due to the use of different synthesis techniques, calcination temperature and duration. Also, similar to Godara and group [8] we observe lattice contraction with increasing Ca substitution [37].
3.2 Microstructural studies
The FESEM micrograph of the typical Ca-substituted barium hexaferrite samples (for x = 0.2, 0.4 and 0.5) is shown in figure 4a, b and c. It is clear from figure 4a that the x = 0.2 sample consists of grains with distinct grain boundaries. Further, (a) most of these grains appear to have plate-like shape and (b) some of them could be noticed to have boundaries varying from four to six. A very little amount of secondary growth could also be noticed by the presence of very small-sized round-shaped grains. The average grain size in this sample is ≈0.21 μm. The secondary grain growth appears to be more dominant in the x = 0.4 sample, as is evident from the presence of a large number of < 300 nm-sized grains. In this sample, the bigger (plate-like grains) with well-defined boundaries are also noticeable in the background. However, the secondary grains are obscuring most of the view. The average grain size (most of the smaller grains opt for the measurement as boundaries of larger grains are difficult to decipher) in this sample is ≈0.25 μm. In the x = 0.5 sample, no distinct grain boundaries between secondary grains and primary grains are visible. The image rather presents a fusion of smaller grains into larger grains. The fused kind of morphology observed in this sample could be a potential hint of maximum tolerable Ca concentration in BFO lattice between 40 and 50 at%. It is to be noticed that the presence of even a large number of secondary grains in x = 0.4 sample and the fusion of smaller and larger grains in the x = 0.5 sample make the estimation of average grain size rather difficult. The histogram giving the size distribution of the prepared samples is shown in figure 5a, b and c.

FESEM images of Ba1−xCaxFe12O19 samples (a) x = 0.2, (b) x = 0.4 and (c) x = 0.5 calcined at 1200°C.

Histogram giving the size distribution of Ba1−xCaxFe12O19 samples: (a) x = 0.2, (b) x = 0.4 and (c) x = 0.5 calcined at 1200°C.
3.3 Energy dispersive X-ray analysis
Energy dispersive X-ray (EDX) spectrum of the typical Ba1−xCaxFe12O19 hexaferrite sample (for x = 0.4) is shown in figure 6. The spectrum confirms the presence of barium, calcium, iron and oxygen in the prepared sample. The atomic percentages of various elements (also x = 0.2 and 0.5) have also been calculated from the EDX spectra and are given in table 3. The table also gives the values of the measured and expected Fe:Ba ratio. It is clear that the measured and expected Fe:Ba ratio exhibits a similar trend with increasing Ca concentration. The small differences between the experimental and expected Fe:Ba ratio could be due to the fact that the experiment values have been obtained using a standardless quantification approach.

EDX spectra of x = 0.4 Ba1−xCaxFe12O19 samples.
3.4 Magnetic studies
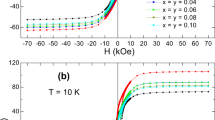
Figure 7a, b depicts the room temperature M–H loops for x = 0.1–0.5 Ba1−xCaxFe12O19 samples. The finite non-saturating loops unambiguously demonstrate that (a) all the samples possess hard ferrite characteristics and (b) have diminishing coercivity [48]. The remnant magnetization (Mr) and coercivity (Hc) of different samples were retrieved from the M–H loops (average of Y-axis intercept and X-axis intercept, respectively). The saturation magnetization (Ms) value was obtained by carrying out a plot between M vs. 1/H2 (law of approach to saturation method) [8,49]. The Y-axis intercept of the linear fit (shown in figure 8) to this data (between 12,000 to 15,000 Oe range) is taken as the Ms value. The magnetic parameters so extracted are given in table 4. The table clearly shows a monotonic increase in the value of Ms from 70.26 to 92.19 emu g−1, and a decrease in Hc from 2326 to 2045 Oe with an increase in the value of ‘x’.

(a) The hysteresis (M–H) loops for x = 0.1–0.5 Ba1−xCaxFe12O19 samples recorded at room temperature and (b) zoomed view of the hysteresis loops for x = 0.1–0.5 Ba1−xCaxFe12O19 samples.

The M vs. 1/H2 plots along with the linear fitted data for x = 0.1–0.5 Ba1−xCaxFe12O19 samples.
The Mr, Ms and Hc values for better elucidation are plotted as a function of increasing Ca-concentration (x) and are shown in figure 9a and b. It is clear from figure 9a that Mr and Ms almost stay constant up to x = 0.2, beyond which they start rising rapidly. The constant value of Mr and Ms up to x = 0.2 could be because Ca substitutes (a) mostly Ba-sites and (b) part of it substitutes equally between up-spin (2a, 2b and 12k) and down-spin (4f1 and 4f2) sites. As a consequence, the net distribution of 12 Fe3+ ions between up-spin and down-spin sites remains unaffected, thereby leading to an almost constant value of Mr and Ms. However, beyond x = 0.2, the Ca2+ ions in addition to occupying Ba sites also start preferably occupying down-spin Fe sites. As a consequence, there is a decrease in the value of magnetic moment contribution from down-spin Fe sites (which decrease the magnetic moment), which results in the enhancement of the value of Mr and Ms [50]. The small increase in the value of Mr and Ms at x = 0.5 could be due to the phase impurity present in this sample (as discussed in the XRD section) [35]. Also, the SEM data for this sample hinted at fused grain growth.

Variation of (a) Ms and Mr, and (b) Hc and k as a function of ‘x’.
The coercivity value meanwhile decreases rapidly in the beginning from 4168 Oe (x = 0.0) to 2326 Oe (x = 0.1) and thereafter it decreases slightly to 2045 Oe (x = 0.4), as shown in figure 9b. The Hc variation can be somewhat explained in terms of varying values of k (magnetocrystalline anisotropy (\(k=\frac{{\mu }_{0}{M}_{\mathrm{s}}{H}_{\mathrm{c}}}{2}\)); (\({\mu }_{0}\) (magnetic permeability of free space) is taken as unity in cgs units)) [51]. The k-values have also given in table 4 and plotted in figure 9b. It is clear from figure 9b that the decrease in Hc is in nearly direct correlation with a varying value of k. However, there is a slight difference between the minima in Hc (at x = 0.4) and the minima of k (at x = 0.3). The difference could be due to the type of atomic site being substituted by Ca. Microscopically, the initial rapid decrease in coercivity may be related to more preference for 4f2 and 2a (octahedral sites which have maximum contribution for magnetocrystalline anisotropy in hexaferrites) sites up to x = 0.1 [2,52,53]. As a result, Mr and Ms do not change but Hc decreases rapidly. Beyond x = 0.1, the other up-spin sites become slightly more preferred in addition to the 4f2 and 2a sites. As a result, Mr and Ms start to increase marginally, whereas Hc keeps decreasing slowly. At still higher values of x (up to x = 0.4), the spin-up sites become increasingly preferred in addition to 4f2 and 2a sites. As a result, Mr and Ms rise rapidly and Hc only decreases marginally. These results hence suggest that the preference of Ca ions for different Fe sites keeps on changing with increasing Ca concentration. The slight anomalous results of the x = 0.5 samples could be due to chemical inhomogeneity, thermal fluctuations and phase impurities [35]. It may be noted that samples with Hc value of ≈600 Oe are potentially used for longitudinal magnetic recording and those with Hc ≥ 1200 Oe are preferred for perpendicular recording media [2]. For this study, Hc is observed to be ≥1200 Oe in most of the samples. Hence, these samples may be better suited for perpendicular recording media and permanent magnets.
The squareness ratio (deduced by taking the ratio of Mr and Ms) tells about the single or multidomain nature of the ferrite material. If this ratio is lower than 0.5, the multidomain grains exist in the randomly oriented fashion in the prepared ferrite material [1,54,55]. In all our samples, this ratio is less than 0.5 implying they are multidomain.
Comparative detailed information of the magnetic parameters obtained by different researchers on Ca-substituted BFO samples (using different synthesis techniques and calcination parameters) is given in table 5. Among these reports, the highest Hc value (≈4752 Oe) has been reported by Huang et al [47] for x = 0.5 in samples synthesized using solid-state reaction method (like ours). In contrast, we observed almost 54% reduction in coercivity (≈2153 Oe) at this composition along with phase impurities. We actually observe a very narrow variation of coercivity from 2326 Oe (at x = 0.1) to 2045 Oe (at x = 0.4). The other researchers have reported an Hc value close to 2000 Oe (similar to ours) at this composition [37]. Godara and group [8] reported an Hc value (≈1729 Oe), which is lowered by 20% as compared to ours (≈2173 Oe). However, the Ms and Mr values reported by us are the highest among the compiled results.
4 Conclusions
M-type calcium-substituted barium hexaferrite samples were successfully synthesized via solid-state reaction technique. X-ray analysis reveals the presence of a single M-type phase only up to x = 0.4 with Fe2O3 as phase impurity beyond x = 0.4. The \(\chi\)2-values ≈1.76, 1.43, 1.84, 1.45, 1.77 and 1.50 were obtained for x = 0.1, 0.2, 0.3, 0.4, 0.5 and 0.6 samples, respectively. The XRD data is indicative of ≈50 at% Ca solubility in BFO at 1200°C. A gradual reduction in the lattice parameter (c) from 23.195 Å (at x = 0.1) to 23.147 Å (at x = 0.5) is observed. This observed trend could be explained on the basis of the relative ionic radii of the host Ba2+ (1.34 Å) and doped Ca2+ (0.99 Å) ions. The structural data further suggested decreasing sample porosity from ≈37% at x = 0.1 to ≈23% at x = 0.5. FESEM micrographs show that at lower Ca concentrations (x = 0.1), the samples consist of discrete grains with insignificant secondary grain growth, while at higher Ca concentrations (x = 0.5), enhanced secondary grain growth and grain amalgamation are noticeable. The M–H investigations reveal nearly constant Mr and Ms up to x = 0.2, after which they start rising quickly. This might be because Ca largely substitutes for Ba-sites and that a portion of it substitutes equally for an up-spin (2a, 2b and 12k) and down-spin (4f1 and 4f2) sites. The coercivity value meanwhile decreases rapidly in the beginning from 4168 Oe (x = 0.0) to 2326 Oe (x = 0.1) and thereafter it decreases marginally to 2045 Oe (x = 0.4). This may be because up to x = 0.1, the octahedral sites (4f2 and 2a, which contribute most to the magnetocrystalline anisotropy in hexaferrites) may be preferred substitutional sites. Both Mr ≈42.65 emu g−1 and Ms ≈92.19 emu g−1 exhibit a maximum value at x = 0.4. The coercivity >1200 Oe observed in our samples could make these materials potentially suitable for perpendicular recording media and permanent magnet applications.
Change history
08 December 2023
A Correction to this paper has been published: https://doi.org/10.1007/s12034-023-03096-x
References
Yasmin N, Zahid M, Khan H M, Hashim M, Islam M U, Yasmin S et al 2019 J. Alloys Compd. 774 962
Khan H M, Islam M U, Xu Y, Ashiq M N, Ali I, Iqbal M A et al 2014 Ceram. Int. 40 6487
Yuping L, Daxin B, Zhangzhong W, Zhang Y, Lizhao C, Guangcheng Z et al 2018 J. Alloys Compd. 734 130
Verma S, Chawla A, Pushkarna I, Singh A, Kumar S, Kumar D et al 2021 Mater. Today Commun. 27 102291
Pullar R C 2012 Prog. Mater. Sci. 57 1191
Mahmood S H, Dushaq G H, Bsoul I, Awawdeh M, Juwhari H K, Lahlouh B I et al 2014 J. Appl. Math. Phys. 02 77
Mosleh Z, Kameli P, Poorbaferani A, Ranjbar M and Salamati H 2016 J. Magn. Magn. Mater. 397 101
Verma S, Singh A, Sharma S, Kaur P, Godara S K, Malhi P S et al 2023 J. Alloys Compd. 930 167410
Singh V P, Jasrotia R, Kumar R, Raizada P, Thakur S, Batoo K M et al 2018 World J. Condens. Matter Phys. 08 36
Abbas W, Ahmad I, Kanwal M, Murtaza G, Ali I, Khan M A et al 2015 J. Magn. Magn. Mater. 374 187
Abdellahi M, Najfinezhad A, Samanadari S S, Khandan A and Ghayour H 2018 Chin. J. Phys. 56 331
Huang L, Huang R, Pang F, Li A, Huang G, Zhou X et al 2020 RSC Adv. 10 18008
Kumar S, Kumar R, Kaur N and Singh P 2021 Results Phys. 22 103903
Habanjar K, Shehabi H, Abdallah A M and Awad R 2020 Appl. Phys. A Mater. Sci. Process. 126 1
Singh H and Rajput J K 2020 SN Appl. Sci. 2 1
Iqbal M J, Ashiq M N and Gomez P H 2009 J. Alloys Compd. 478 736
Ounnunkad S 2006 Solid State Commun. 138 472
Kumar S, Supriya S, Pandey R, Pradhan L K, Singh R K and Kar M 2018 J. Magn. Magn. Mater. 458 30
Sláma J, Grusková A, Papánová M, Kevická D, Dosoudil R, Jančárik V et al 2004 J. Magn. Magn. Mater. 272–276 385
Trukhanov A V, Trukhanov S V, Panina L V, Kostishyn V G and Kazakevich I S 2017 J. Magn. Magn. Mater. 426 487
Tehrani M K, Ghasemi A, Moradi M and Alam R S 2011 J. Alloys Compd. 509 8398
Sözeri H, Küçük I and Özkan H 2011 J. Magn. Magn. Mater. 323 1799
Corral-Huacuz J C and Mendoza-Suárez G 2002 J. Magn. Magn. Mater. 242–245 430
Winotai P, Thongmee S and Tangab I M 2000 Mater. Res. Bull. 35 1747
Soman V V, Nanoti V M and Kulkarni D K 2013 Ceram. Int. 39 5713
Khademi F, Poorbafrani A, Kameli P and Salamati H 2012 J. Supercond. Nov. Magn. 25 525
Tokunaga Y, Kaneko Y, Okuyama D, Ishiwata S, Arima T, Wakimoto S et al 2010 Phys. Rev. Lett. 105 17
Leccabue F, Panizzieri R, Garcia S, Suarez N, Sanchez J L, Ares O et al 1990 J. Mater. Sci. 25 2765
Gregori M L, Pinho M S, Lima R C, Leandro J C S and Ogasawara T 2004 Key Eng. Mater. 264–268 1229
Belous A G, V’yunov O I, Pashkova E V, Ivanitskii V P and Gavrilenko O N 2006 J. Phys. Chem. B 110 26477
Kaur T, Kaur B, Bhat B H, Kumar S and Srivastava A K 2015 Phys. B Condens. Matter 456 206
Dhage V N, Mane M L, Keche A P, Birajdar C T and Jadhav K M 2011 Phys. B Condens. Matter 406 789
Popa P D, Rezlescu E, Doroftei C and Rezlescu N 2005 J. Optoelectron. Adv. Mater. 7 1553
Ali I, Islam M U, Awan M S, Ahmad M and Iqbal M A 2013 J. Supercond. Nov. Magn. 26 3277
Ashima, Sanghi S, Agarwal A and Reetu 2012 J. Alloys Compd. 513 436
Ashima Sanghi S, Agarwal A and Reetu, 2011 AIP Conf Proc. 1393 137
Kumar S, Singh M, Kaur V, Narang S B and Kumar A 2021 Ceram. Int. 47 19271
Toby B H 2006 Powder Diffr. 21 67
Jacob K T, Raj S and Rannesh L 2007 Zeitschrift Fuer. Met. Res. Adv. Tech. 98 776
Mamatha C, Krishnaiah M and Sreedhar B 2017 Procedia Eng. 215 130
Yadav R K and Chauhan P 2020 Indian J. Pure Appl. Phys. 57 881
Abasht B, Beitollahi A and Mirkazemi S M 2016 J. Magn. Magn. Mater. 420 263
Jin S, Yang Y, Medvedeva J E, Ireland J R, Metz A W, Ni J et al 2004 J. Am. Chem. Soc. 126 13787
Ji L, Jiang G, Wu D and Chen J 2020 Mater. Res. Express 7 066103
Wagner T R and Styranec T J 1998 J. Solid State Chem. 138 313
Maria K H, Choudhury S and Hakim M A 2013 Int. Nano Lett. 3 1
Huang K, Yu J, Zhang L, Xu J, Li P, Yang Z et al 2020 J. Alloys Compd. 825 154072
Chauhan C C, Kagdi A R, Jotania R B, Upadhyay A, Sandhu C S, Shirsath S E et al 2018 Ceram. Int. 44 17812
Brown W F 1940 Phys. Rev. 58 736
Yoon D H, Muksin and Raju K 2016 J. Supercond. Nov. Magn. 29 439
Lee J, Lee E J, Hwang T Y, Kim J and Choa Y H 2020 Sci. Rep. 10 1
Bhandari C, Flatté M E and Paudyal D 2021 Phys. Rev. Mater. 5 9
Jalili H, Aslibeiki B, Varzaneh A G and Chernenko V A 2019 Beilstein J. Nanotechnol. 10 1348
Katlakunta S, Meena S S, Srinath S, Bououdina M, Sandhya R and Praveena K 2015 Mater. Res. Bull. 63 58
Vinnik D A, Klygach D S, Zhivulin V E, Malkin A I, Vakhitov M G, Gudkova S A et al 2018 J. Alloys Compd. 755 177
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Verma, S., Singh, A., Godara, S. et al. Ca-induced changes in the crystal structure and magnetic properties of barium hexaferrite. Bull Mater Sci 46, 196 (2023). https://doi.org/10.1007/s12034-023-03031-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12034-023-03031-0