
Abstract
Structural, mechanical and optoelectronic features of zinc-blende Cd1−x−yZnxHgySe quaternary alloys as well as allied binary compounds and ternary alloys have been investigated through first-principle calculations. Computed elastic stiffness constants ensure that each specimen is mechanically stable, ductile, elastically anisotropic and compressible. The covalent bonding plays a superior role over ionic bonding in each specimen. The phonon dispersion spectra ensure that each binary compound is dynamically stable, while each ternary or quaternary alloy exhibits dynamical instability. Each semiconductor alloy exhibits a direct (Γ–Γ) bandgap. Electrons possess lower effective mass compared to holes. Electrons excited from the Se-4p state of valence band to Zn-5s, Cd-6s and Hg-7s states of conduction band contribute peaks in dielectric function spectra of the considered specimens. Computed oscillator strength of quaternary alloys reveals availability of adequate number of electrons in respective conduction bands. On the basis of computed optoelectronic properties, the alloys would be suitable to fabricate near-UV and UV optoelectronic devices.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Tuning of physical properties of some elemental or compound semiconductors through formation of alloys is necessary in materials engineering to get the desired properties for fabricating a specific device. Formation of quaternary alloys of a set of more than two compounds is treated as advanced procedure in such tuning process compared to that of ternary alloys, where properties of only a couple of compounds can be tuned. In the last couple of decades, AxByC1−x−yD quaternary alloys have been extensively designed for diverse target-oriented applications. They are enclosed with three systems AxByC, AxC1−xD and ByC1−yD of ternary alloys and synthesized with three binary compounds AD, BD and CD, which should be selected appropriately to achieve the desired goal.
The binary compounds under group IIB-VIA, the zinc-blende (B3) diatomic cadmium, zinc and mercury selenides CdSe, ZnSe and HgSe are treated as very important optoelectronic materials [1]. Experiments on CdSe and ZnSe reveal that they possesses wide and direct-bandgap (Γ–Γ) semiconductor properties. In contrast, different experiments have suggested that HgSe possess zero-gap [2], negative bandgap [3] and marginally positive fundamental bandgap [4]. Their mixed ternary and quaternary alloys, thin films and miscellaneous nanostructures have diversified photovoltaic, optoelectronic, spintronic and luminescence applications [5,6,7,8,9,10,11,12,13,14].
In the past several decades, diverse properties of CdSe, ZnSe and HgSe have been investigated experimentally [15,16,17,18,19,20,21,22,23,24,25,26]. Such properties were also studied theoretically through several first-principle calculations [27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45]. In addition, thin films of CdZnSe ternary alloys [46], Zn1−xCdxSe/ZnSe heterostructures [47], quantum dots [48,49], ZnCdSe/ZnSe single-quantum wells [50], Zn1−xCdxSe/MgSe multiple quantum wells [51], etc. were prepared and their optoelectronic properties were characterized experimentally. Gas source molecular beam epitaxial growth of wide-bandgap Zn1−xHgxSe alloys [52] was investigated. Band structure of Hg1−xCdxSe mixed crystals was also investigated from the inter-band magneto-absorption experiment [53].
On the theoretical side, several significant calculations on Cd and Zn chalcogenides based on diverse clusters, mono-layers and quantum-dots have been performed so far [54,55,56,57,58,59]. Density-functional calculations of structural and electronic properties of stoichiometric zinc-blende, wurtzite and rocksalt CdSe clusters as well as nonstoichiometric zinc-blende CdSe clusters [54] and later the structural, electronic and optical properties of ligand-free ZnmSen clusters [55] were performed. The effect of surface passivation on structural, electronic and optical properties of stoichiometric ZnnSen clusters were studied theoretically [56]. Self-consistent charge density-functional tight-binding method was applied to explore tight-binding parameters for Zn-X (X = Zn, O, S, Se, Te, Cd, H, C and N) for large-scale quantum-mechanical simulation of various zinc-chalcogenides nanostructures [57]. Using quantum confinement, controlling of biexciton recombination in quasi-type-I and II CdSe/CdS core/shell spherical quantum dots were studied theoretically [58]. First-principle calculations were also carried out to investigate stability and electronic structure of two-dimensional CP3 monolayer to find the area of their potential applications [59]. Moreover, optoelectronic features of bulk CdZnSe [60], HgCdSe [61] and HgZnSe [62,63] ternary specimens were also investigated from first-principle calculations.
It is to be noted that none of the experimental or theoretical investigation is carried out on any of the said features of Cd1−x−yZnxHgySe quaternary alloys to search their probable applications. Therefore, experimental or theoretical studies on structural, elastic and optoelectronic characteristics of cubic Cd1−x−yZnxHgySe quaternary specimens would show a new horizon in their diversified mechanical and optoelectronic applications. Motivated by these facts, we have carried out the present calculations, which is the introductory initiative for exploring the said characteristics of zinc-blende Cd1−x−yZnxHgySe quaternary specimens. The same for the allied zinc-blende binary CdSe, ZnSe and HgSe as well as CdZnSe, HgCdSe and HgZnSe ternary specimens are also calculated. Change in structural, mechanical, electronic and optical features in Cd1−x−yZnxHgySe triangular quaternary system within Zn concentration (x) at each considered Hg concentration (y) and vice versa are computed elaborately.
2 Method of calculations
All-electron density functional theory (DFT) [64,65] based FP-LAPW [66] calculations is carried out with the WIEN2K code [67]. The Phonopy code [68] is employed to investigate dynamical stability/instability of the cubic crystals with computation of respective phonon dispersion spectra. The cubic-elastic code [69] is employed to calculate elastic properties of the cubic crystals.
The PBE-GGA [70] is applied in structural and elastic properties, while mBJ-GGA [71] and PBE+U [72,73] methods are applied in electronic and optical properties to compute exchange-correlation (XC) potentials. Electronic features with PBE-GGA are modified with Coulomb repulsion between valence d-electrons of transition elements through Hubbard parameter U [74] in the PBE+U approach. At U = 10.0, 8.0 and 6.0 eV, calculated bandgap of ZnSe, CdSe, and HgSe and their respective experimentally measured data are observed to be closest to each other during the variation of respective calculated bandgaps with U. We have selected them as U for 3d, 4d and 5d electrons of zinc, cadmium and mercury atoms, respectively, in case of calculations of electronic and optical features with PBE+U.
On the basis of the muffin-tin (MT) model, the FP-LAPW approach considers that atoms exist inside the MT spheres. The wavefunction in this region is expanded with spherical harmonics by selecting lmax = 10 as the largest angular momentum. Considering largest cut-off wave vector Kmax = 8.0/RMT and magnitude of largest vector Gmax = 16 Ry1/2 in the Fourier expansion of the charge density, the plane wave basis is used to expand the wavefunction in the interstitial region. The selected MT-sphere radius holding cadmium, zinc, mercury and selenium atoms as 2.4, 2.3, 2.5 and 2.3 a.u., respectively, ensures non-overlapping atomic spheres as well as preventing any charge leakage from the respective core. The optimum k-mesh is selected as 10 × 10 × 10 by varying total energy as a function of the number of k-points. The core states are separated from valence states with a core-cut-off energy –6.0 Ry. The fully relativistic approximation for core, but scalar relativistic approximation for valence electrons is employed. Total energy convergence is acquired through self-consistent-field approximation. In such an approximation, the total energy difference 10−5 Ry between two consecutive steps is selected to be the threshold for the termination of iterations.
The designing processes of diverse unit cells of the specimens under the Cd1−x−yZnxHgySe system have been described in the Section-I of the supplementary materials. For visualization of designed crystal structures, the graphic code XCrySDen [75] is used. Optimized zinc-blende crystal structures of Cd0.50Zn0.25Hg0.25Se, Cd0.25Zn0.25Hg0.50Se and Cd0.25Zn0.50Hg0.25Se quaternary alloys are presented as supplementary figure S1a–c, respectively, in the supplementary materials.
3 Results and discussions
3.1 Structural properties
3.1a Lattice constant and bulk modulus a0 and B0: The optimized lattice constant (a0) and bulk modulus (B0) for each designed unit cell are evaluated from fitting of self-consistently computed respective energy–volume curve to Murnagran’s equation of state [76]. Computed a0 and B0 are presented in table 1. In the case of diatomic specimens, excellent closeness between computed a0 and B0 and respective experimental data are observed [1,17]. They also agree fairly well with a few respective earlier theoretically calculated data [27,28,29,30,35,39,62]. Structural data for any type of alloy does not exist in the literature.
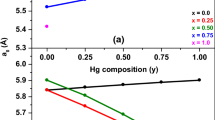
3.1b Concentration dependence of lattice constant and bulk modulus: Figure 1a displays the variations of computed a0 as function of Hg-concentration (y) at each Zn-concentration (x) and vice-versa, while figure 1b displays the same of computed B0 in the Cd1−x−yZnxHgySe triangular quaternary system. Here, a0 increases almost linearly in figure 1a, while B0 decreases nonlinearly in figure 1b with increasing y at any particular x. We have observed a completely reverse picture with increasing x at any particular y. Repeated replacement of cadmium atom(s) of lower diameter (0.322 nm) with mercury atom(s) of relatively bigger diameter (0.342 nm) causes the increase in a0 in the former. In contrast, successive replacement of cadmium atom(s) of bigger diameter (0.322 nm) with zinc atom(s) of relatively smaller diameter (0.284 nm) reduces a0 in the latter.

(a) Hg-composition (y) dependence curves of computed a0 for the compounds under Cd1−x−yZnxHgySe quaternary system. (b) Hg-composition (y) dependence curves of computed B0 for the compounds under Cd1−x−yZnxHgySe quaternary system. (c) Composition (x/y) dependence curves of computed a0 for the ternary alloys. (d) Composition (x/y) dependence curves of computed B0 for the ternary alloys.
Figure 1c shows decrease in a0 with increasing x in the ZnxCd1−xSe system and with increasing x or reducing y (= 1–x) in the ZnxHgySe/ZnxHg1−xSe system due to replacement of larger cadmium and mercury, respectively, with smaller zinc atom(s). Also, a0 enhances with increasing y in the HgyCd1−ySe system due to the replacement of smaller cadmium with larger mercury atom(s). The compositional variation of a0 is opposite to that of B0 in any of the aforesaid variations in figure 1d because change in B0 is inversely proportional to that of equilibrium volume V0 = \({a}_{0 }^{3}\) of any cubic cell.
Figure 1c and d shows slight deviation of composition dependence curves of a0 and B0, respectively, from the corresponding linear dependence curve [77] for the ZnxCd1−xSe, HgyCd1−ySe and ZnxHg1−xSe systems. The following quadratic equations represent such marginal nonlinearity in a0(x) or a0(y) curve for each ternary system
Here, lattice constant of ZnSe, CdSe and HgSe is \({a}_{0 }^{\mathrm{ZnSe}}\), \({a}_{0 }^{\mathrm{CdSe}}\) and \({a}_{0 }^{\mathrm{HgSe}}\), respectively. Also, \({b}_{\mathrm{ZnCdSe }}^{a}\), \({b}_{\mathrm{HgCdSe} }^{a}\) and \({b}_{\mathrm{ZnHgSe} }^{a}\) is the bowing parameter in composition dependence curve of lattice constant for the ZnxCd1−xSe, HgyCd1−ySe and ZnxHg1−xSe ternary system, respectively. Similar quadratic equations also represent the marginal nonlinearities in B0(x) or B0(y) curves for the three ternary systems. In this case, \({b}_{\mathrm{ZnCdSe} }^{B}\), \({b}_{\mathrm{HgCdSe} }^{B}\) and \({b}_{\mathrm{ZnHgSe} }^{B}\) are treated as bowing parameter in the composition dependence curve of bulk modulus for ZnxCd1−xSe, HgyCd1−ySe and ZnxHg1−xSe ternary system, respectively.
The a0 vs. composition (x or y) curve for ZnxCd1−xSe and ZnxHg1−xSe systems show marginally upward deviation, while HgyCd1−ySe system shows marginally downward deviation from respective linear variation as per Vegard’s law [77]. In contrast, the compositional (x or y) variation of the B0 curve in case of each of the ZnxCd1−xSe, ZnxHg1−xSe and HgyCd1−ySe systems shows very small upward deviation from the corresponding linear variation. From quadratic fit of each of the a0(x) or a0(y) curve, calculated lattice constant bowing parameter is found as \({b}_{\mathrm{ZnCdSe} }^{a}\)= –0.1223 Å, \({b}_{\mathrm{ZnHgSe} }^{a}\)= –0.1611 Å and \({b}_{\mathrm{HgCdSe} }^{a}\)= 0.0137 Å for the ZnxCd1−xSe, ZnxHg1−xSe and HgyCd1−ySe system, respectively. From the quadratic fit of the B0(x) or B0(y) curve, the calculated bulk modulus bowing parameter is \({b}_{\mathrm{ZnCdSe} }^{B}\)= –1.206 GPa, \({b}_{\mathrm{HgCdSe} }^{B}\)= –2.618 GPa and \({b}_{\mathrm{ZnHgSe} }^{B}\)= –0.7669 GPa for the ZnxCd1−xSe, ZnxHg1−xSe and HgyCd1−ySe system, respectively.
3.2 Thermodynamic and dynamical stability/instability
3.2a Thermodynamic stability/instability: Thermodynamic stability/instability of a crystalline alloy can be investigated through calculation of its formation energy. The energy of formation of AxB1−xC (i.e., ZnxCd1−xSe, ZnxHg1−xSe and HgyCd1−ySe) ternary alloys and for Cd1−x−yZnxHgySe (i.e., A1−x−yBxCyD) quaternary alloys are calculated by the relations, given below
Clearly, \({E}_{0 }^{{A}_{x}{B}_{1-x}C}\) indicates total energy of any of the said ternary alloys in equation (4), while \({E}_{0 }^{{A}_{1-x-y}{B}_{x}{C}_{y}D}\) is the total energy of the Cd1−x−yZnxHgySe quaternary specimens at any composition pair (0.25, 0.25), (0.25, 0.50) and (0.50, 0.25) in equation (5). The \({E}_{0 }^{AC}\), \({E}_{0 }^{BC}\), \({E}_{0 }^{BD}\), \({E}_{0 }^{CD}\) and \({E}_{0 }^{AD}\) are the total energy of any of the diatomic ZnSe, HgSe and CdSe, involved in each ternary or quaternary system. Computed formation energy vs. compositions (x, y) contour plots for all the alloys of the Cd1−x−yZnxHgySe system are presented in figure 2. The formation of any thermodynamically stable ternary alloy at each x or y = 0.25, 0.50, 0.75 as well as quaternary alloy at (0.25, 0.25), (0.25, 0.50) and (0.50, 0.25) composition pair is ensured with negative value of formation energy, calculated using equations (4) and (5), respectively.

Computed formation energy vs. compositions contour plot for the ternary and quaternary alloys under the Cd1−x−yZnxHgySe system.
3.2b Dynamical stability/instability: The dynamical stability or instability of any specimen allied to the Cd1−x−yZnxHgySe system is investigated by calculating respective phonon dispersion spectra. The computed phonon dispersion spectra for CdSe, ZnSe and HgSe in the W-L-Γ-X-W-K direction of high symmetry are presented in figure 3a–c, respectively. The same for Cd0.50Zn0.25Hg0.25Se, Cd0.25Zn0.25Hg0.50Se and Cd0.25Zn0.50Hg0.25Se quaternary alloys along the R-Γ-X-M-Γ direction are presented in figure 4a–c, respectively. For x or y = 0.25, 0.50 and 0.75, the calculated phonon dispersion spectra for the ternary alloys under the ZnxCd1−xSe, HgyCd1−ySe and ZnxHg1−xSe systems are presented as supplementary figures S2a–c, S3a–c and S4a–c, respectively.

Computed phonon dispersion spectra of zinc-blende binary: (a) CdSe, (b) ZnSe and (c) HgSe.

Computed phonon dispersion spectra of zinc-blende: (a) Cd0.50Zn0.25Hg0.25Se, (b) Cd0.25Zn0.25Hg0.50Se and (c) Cd0.25Zn0.50Hg0.25Se quaternary alloy.
Clearly, the dynamical stability of zinc-blende CdSe, ZnSe and HgSe under the ambient conditions are confirmed because no imaginary/negative frequency is present in the respective phonon dispersion spectra. In contrast, the presence of negative frequencies in the phonon dispersion spectra of each ternary or quaternary alloy under the same conditions is the signature of respective dynamical instability.
3.3 Elastic properties
3.3a Elastic stiffness constants (Cij) of cubic specimens: Generally, 36 independent elastic stiffness constants are associated with any crystal. But the lattice symmetry due to identical axes in a cubic crystal reduces the number to three only. The \(C_{11}\) is associated with elasticity along the length, while both the \(C_{12}\) and \(C_{44}\) are associated with the elasticity related to the shape of a cubic lattice.
In tables 1–3, calculated elastic stiffness constants (C11, C12 and C44) and related mechanical features of all the cubic specimens are available. Tables 1–3 also contain experimentally measured and previously calculated data of some of the parameters of binary compounds for comparison, while any such data for ternary and quaternary alloys do not exist in the literature. For each specimen, the observed trend is C11 > C12 > C44 in table 1, but it could be validated only with experimental data for HgSe [15,16,18,19] and ZnSe [17,20] as well as previous calculated data for CdSe [29,30,31]. Each specimen satisfies Born’s mechanical stability criteria for a cubic crystal [69] and hence exhibits mechanical stability. Inequality in C44 and calculated shear constant C/= (C11–C12)/2 with C44 > C/ in table 1 has established elastic anisotropy of each cubic crystal [69]. Calculated constants C11, C12 and C44 reduce, in figure 5a–c, respectively, in the Cd1−x−yZnxHgySe system with increasing mercury concentration (y) at each selected zinc concentration (x). Conversely, any of them enhances with increasing zinc concentration (x) at each selected mercury concentration (y). In the case of HgSe and ZnSe, computed C11, C12 and C44 and the corresponding experimentally observed data are extremely close to each other [15,16,17,18,19,20]. The same for CdSe shows proximity to some corresponding earlier calculated data [29,30,31,32,33].

Hg-composition (y) dependence curve of computed (a) C11, (b) C12, (c) C44 for the compounds under Cd1−x−yZnxHgySe quaternary system.
3.3b Elastic moduli, constants and isotropy/anisotropy: Directly or indirectly in terms of C11, C12 and C44, we have computed shear modulus (G) proposed by Voigt (GV), Reuss (GR) and Hill (GH) along with bulk modulus (B0) and Young’s modulus (Y) of each cubic crystal. Larger value of each of them of a material is a signature of its superior rigidity and vice versa [69]. Calculated diverse shear modulus for all the considered specimens are also included in table 1, but B0 and Y are included in table 2. In the case of our considered specimens, the minimum stiffness is shown by HgSe and maximum by ZnSe. Again, C11 > B0 > C12 for each specimen in tables 1 and 2 is indicating respective mechanical stability [69]. Computed B0 of each specimen from the energy–volume curve and that with corresponding C11 and C12 are nearly equal. Any of the calculated GV, GR, GH, B0 and Y in the Cd1−x−yZnxHgySe system reduces, as shown in supplementary figure S5a–c, respectively, with growing mercury concentration (y) at each selected zinc concentration (x). Conversely, each of them enhances with growing zinc concentration (x) at each selected mercury concentration (y).
The ratio B0/C44 calculates the plastic behaviour [78], while both Cauchy’s pressure (C// = C12–C44) and Pugh’s ratio (B0/GH) calculate the ductile or brittle behaviour of a material [79]. The calculated B0/C44, B0/GH and C// for each specimen are presented in table 2. The B0/C44 >1.5 indicate that all the specimens are plastic. The computed B0/GH > 2.1 and the positive sign of C// indicate their ductility [69]. In supplementary figure S6, the correlation between C// and B0/GH again confirms ductility of all the specimens.
The Poisson’s ratio (σ) is calculated with GH and B0 of a material [69]. For a cubic specimen, Kleinman parameter (ξ) of a cubic crystal is evaluated by C11 and C12 [69]. In table 2, calculated σ in the range 0.300–0.343 ensures that each specimen is highly compressible, ductile, plastic and contains a central type of inter-atomic bonding force [69]. The calculated ξ in the range 0.704–0.739 ensure that bending plays major role over stretching in chemical bonds in all the specimens [69]. In terms of C11, C12 and C44, computed Zener anisotropy factor (AZ) with AZ ≠ 1 stands for a varying mode of anisotropy, while AZ = 1 stands for total isotropy of a crystal [69]. The computed AZ > 1.9 for all the considered specimens in table 2 is a clear indication of elastically anisotropic nature with the largest inflexibility along the <111> diagonal of the cubic crystal [69].
3.3c Bonding characteristics: The C11/C12 having values 1.0, 2.0, and in between them indicate totally ionic, totally covalent and a mixture of covalent and ionic bonding, respectively, in a zinc-blende crystal [80]. Computed C11/C12 in between 1.562 and 1.676, presented in table 2, is indicating mixture of covalent and ionic bonding with the supremacy of the former in any specimen. The Phillips ionicity (fi) for all the zinc-blende crystals is calculated in terms of respective C11 and C12 [79] and presented in table 3. They are found in the range 0.632–0.685. For CdSe, HgSe and ZnSe, computed fi is extremely close to the corresponding experimentally measured fi for CdSe [22], HgSe [23] and ZnSe [22].
3.3d Acoustic phonon velocities and thermal properties: In a crystal, velocities of longitudinal (vl) and transverse (vt) acoustic phonon waves are calculated with GH and B0 of the crystal, while that for mean acoustic phonon wave (vm) is calculated with vl and vt [79]. The Debye temperature (ΘD) differentiates the quantum and classical characteristics of phonons in a solid. Since acoustic phonon vibrations result in their vibrational excitations at low temperatures, the ΘD is computed in terms of vm at T<ΘD [79]. In the thermal transport phenomenon, the Debye frequency (ωD) of a solid is the uppermost boundary of frequency of phonon vibration and it is calculated as a function of ΘD [81]. The minimum thermal conductivity of a material (Kmin) is calculated with mean acoustic phonon wave velocity (vm) using an empirical relationship [82]. Another empirical formula has been suggested to calculate the melting temperature (Tm) of a cubic material in terms of its C11 [83]. Table 3 includes computed vl, vt, vm, ΘD, ωD, Kmin and Tm for all the considered specimens. The computed vl and vt for HgSe show fair closeness to the respective earlier calculated data [37,42]. The calculated ΘD for HgSe shows fair closeness to the corresponding experimental data [16]. Table 3 again indicates that calculated Kmin for HgSe exists within the experimentally investigated range [16]. The computed Tm for HgSe exhibits fair closeness to the corresponding experimentally investigated data [16]. In the Cd1−x−yZnxHgySe system, supplementary figure S7a–d, displays reduction in computed ΘD, ωD, Kmin and Tm, respectively, with growing Hg concentration (y) at any particular Zn concentration (x). In contrast, each of them shows enhancement with growing Zn concentration (x) at any particular Hg concentration (y).
In most of the cases, we have observed that if a crystal system shows dynamical stability, it also shows mechanical stability. If a crystal system exhibits both mechanical and dynamical stability in a particular crystallographic phase, then it is the stable phase of that crystal. But in a particular phase, if a crystal system shows both mechanical and dynamical instability then it is the unstable phase of that crystal and those systems are insignificant to study.
On the other hand, if a crystal system is mechanically stable but dynamically unstable and vice-versa in a particular phase, then it can be treated as a possible meta-stable phase of the crystal. In our case, all the ternary and quaternary specimens investigated are mechanically stable but dynamically unstable so that zinc-blende is their meta-stable phase.
Dynamical instability takes place at a non-zero critical frequency and involves all the mechanical problems in which the time is included. In the equilibrium state, this is subjected to a dynamic loading on structural–mechanical systems either by sudden shock and impact loading or hydro-elastic forces or pulsating parametric forces. In this case, the sudden instabilities of periodic motions can occur when a structural system is loaded by periodic excitation, such as vibration of atoms in crystals, etc. So, it is basically a highly multidisciplinary problem, which has an immense impact on diverse fields of materials science and structural engineering. Therefore, though our ternary alloys are dynamically unstable but mechanically stable, we have studied the electronic and optical properties of these meta-stable zinc-blende ternary and quaternary alloys.
3.4 Electronic properties
3.4a Band structure and bandgap: In case of the quaternary system Cd1−x−yZnxHgySe, though mBJ-GGA caters band structure profile of any alloy with direct bandgap (Γ–Γ) qualitatively similar to those with PBE+U scheme, the former provides larger bandgap compared to the latter. Since atomic orbitals of all the constituent atoms contribute to the band structure of only the Cd0.50Zn0.25Hg0.25Se, Cd0.25Zn0.25Hg0.50Se and Cd0.25Zn0.50Hg0.25Se quaternary alloys, their mBJ-GGA oriented band structures are presented as supplementary figure S8a–c, respectively. With both the potential schemes, calculated minimum bandgap (Eg) of each specimen is given in table 4.
Calculations show direct bandgap semiconductor (Γ–Γ) features of CdSe and ZnSe irrespective of employed potential scheme and it is in concordance with respective experimental observations [1,24]. Though the computed fundamental bandgap (Eg) of CdSe and ZnSe with mBJ-GGA is almost identical to the respective experimental data [1,24], the same with PBE+U is smaller than the respective experimental data [1,24]. The HgSe under the mBJ-GGA scheme exhibits semiconductor character with narrow and direct bandgap (Γ–Γ). In contrast, the HgSe under the PBE+U scheme shows its metallic nature with coinciding valence band maximum and conduction band minimum along the high symmetry line (Γ–Γ). Though the former character of HgSe is supported by an experiment [4] and the latter one by another [3], calculations could not establish its experimentally observed semi-metal character with zero bandgap [2]. The discrepancy between mBJ-GGA based positive Eg and the corresponding experimental data is 0.363 eV [4], while the same between PBE+U based computed negative Eg and corresponding experimental data is nearly –0.12 eV [1,25].
From the aforesaid discussion, it is clear that though mBJ-GGA induced band structure and bandgap of CdSe and ZnSe are extremely close to the respective experimental observations, the same for the HgSe completely contradicts the corresponding experimental observation and its bandgap is found to be marginally positive. On the other hand, the PBE+U scheme is able to incorporate the interactions between localized d-electrons of transition metal elements and hence improve the accuracy of calculated electronic properties. In the present study, the PBE+U is successfully able to provide correct band structure and accurate bandgap of all the binary specimens. The PBE+U induced bandgaps of CdSe and ZnSe are only 0.132 and 0.197 eV, respectively, smaller than the corresponding mBJ-GGA induced data. The PBE+U also possesses a negative fundamental bandgap of HgSe, which agrees excellently well with the corresponding experimental observation. Moreover, though the qualitative nature of band structures of each ternary/quaternary alloy with both the potential schemes are almost identical, we have observed marginal discrepancy between the PBE+U and mBJ-GGA based computed bandgap data for each alloy. Therefore, the PBE+U potentials are able to show better performance compared to mBJ-GGA scheme in describing electronic properties of all the specimens within the Cd1−x−yZnxHgySe system.
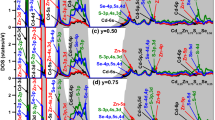
3.4b Density of states: Calculations of density of states (DOS) for each quaternary alloy, especially the total (TDOS) for an alloy and the partial (PDOS) for its constituent atoms help us to achieve information regarding involvement of atomic orbitals constituting diverse electronic levels in different energy segments of respective band profile. Since calculated total and partial DOS profiles for mBJ-GGA and PBE+U become identical, the same for Cd0.50Zn0.25Hg0.25Se, Cd0.25Zn0.50Hg0.25Se and Cd0.25Zn0.25Hg0.50Se quaternary alloys with the former potential scheme are presented as supplementary figure S9a–c, respectively. Electronic excitations between diverse electronic levels of valence and conduction band adjacent to Fermi level of band structure of a material are responsible for its different optical features. Therefore, the analyses of TDOS and hence PDOS of these regions have utmost importance. Close to the Fermi level, valence electronic states in the region from –2.5 to 0.0 eV comprises mostly Se-4p and marginally of Zn-3p, 3d; Cd-4p, 4d; Hg-5p, 5d states. The same in the conduction band region from 0.0 to 5.0 eV comprises mostly of Hg-7s, Zn-5s, Cd-6s and Se-4p, 5s, 4d states. Therefore, diverse optical features of considered compounds and alloys are resulting from highly probable electronic excitations from the filled Se-4p valence state to vacant Hg-7s, Cd-6s and Zn-5s states of conduction band nearest to the Fermi level.
3.4c Concentration dependence of bandgap: In the case of Cd1−x−yZnxHgySe system and with mBJ-GGA and PBE+U, reduction in calculated Eg with growing mercury concentration (y) at each considered zinc concentration (x) are displayed in figure 6a and b, respectively. But, calculated Eg with both the potential schemes enhances with growing zinc concentration (x) at any particular mercury concentration (y). Calculated Eg nonlinearly increases with growing zinc concentration (x) in ZnxCd1−xSe and ZnxHg1−xSe ternary systems, but reduces in the HgyCd1−ySe ternary system with growing mercury composition (y). For mBJ-GGA and PBE+U schemes, such variations in the ternary systems are presented in figure 6c and d, respectively. In case of these ternary systems, the following quadratic equations express the nonlinear variation in Eg(x) or Eg(y) curve

(a) Hg-composition (y) dependence curves of mBJ-GGA based computed Eg for the compounds under Cd1−x−yZnxHgySe quaternary system. (b) Hg-composition (y) dependence curves of PBE+U based computed Eg for the compounds under Cd1−x−yZnxHgySe quaternary system. (c) Composition (x or y) dependence curves of mBJ-GGA based computed Eg for the ternary alloys. (d) Composition (x or y) dependence curves of PBE+U based computed Eg for the ternary alloys.
In the ZnxCd1−xSe system, the Eg(x) curve shows marginal downward deviation from its ideal linear variation and calculated bandgap bowing parameter is \({b}_{\mathrm{g}}^{\mathrm{ZnCdSe}}\) = 0.1554 eV with mBJ-GGA and 0.0709 eV with PBE+U scheme. In contrast, marginal upward deviation from ideal linear variation with any XC potential is observed in the Eg(y) curve of the HgyCd1−ySe as well as in the Eg(x) curve of the ZnxHg1−xSe system. Our calculated mBJ-GGA based bandgap bowing parameter for HgyCd1−ySe and ZnxHg1−xSe system is \({b}_{\mathrm{g}}^{\mathrm{HgCdSe}}\)= – 0.2286 eV and \({b}_{\mathrm{g}}^{\mathrm{ZnHgSe}}\)= – 0.3497 eV, respectively, while the same with the PBE+U potentials is \({b}_{\mathrm{g}}^{\mathrm{HgCdSe}}\)= – 0.1029 eV and \({b}_{\mathrm{g}}^{\mathrm{ZnHgSe}}\)= – 0.1760 eV, respectively.
3.4d Effective mass of charged particles: The mass (m*) of an accelerated charged particle due to periodically varying potential is called its effective mass. Therefore, free-particle mass (m0) and effective mass (m*) of a particle are completely different from each other. It provides information regarding the nature of dominant carriers (electron or hole) accountable for transport properties exhibited by a semiconductor. Table 5 contains computed effective mass of electrons (\({m}_{e}^{*}/{m}_{0}\)), heavy-hole (\({m}_{\mathrm{hh}}^{*}/{m}_{0}\)), and light hole (\({m}_{\mathrm{lh}}^{*}/{m}_{0}\)) of each specimen (in the unit of m0). We have observed m*(mBJ-GGA)>m*(PBE+U) in the case of each specimen. Clearly, \({m}_{\mathrm{hh}}^{*}/{m}_{0}\) and \({m}_{\mathrm{lh}}^{*}/{{m}_{0}}\) of any sample is larger than the corresponding \({m}_{\mathrm{e}}^{*}/{m}_{0}\). Therefore, the transport phenomenon in any considered specimen is due to the predominant role of electrons compared to the holes. In the Cd1−x−yZnxHgySe system, \({m}_{\mathrm{e}}^{*}/{m}_{0}\), \({m}_{\mathrm{lh}}^{*}/{m}_{0}\) and \({m}_{\mathrm{hh}}^{*}/{m}_{0}\) reduces with growing mercury concentration (y) at each considered zinc concentration (x). On the other hand, each of them enhances with growing zinc concentration (x) at any particular mercury concentration (y). We have displayed such variations with mBJ-GGA as supplementary figure S10a–c and the same with PBE+U as supplementary figure S10d–f, respectively.
3.5 Optical properties
3.5a Frequency dependence of diverse optical parameters: Optical characteristics of each specimen allied to the Cd1−x−yZnxHgySe system are analysed by calculating mBJ-GGA and PBE+U based frequency response spectra of different optical parameters up to 30.0 eV of incident photon energy. The optical properties of any material are analysed mainly in terms of its complex dielectric function \(\varepsilon (\omega )\) and it provides complete behaviour of any compound during its interaction with the incident electromagnetic radiation. The expressions for real part of dielectric function ε1(ω), imaginary part of dielectric function ε2(ω), refractive index n(ω), extinction coefficient k(ω), normal incidence reflectivity R(ω), optical conductivity σ(ω), optical absorption coefficient α(ω), electron energy loss function L(ω) and optical transmission coefficient T(ω), used in the present study, are provided in Section-II of supplementary materials.
The real part \(\varepsilon_{1} (\omega )\) describes the dispersion and the imaginary part \(\varepsilon_{2} (\omega )\) describes the energy absorption of the incident electromagnetic radiation by the material. A material behaves as an optically rarer medium when its refractive index is greater than unity. It behaves as fully transparent material when the refractive index becomes unity. The negative value of refractive index makes a material optically denser and hence behaves as an opaque material. A material having higher extinction or attenuation coefficient offers higher resistance to the incident photons to penetrate through the material and vice versa. For calculation of reflection losses from optical instruments, it is necessary to calculate the reflectivity at normal incidence. The normal incidence reflectivity depends upon refractive indices of two mediums. Optical conductivity is the property of a material that gives the relationship between the induced current density in the material and the magnitude of the inducing electric field for arbitrary frequencies. Therefore, significantly high optical conductivity of a material implies its metallic behaviour and vice versa. The optical absorption coefficient \(\alpha (\omega )\) is the measurement of the length penetrated by an incident photon inside a semiconductor material before its complete absorption when its associated energy is greater than roughly the electronic bandgap of the material. Electron energy loss function of a material is useful in calculating energy loss due to inelastic scattering of electrons by atoms in a lattice and its frequency response is opposite to the \(\varepsilon_{2} (\omega )\) of that material. Optical transmission refers to the proportion of light that is transmitted through an optically clear medium. Applications such as fibre-optic communications, optoelectronic sensors and photonics require the highest levels of transmission.
For Cd0.50Zn0.25Hg0.25Se, Cd0.25Zn0.25Hg0.50Se and Cd0.25Zn0.50Hg0.25Se quaternary alloys, the variation of ε1(ω), ε2(ω), n(ω), k(ω), with frequency of the incident radiation under the mBJ-GGA are displayed as figure 7a–d and with PBE+U as figure 7e–h, respectively. On the other hand, variation of R(ω), σ(ω), α(ω), L(ω) with frequency of the incident radiation under the mBJ-GGA are displayed as figure 8a–d and with PBE+U as figure 8e–h, respectively. Moreover, dependence of each optical parameter on wavelength (λ) of the incident photon energy is displayed as inset in the corresponding frequency dependence spectra. They exhibit contributions of these optical parameters in the UV–Vis and IR regions. Mutual frequency dependence of transmission coefficient T(ω) and absorption edge α(ω) in case of quaternary alloys under mBJ-GGA and PBE+U are displayed in figure 9a and b, respectively. Diverse optical constants of each specimen are calculated and presented. Though frequency dependence of any optical feature of a specimen is qualitatively similar to that with PBE+U, they cater quantitatively to different optical constants.

Computed frequency response spectra of (a) ε1(ω), (b) ε2(ω), (c) n(ω), (d) k(ω) with mBJ-GGA and (e) ε1(ω), (f) ε2(ω), (g) n(ω), (h) k(ω) with PBE+U for the quaternary alloys. The variation of each optical parameter with wavelength is displayed as inset.

Computed frequency response spectra of (a) R(ω), (b) σ(ω), (c) α(ω), (d) L(ω) with mBJ-GGA and (e) R(ω), (f) σ(ω), (g) α(ω), (h) L(ω) with PBE+U for the quaternary alloys. The variation of each optical parameter with wavelength is displayed as inset.

Mutual variation of computed frequency response spectra of transmission coefficient T(ω) and absorption edge α(ω) of the Cd0.50Zn0.25Hg0.25Se, Cd0.25Zn0.50Hg0.25Se and Cd0.25Zn0.25Hg0.50Se quaternary alloys with (a) mBJ-GGA and (b) PBE+U potentials.
The ε1(ω) spectra of each quaternary specimen show peaks and hence prominent dispersion of incident radiation below 5.0 eV, while negative ε1(ω) ensures their metallic character in 7.5–12.0 eV energy region. The ε2(ω) spectra of each quaternary specimen show peaks in the 3.0–9.0 eV energy region and they originate as a result of collective effort of electronic transitions from filled Se-4p valence state to empty Hg-7s, Zn-5s, Cd-6s states of conduction band.
The n(ω) spectra of each quaternary specimen show the largest refractive index n(ω)max in the incident photon energy region below 8.0 eV. The n(ω)max for Cd0.50Zn0.25Hg0.25Se, Cd0.25Zn0.25Hg0.50Se and Cd0.25Zn0.50Hg0.25Se quaternary alloy is calculated as 2.940, 3.109 and 2.978, respectively, with mBJ-GGA, and 3.213, 3.318 and 3.229, respectively, with PBE+U. Above 9.0 eV, the n(ω) for each specimen below unity (c/v<1) is a signature of opacity of each material and it ensures larger group velocity of wave packet through the semiconductor specimens vg = c/n than the velocity of light (c) in free-space [84]. Significantly high k(ω) with peaks is observed in the k(ω) spectra of the quaternary alloys in the energy region 3.0–13.0 eV and the peak value kmax(ω) for all the specimens is very close to 2.0. It indicated that extremely high resistance is offered by these semiconductor specimens to those photons having energy in the 3.0–13.0 eV compared to the photons having energy beyond this range [84].
Considerably high reflectivity [R(ω)>0.20] in the wide energy range 0.0–15.0 eV is observed in the R(ω) spectra of each quaternary specimen. Few peaks exist in this energy region with maximum reflectivity Rmax(ω) > 0.40. We have observed a substantially high σ(ω) having a pair of peaks in the narrow energy range 5.5–8.5 eV in the σ(ω) spectra of the quaternary alloys and it ensures metallic character of quaternary specimens in this energy region. Computed σmax(ω), i.e., largest optical conductivity for the Cd0.50Zn0.25Hg0.25Se, Cd0.25Zn0.25Hg0.50Se and Cd0.25Zn0.50Hg0.25Se specimen with mBJ-GGA are 7290, 7242 and 7050 (Ohm cm)–1, respectively, and with PBE+U are 7000, 7036 and 6951 (Ohm cm)–1, respectively. The α(ω) of each quaternary specimen becomes substantially high with peaks having significant intensity in the region 7.0–10.0 eV in the respective α(ω) spectra. The αmax(ω), i.e., maximum absorption of incident energy by the three quaternary specimens exist in between 150 and 160 cm–1.
It is observed that the frequency responses of the electronic energy loss function L(ω) and ε2(ω) of a specimen are opposite in nature. Each quaternary specimen shows negligibly small L(ω) but significantly high ε2(ω) up to 10.0 eV of energy. Conversely, L(ω) of each specimen becomes significantly high with distinct peaks but ε2(ω) becomes negligibly small in the energy region 12.0–26.0 eV. For each specimen, plasma resonance frequency is the frequency corresponding to the energy at which a peak with maximum intensity in the corresponding L(ω) spectrum occurs.
Figure 9a and b shows significantly high transmission coefficient T(ω) of any quaternary alloy with mBJ-GGA and PBE+U, respectively, in comparison with respective reflection and absorption coefficients in the extremely low energy region. Conversely, significantly, dominating absorption compared to the reflection and transmission is observed in the high-energy region. Nearly equally intense absorption, transmission and reflection take place simultaneously in the 1.0–1.5 eV narrow energy region.
3.5b Optical constants: The static dielectric constant ε1(0), refractive index n(0) and reflectivity R(0) for each specimen are calculated from the corresponding spectra and presented in table 6. Each of them shows strong bandgap dependence. In a semiconductor, the ε1(0) changes approximately as a function of bandgap according to the Penn model [85] in the following way:
Moreover, static refractive index and static reflectivity of a semiconductor are connected with its \({\varepsilon }_{1}(0)\) as \(n\left(0\right)\approx \sqrt{{\varepsilon }_{1}\left(0\right)}\) and \(R\left(0\right)={[1-n\left(0\right)]}^{2}/{[1+n\left(0\right)]}^{2}\). Therefore, a semiconductor with larger minimum bandgap (Eg) possesses a smaller value of static optical constants and vice versa. Calculated static optical constants of the specimens increases according to table 6, while the corresponding Eg reduces in table 4 with growing mercury concentration (y) at each considered zinc concentration (x). A completely reverse picture is observed with growing zinc composition (x) at each particular mercury composition (y). These changes in mBJ-GGA based ε1(0), n(0) and R(0) are presented as supplementary figure S11a–c and with PBE+U as supplementary figure S11d–f, respectively. Moreover, each of the PBE+U based computed optical constants of any specimen is higher than the respective mBJ-GGA based data, while the bandgap with the latter is higher than that with the former. In case of CdSe and ZnSe, available experimentally observed ε1(0) [26] are extremely close to the mBJ-GGA based corresponding calculated data.
In ε2(ω), k(ω), σ(ω) and α(ω) spectrum, critical point energy (Ec) is the minimum incident energy needed to initiate interaction between these parameters and incident radiation in a material. Our calculated Ec for all the specimens are presented in table 7 and each of them shows strong bandgap dependence. Calculated Ec in each of the ε2(ω), k(ω), σ(ω) and α(ω) spectra of the compounds in table 7 and their calculated Eg in table 4 decrease with growing mercury concentration (y) at each considered zinc concentration (x). A completely reverse picture is observed with growing zinc composition (x) at each considered mercury composition (y). These type of variation in mBJ-GGA based computed Ec in the ε2(ω), k(ω), σ(ω) and α(ω) spectra are presented as supplementary figure S12a–d and with PBE+U as supplementary figure S12e–h, respectively. Moreover, the mBJ-GGA based calculated Eg of a specimen and calculated Ec in its any of the said spectra is higher than the PBE+U based respective data. The Kramers–Kronig transformations between ε1(ω) and ε2(ω), between n(ω) and k(ω) as well as the relation σ(ω) = ωε2(ω)/4π and α(ω) = 2ωk(ω)/c [84] authenticate the similarity in the compositional variation of calculated bandgap Eg and that of calculated Ec in the ε2(ω), k(ω), σ(ω) and α(ω) spectra, respectively, and vice versa.
3.5c Oscillator strength: For Cd0.50Zn0.25Hg0.25Se, Cd0.25Zn0.25Hg0.50Se and Cd0.25Zn0.50Hg0.25Se quaternary alloys, it is calculated with the effective number of electrons (Neff) reaching their respective conduction band at the time of any highest occupied molecular orbital–lowest unoccupied molecular orbital optical transition. With mBJ-GGA, variation of oscillator strength (Neff) of each quaternary alloy with incident energy is presented in figure 10. No electron is able to reach the conduction band (Neff = 0) of any quaternary alloy with incident energy smaller than the respective bandgap. However, electrons start reaching the conduction band when incident energy is nearly close to the bandgap of the material. The oscillator strength (Neff) for each specimen sharply increases with increasing incident energy. Beyond 26.0 eV of incident energy, we have observed saturation in the oscillator strength. It is indicating the maximum oscillator strength (Neff) shown by the respective quaternary alloy during an optical excitation. We have observed that at saturation, the available Neff in the conduction band is 40, 41 and 38 for Cd0.50Zn0.25Hg0.25Se, Cd0.25Zn0.25Hg0.50Se and Cd0.25Zn0.50Hg0.25Se quaternary alloy, respectively.

Computed mBJ-GGA based oscillator strength (Neff) vs. incident energy curve for the Cd0.50Zn0.25Hg0.25Se, Cd0.25Zn0.50Hg0.25Se and Cd0.25Zn0.25Hg0.50Se quaternary alloys. The magnified view of variation of Neff with energy of the incident photon is displayed as inset for low energy region 0.0–6.0 eV.
4 Conclusion
The present first-principle calculations on cubic compounds and alloys under the Cd1−x−yZnxHgySe triangular quaternary system show that each of them exhibits thermodynamic and mechanical stability, ductility, elastic anisotropy and compressibility. Majority of covalent character over ionic is observed in the chemical bonding of each specimen. Bending in chemical bonds shows superiority over stretching in each specimen. In each specimen, inter-atomic forces are observed to be central in nature. Though zinc-blende binary compounds are dynamically stable, ternary and quaternary alloys in zinc-blende phase show dynamical instability. Therefore, mechanical stability, but dynamical instability is indicating zinc-blend as a meta-stable phase of the ternary and quaternary alloys. Calculations of Debye temperature, thermal conductivity and melting temperature of each specimen are also a part of the present study. Irrespective of nature of the employed XC potential scheme, ZnSe, CdSe and any kind of alloy are direct (Γ−Γ) fundamental bandgap semiconductor. Computations with PBE+U confirm the metallic character of HgSe, while mBJ-GGA reveals its semiconductor character with narrow and direct (Γ−Γ) bandgap. The mBJ-GGA scheme provides a comparatively larger bandgap of any specimen compared to the PBE+U. Transport phenomenon in any considered specimen is due to the predominant role of electrons having lower effective mass compared to the holes. The peaks having significant intensity in the spectrum of dielectric function of any considered sample originate from Se-4p to Zn-5s, Cd-6s and Hg-7s electronic transitions. Semiconductors with superior bandgap possesses lower static optical constants, while they require greater threshold energies (Ec) to initiate response in the ε2(ω), k(ω), σ(ω) and α(ω) spectra and vice versa. The availability of sufficient amount of electrons in the conduction band of any quaternary alloy during any optical excitation is ensured beyond 26.0 eV of incident energy with respective computed oscillator strength.
Based on calculated mechanical properties, e.g., strength/hardness, ductility, anisotropy, compressibility, etc., the cubic specimens allied to the Cd1−x−yZnxHgySe system would be suitable for load deflection, thermo-elastic stress, etc., related mechanical applications. Computed thermal properties, e.g., thermal conductivity and melting temperature reveal that they would be potential materials for thermal resistant coating, thermal barrier and thermal management-related applications.
Zinc-blende ternary and quaternary semiconductors of the Cd1−x−yZnxHgySe system possess direct fundamental bandgap (Γ–Γ). Optical properties of these alloys are regulated by their respective direct bandgap centred at Γ-point. Transport properties in these alloys are predominantly due to electrons and they exhibit high mobility. Moreover, substantial variation of the fundamental optical absorption edge of these alloys is observed over the near-UV and UV ranges of the electromagnetic spectrum with concentrations x and y. Such striking optoelectronic features properties of these semiconductor alloys would lead them as potential candidates in diverse near-UV and UV optoelectronic applications.
References
Medelung O (ed) 1982 Landolt Bornstein: numerical data and functional relationship in science and technology (Springer)
Von Truchseβ M, Pfeuffer-Jeschke A, Becker C R, Landwehr G and Batke E 2000 Phys. Rev. B 61 1666
Szuszkiewi W 1979 Phys. Status Solidi (b) 91 361
Gawlik K U, Kipp L, Skibowski M, Orłowski N and Manzke R 1997 Phys. Rev. Lett. 78 3165
Wang J and Isshiki M 2006 Wide-bandgap II–VI semiconductors: growth and properties, Springer handbook of electronic and photonic materials (Berlin: Springer) p 325
Hasse M A, Qui J, De Puydt J M and Cheng H 1991 Appl. Phys. Lett. 59 1272
Tamargo M C, Brasil M J S P, Nahory R E, Martin R J, Weaver A L and Gilchrist H L 1991 Semicond. Sci. Technol. 6 A8
Shieh F, Saunders A E and Korgel B A 2005 J. Phys. Chem. B 109 8538
Peng Z A and Peng X G 2001 J. Am. Chem. Soc. 123 183
Xi L F, Chua K H, Zhao Y Y, Zhang J, Xiong Q H and Lam Y M 2012 RSC Adv. 2 5243
Wissmann H, Tran-Anh T, Rogaschewski S and von Ortenberg M 1990 J. Cryst. Growth 201/202 619
Kuno M, Higginson K A, Bonevich J E, Qadri S B, Yousuf M and Mattoussi H 2002 Proc. SPIE 4808 146
Esmaeili-Zare M, Salavati-Niasari M and Sobhani A 2012 Ultrason. Sonochem. 19 1079
Bazarganipour M, Sadri M, Davar F and Salavati-Niasari M 2011 Polyhedron 30 1103
Medelung O (ed) 1996 Landolt Bornstein: semiconductors basic data in science & technology (Berlin: Springer)
Adachi S 2009 Properties of semiconductor alloys (West Sussex: Wiley)
Lee B H 1970 J. Appl. Phys. 41 2988
Lehoczky A, Nelson D A and Whitsett C R 1969 Phys. Rev. 188 1069
Ford P J, Miller A J, Saunders G A, Yourtçu Y K, Furdyna J K and Jaczynski M 1982 J. Phys. C: Sol. Stat. Phys. 15 657
Berlincourt D, Jafee H and Shlozawa L R 1963 Phys. Rev. 129 1009
Cline C F, Dunegan H L and Henderson G W 1967 J. Appl. Phys. 38 1944
Phillips J C 1970 Rev. Mod. Phys. 42 317
Kumazaki K 1976 Phys. Status Solidi A 33 615
Strehlow W H and Cook E L 1973 J. Phys. Chem. Ref. Data 2 163
Einfeldt S, Goschenhofer F, Becker C R and Landwehr G 1995 Phys. Rev. B 51 4915
Manabe A, Mitsuishi A and Yoshinaga H 1967 Jpn. J. Appl. Phys. 6 593
Chen X J, Mintz A, Hu J S, Hua X L, Zinck J and Goddard W A I I I 1995 J. Vac. Sci. Technol. B 13 1715
Heyd J, Peralta J E and Scuseria G E 2005 J. Chem. Phys. 123 174101
Deligoz E, Colakoglu K and Ciftci Y 2006 Physica B 373 124
Ouendadji S, Ghemid S, Meradji H and El Haj Hassan F 2011 Comp. Matr. Sci. 50 1460
Guo L, Zhang S, Feng W, Hu G and Li W 2013 J. Alloys Comp. 579 583
Sharma S, Verma A S, Sarkar B K, Bhandari R and Jindal V K 2011 AIP Conf. Proc. 1393 229
Rabani E 2002 J. Chem. Phys. 116 258
Kamran S, Chen K and Chen L 2008 Phys. Rev. B 77 094109
Ullah N, Murtaza G, Khenata R, Wong K M and Alahmed Z A 2014 Phase Transit. 87 571
Al Shafaay B, El Haj Hassan F and Korek M 2014 Comp. Mater. Sci. 83 107
Duz I, Erdem I, Ozdemir Kart S and Kuzucu V 2016 Arch. Mater. Sci. Eng. 79 5
El Haj Hassan F, Al Shafaay B, Meradji H, Ghemid S, Belkhir H and Korek M 2011 Phys. Scr. 84 065601
Khenata R, Bouhemadou A, Sahnoun M, Reshak A H, Baltache H and Rabah M 2006 Comp. Mater. Sci. 38 29
Casali R A and Christensen N E 1998 Sol. Stat. Commun. 108 793
Bilal M, Shafiq M, Ahmad I and Khan I 2014 J. Semicond. 35 0720011
Varshney D, Shriya S and Khenata R 2012 Mater. Chem. Phys. 135 365
Fleszar A and Hankew, 2005 Phys. Rev. B 71 045207
Zakharov O, Rubio A, Blasé X, Cohen M L and Loui S G 1994 Phys. Rev. B 50 10780
Boutaiba F, Zaoui A and Ferhat M 2009 Superlattices Microst. 46 823
Benkert A, Schumacher C and Brunner K 2007 Appl. Phys. Lett. 90 162105
Tournie E, Ongaretto C, Laugt M and Faurie J P 1998 Appl. Phys. Lett. 72 217
Rafipoor M, Dupont D, Tornatzky H, Tessier M D, Maultzsch J, Tessier M D et al 2018 Chem. Mater. 30 4393
Yeh C-W, Chen G-H, Ho S-J and Chen H-S 2019 Nano Mater. https://doi.org/10.1021/acsanm.9b01213
Sparing L M, Mintairov A M, Hodak J H, Martini I B, Hartland G V, Bindley U et al 2000 J. Appl. Phys. 87 3063
Wu J D, Huang Y S, Li B S, Shen A, Tamargo M C and Tiong K K 2010 J. Appl. Phys. 108 123105
Hara K, Machimura H, Usui M, Munekata H, Kukimoto H and Yoshino J 1995 Appl. Phys. Lett. 66 3337
Mycielski A, Kossutt J, Dobrowolskat M and Dobrowolskit W 1982 J. Phys. C: Sol. Stat. Phys. 15 3293
Sarkar P and Springborg M 2003 Phys. Rev. B 68 235409
Goswami B, Pal S, Sarkar P, Seifert G and Springborg M 2006 Phys. Rev. B 73 2053124
Goswami B, Pal S and Sarkar P 2007 Phys. Rev. B 76 045323
Saha S, Pal S, Sarkar P, Rosa A L and Frauenheim T 2012 J. Comput. Chem. 33 1165
Kaledin A L, Kong D, Wu K, Lian T and Musaev D G 2018 J. Phys. Chem. C 122 18742
Kar M, Sarkar R, Pal S and Sarkar P 2020 Phys. Rev. B 101 195305
Benchikh K, Abid H and Benchehima M 2017 Mater. Sci. Pol., https://doi.org/10.1515/msp-2017-0005
Debbarma M, Sarkar U, Debnath B, Ghosh D, Chanda S, Bhattacharjee R and Chattopadhyaya S 2018 Curr. Appl. Phys. 18 698
Murtaza G, Ullah N, Rauf A, Khenata R, Omran S B, Sajjad M et al 2015 Mater. Sci. Semicond. Process. 30 462
Debbarma M, Sarkar U, Debnath B, Ghosh D, Chanda S, Bhattacharjee R et al 2018 J. Alloys Comp. 748 446
Hohenberg P and Kohn W 1964 Phys. Rev. B 136 864
Kohn W and Sham L J 1965 Phys. Rev. 140 A1133
Andersen O K 1975 Phys. Rev. B 42 3063
Blaha P, Schwarz K, Madsen G H, Kbasnicka D and Luitz J 2001 in Schwarz K (ed.) FP-LAPW+lo program for calculating crystal properties, 2nd edn. Vienna University of Technology, Vienna
Togo A, Oba F and Tanaka I 2008 Phys. Rev. B 78 134106
Jamal M, Asadabadi S J, Ahmed I and Aliabad H A R 2014 Comp. Mater. Sci. 95 592
Perdew J P, Burke K and Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Tran F and Blaha P 2009 Phys. Rev. Lett. 102 226401
Anisimov V I, Zaanen J and Andersen O K 1991 Phys. Rev. B 44 943
Anisimov V I, Solovyev I V, Korotin M A, Czyyk M T and Sawatzky G A 1993 Phys. Rev. B 48 16929
Hubbard J 1963 Proc. Roy. Soc. Lond. A 276 238
Kokalj A 2003 Comp. Mat. Sci. 28 155
Murnaghan F D 1944 Proc. Natl. Acad. Sci. USA 30 244
Vegard L 1921 Z. Phys. 5 17
Pettifor D G 1992 Mater. Sci. Technol. 8 345
Li S, Li S and Ju X 2017 J. Alloys Comp. 695 2916
Potter R Y 1957 J. Phys. Chem. Solids 3 223
Huang W and Yang L 2015 Can. J. Phys. 93 1
Liu B, Wang J Y, Li F Z and Zhou Y C 2010 Acta Mater. 58 4369
Fine M E, Brown L D and Marcus H L 1984 Scr. Metall. 18 951
Fox M 2001 Optical properties of solids (UK: Oxford University Press)
Penn D R 1962 Phys. Rev. 128 2093
Acknowledgements
The DST-INSPIRE-SRF [Ref. No DST/INSPIRE/03/2017/002068] of the DST, Government of India, is gratefully acknowledged by Sayantika Chanda.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chanda, S., Debbarma, M., Ghosh, D. et al. Investigation of structural, mechanical and optoelectronic properties of cubic Cd1−x−yZnxHgySe quaternary alloys through first-principle calculations. Bull Mater Sci 45, 34 (2022). https://doi.org/10.1007/s12034-021-02610-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12034-021-02610-3