
Abstract
Herein, reliable work has been carried out on the synthesis of cerium-oxide (\(\hbox {CeO}_{2})\) nanoparticles (NPs) via hydrothermal and co-precipitation methods. We emphasize the highly adequate hydrothermal method for synthesizing \(\hbox {CeO}_{2}\) NPs for biomedical applications. Absorption spectra showed peaks at 283 and 274 nm, which confirm the formation of \(\hbox {CeO}_{2}\) NPs for hydrothermal (HYNPs) and co-precipitation (CONPs) methods, respectively. Functional group analysis firmly showed the presence of organic and inorganic species, which revealed similar characteristics of both HYNPs and CONPs. The cubic structure and the average crystallite size of the synthesized NPs are determined using Scherrer’s and Williamson–Hall methods. The obtained average particle size is compared by using high-resolution transmission electron microscopy, which is around 10 and 5 nm for HYNPs and CONPs, respectively. Further, we studied their antimicrobial activities and consequently, the synthesized \(\hbox {CeO}_{2}\) NPs showed excellent antimicrobial activities. Moreover, HYNPs yielded promising antioxidant activity with the lowest \(\hbox {IC}_{50}\) as compared with CONPs. Owing to the biological activity of \(\hbox {CeO}_{2}\) NPs, HYNPs act as best therapeutic agents in biomedical applications as well as its vital role in antibiotics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Nanostructural rare-earth materials have extensive applications because of their unique and exceptional characteristics. The interfacial properties of rare-earth inorganic nanoparticles (NPs), and effects on the rate of reaction mediated on the surface and size-dependent change in crystallinity of NPs are some of them. Hence, these materials with a high surface-to-volume ratio and quantum size effect at the nanoscale enhance interfacial reactivity [1] and show good optical, mechanical, biomedical, physical and catalytic applications [2, 3]. Among them, cerium is one of the best rare-earth elements. Generally, nanoceria is prevalent, because of its unique properties such as being a good oxide ion conductor in solid oxide fuel cells [4] and as gas sensors [5]. It also has wide band gap energy which in turn leads to its UV absorbing ability, high-thermal stability, hard in nature and catalytic reactivity [6, 7]. However, upon variation in its morphologies, ceria exhibits physicochemical properties, which enables application as catalysts in an emission control system, ultraviolet blocking materials, solar cells and sunscreen cosmetics [8].
Recently, nanoceria has been synthesized using various methods and also to improve its physical–chemical properties by different methods. Some of the important methods are hydrothermal [9, 10], co-precipitation [11], reversed micelle reaction [12], template-assisted precipitation [12], sol–gel [13], spray drying system [14], plasma spray technique [15], microemulsion procedure [16], self-assembled system [17], solvothermal [18] and thermal decomposition [19]. Employing these methods over the past few years, cerium oxide \((\hbox {CeO}_{2})\) nanostructures such as NPs, nanorods, nanowires, nanotubes, nanocubes and nanospheres have been synthesized. \(\hbox {CeO}_{2}\) NPs synthesized by sol–gel were studied for their catalytic activities and electrochemical characteristics to develop high-sensitivity, third-generation biosensors [20, 21]. Electrochemically synthesized \(\hbox {CeO}_{2}\) NPs, or a part of them, are used as transducers in sensors [22]. The robust solution synthesized ultrathin \(\hbox {CeO}_{2}\) NPs are used for high oxygen storage capacity because of their high surface area-to-volume ratio [23]. At the surface, \(\hbox {CeO}_{2}\) NPs have high-oxygen mobility and a large oxygen diffusion coefficient, which help in the conversion of the valence state of cerium and then allow oxygen to be released or stored [23]. Using a green approach, monodisperse ceria hollow spheres are synthesized and their photocatalytic activity is enhanced [24]. \(\hbox {CeO}_{2}\) NPs were synthesized and benefits of their physical–chemical properties were reviewed in many fields. Over the past few years, \(\hbox {CeO}_{2}\) NPs have been synthesized and their emerging properties have been reported; based on these properties \(\hbox {CeO}_{2}\) NPs have found application in bio-analysis and biomedical fields [22]. The solution combustion synthesized \(\hbox {CeO}_{2}\) NPs have application in retinopathy, biosensors and cancer therapy [25]. Further, the synthesis of different types of nanoceria shows the interactions with molecules such as proteins, anions and lipids [26].
Over the past decade, scientists have showed significant interest in the medical field using \(\hbox {CeO}_{2}\) NPs because of their promising antimicrobial activities and regenerating antioxidant properties which have been discussed earlier [27]. Fine crystallites of \(\hbox {CeO}_{2}\) with a nano-nature make them very potent and useful materials in various fields. However, there are some difficulties in synthesizing \(\hbox {CeO}_{2}\) NPs, namely, expensive raw materials, complex processes, processing parameters, low-yield production, high-energy consumption, non-eco-friendly and high-cost of production. These cumbersome processes involved had motivated us to synthesize \(\hbox {CeO}_{2}\) NPs by using very simple, one-step and economic methods.
Herein, we focus on hydrothermal and co-precipitation methods to synthesize \(\hbox {CeO}_{2}\) NPs. We report a simple and eco-friendly method. After the synthesis of \(\hbox {CeO}_{2}\) NPs we subjected samples to the spectral characteristic analysis of optical, functional groups and morphological, compositional and thermal behavioural studies. Later, based on these results both HYNPs and CONPs are evaluated for their biological activities particularly antimicrobial and antioxidant studies with an aim to select the most suitable \(\hbox {CeO}_{2}\) NPs for biomedical applications.
2 Materials and methods
\(\hbox {CeO}_{2}\) NPs were synthesized using hydrothermal and co-precipitation methods. The materials used are as follows:
-
cerium(III) nitrate hexahydrate (Ce(\(\hbox {NO}_{3})_{3}\cdot 6\hbox {H}_{2}\)O)
-
sodium hydroxide pellets (NaOH)
-
ethanol (\(\hbox {CH}_{3}\hbox {CH}_{2}\)OH).
Materials were purchased from HIMEDIA Laboratories Pvt. Ltd., Mumbai, India. All chemicals were of AR grade and used without any further purification. Deionized water was used for the preparation of suspension and solution.
2.1 Synthesis of \(\hbox {CeO}_{2}\) NPs
2.1a Hydrothermal method: In brief, the procedure is as follows: 4.34 g of Ce(\(\hbox {NO}_{3})_{3}\!\cdot \!6\hbox {H}_{2}\)O (1 M) was dissolved in 10 ml of deionized water. To prepare NaOH solution, 0.04 g of NaOH (0.1 M) pellets was dissolved in 10 ml of deionized water. The prepared NaOH solution was added dropwise to a cerium precursor with continuous stirring. The mixtures of both solutions were stirred at ambient temperature for a few minutes. Then the obtained light brown-coloured solution was transferred to a 25 ml Teflon-lined stainless-steel autoclave; the autoclave was sealed and placed in an oven for 3 h at a temperature of \(180^{\circ }\hbox {C}\) and on cooling the precipitate obtained was centrifuged several times continuously using deionized water and once with ethanol to remove organic molecules present on the surface of the product. Finally, the product was heated at \(60^{\circ }\hbox {C}\) for 60 min using a laboratory oven to evaporate the water content. The resulting hydrothermally synthesized light brown powder was further used for various characterization techniques.
2.1b Co-precipitation method: Initially, to synthesize \(\hbox {CeO}_{2}\) NPs using the co-precipitation method, the cerium precursor was prepared by dissolving 2.17 g of Ce(\(\hbox {NO}_{3})_{3}{\cdot }6\hbox {H}_{2}\)O (0.5 M) in 20 ml of deionized water, the solution was subjected to stirring and then 0.04 g of NaOH (0.1 M) was dissolved in 5 ml of deionized water. This NaOH solution was added dropwise to the stirring cerium precursor. The mixture of precursor and NaOH solution was being continuously stirred for 30 min. The obtained antique white solution was kept overnight for particles to settle down and then the precipitate was centrifuged and washed several times with deionized water and once with ethanol to remove organic impurities. The final product was heated at \(60^{\circ }\hbox {C}\) in a laboratory oven for 60 min to obtain \(\hbox {CeO}_{2}\) nanopowder which was used for further characterization techniques.
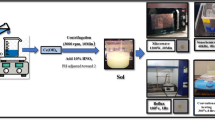
A schematic diagram of the synthesis of \(\hbox {CeO}_{2}\) NPs using hydrothermal and co-precipitation methods is shown in figure 1.

Graphical representation of the synthesis of HYNPs and CONPs.
2.2 Characterization techniques
2.2a Energy band gap studies: UV–Visible spectroscopy was performed for the study of the absorption maxima and energy band gap, which were calculated by using UV–Vis absorption spectra in the wavelength range of 200–600 nm using a UV–Vis spectrometer (model: V-670 Jasco) at Karnatak University, Dharwad, Karnataka, India.
2.2b Functional group analysis: Fourier-transform infrared (FT-IR) absorption bands for the synthesized \(\hbox {CeO}_{2}\) NPs were recorded using KBr powder. The FT-IR spectra were recorded in the range of 4000–400 cm\(^{-1}\) using a FT-IR spectrophotometer (model: Nicolet 6700) at USIC, Karnatak University, Dharwad, Karnataka, India.
2.2c Grain size and structural studies: The crystal structure or phase evolution and crystallite size of the synthesized \(\hbox {CeO}_{2}\) NPs were analysed at a scanning rate of \(0.02^{\circ } \hbox {s}^{-1}\) in the range of 20–80\(^{\circ }\) using Cu-K\(\upalpha \) radiation (\(\lambda \) = 1.5406 Å) from a powder X-ray diffractometer (model: Shimadzu 7000) at Tumkur University, Tumkur, Karnataka, India. The peaks of XRD were compared and analysed by using PCPDFWIN software.
2.2d Surface morphological studies: The particle size and morphology of the synthesized \(\hbox {CeO}_{2}\) NPs were examined at an operating voltage of 200 kV using transmission electron microscopy (TEM) (model: JEOL/JEM 2100) at STIC, Cochin, Kerala, India. Further, by using PIXEL software TEM images were analysed.
2.2e Thermal and stability analysis: Thermal stability and decomposition of \(\hbox {CeO}_{2}\) NPs were analysed using thermogravimetric analysis (TGA) (model: SDT Q600) and differential scanning calorimetry (DSC) (model: Q20 V24.10 Build 122). TGA was recorded in the temperature range of room temperature to \(700^{\circ }\hbox {C}\). DSC was recorded from room temperature to \(500^{\circ }\hbox {C}\) at USIC, Karnatak University, Dharwad, Karnataka, India.
2.3 Biological activities
2.3a Antimicrobial activity: An agar well diffusion method is used for antimicrobial analysis. Herein, we have considered five microorganisms for antimicrobial assessment: for antibacterial studies, Staphylococcus aureus and Bacillus subtilis as Gram positive bacteria and Escherichia coli and Pseudomonas aeruginosa as Gram negative bacteria are considered. For antifungal studies, yeast Candida albicans was used. Initially, 100 \(\upmu \)l of test organisms were swab inoculated onto the sterile nutrient agar Petri plates for bacteria and potato dextrose agar for fungi, for which four wells were bored. Four different concentrations (125, 250, 375 and 500 \(\upmu \)g ml\(^{-1}\) in sterile distilled water) of the synthesized \(\hbox {CeO}_{2}\) NPs were deposited onto the labelled wells, respectively. After that, the plates were incubated at \(37^{\circ }\hbox {C}\) for 24 h for bacteria and at \(27^{\circ }\hbox {C}\) for 48 h for fungi. Later, we measured the antimicrobial activity of the synthesized \(\hbox {CeO}_{2}\) NPs by measuring the zones of inhibition in millimetres using a scale.
2.3b Evaluation of the antioxidant activity of CeO\(_{2}\) NPs using the 1,1-diphenyl 2-picrylhydrazyl (DPPH) radical scavenging method: The DPPH method was initiated for studying antioxidant activities of the synthesized \(\hbox {CeO}_{2}\) NPs and it was analysed on the basis of the free radical-scavenging activity (RSA). The solution of DPPH was prepared according to our previously published paper [28]. Here, 2 ml of DPPH solution was added to 1 ml of different concentrations (100, 200, 300, 400 and 500 \(\upmu \)g ml\(^{-1})\) of the synthesized \(\hbox {CeO}_{2}\) NPs. Ascorbic acid (100, 200, 300, 400 and 500 \(\upmu \)g ml\(^{-1})\) was used as a reference. The mixture of 1 ml distilled water and 2 ml DPPH solution was used as the control. The reaction mixture was mixed and incubated at room temperature in the dark for 30 min. The absorbance was analysed spectrophotometrically at 517 nm.
The antioxidant activity of the synthesized \(\hbox {CeO}_{2}\) NPs was estimated based on the percentage of DPPH RSA by the following equation:
SPSS software (version 20.0) was used for statistical analysis. Experiments were carried out in triplicates, and the data were expressed as mean ± standard deviation by one-way analysis of variance. Tukey’s multiple comparison tests were used to determine significant differences between the standard and synthesized compounds. Correlation analysis was carried out using Pearson’s correlation analysis using p = 0.01.
3 Results and discussion
3.1 Energy band gap studies
UV–Vis spectrophotometry was carried out to study the energy band gap and also for preliminary confirmation of the presence of \(\hbox {CeO}_{2}\) material, the absorption maxima (\({\lambda }_{\mathrm{max}})\) were observed for both HYNPs and CONPs. For both the cases, UV–Vis spectra of \(\hbox {CeO}_{2}\) NPs were recorded using ethanol as a solvent at ambient temperature and absorption maxima were found to be 283 and 274 nm for the hydrothermal and co-precipitated methods, respectively (figure 2a).

(a) UV–Vis spectra of HYNPs and CONPs and (b) Tauc’s plot for HYNPs and CONPs.
The band gap energy of the \(\hbox {CeO}_{2}\) NPs was estimated by the Kubelka–Munk transformation method. Optical absorption (\(\alpha \)) is used as a function of band gap energy (\(h\nu \)) of the synthesized \(\hbox {CeO}_{2}\) NPs and is expressed by the following equation:
where \(\alpha \) is the absorption coefficient; \(h\nu \) is the photon energy; A is the constant; \(E_{\mathrm{g}} \) is the band gap energy and n is the nature of transition; n = 1/2 for allowed direct transition and n = 2 for allowed indirect transition [29, 30].
For the \(\hbox {CeO}_{2}\) NPs, the direct band gap energy \((E_{\mathrm{g}} )\) was determined by fitting the absorption data to the direct transition equation and the graph of (\(\alpha h\nu )^{2}\) vs. \(h\nu \) is plotted. The band gaps of \(\hbox {CeO}_{2}\) NPs synthesized by the hydrothermal and co-precipitation methods were estimated by extrapolation of the linear portion of the curve towards absorption equal to zero, which gives \(E_\mathrm{g}\) for direct transitions and is shown in figure 2b. This is also known as Tauc’s plot. The estimated direct band gaps of the HYNPs and CONPs were found to be 3.85 and 2.69 eV, respectively.
3.2 Functional group analysis
FT-IR spectroscopy was performed to analyse the presence of functional groups, which exhibits the characteristic vibration bands of organic and inorganic species. The IR spectra of HYNPs and CONPs were recorded by the KBr pellet method in the range of 4000–400 cm\(^{-1}\) at a resolution of 2 cm\(^{-1}\). The FT-IR spectra of HYNPs and CONPs are shown in figure 3. The observed IR bands are tabulated in table 1. Both spectra are analysed and all vibration modes are in good agreement with the literature [8, 31,32,33].

FT-IR spectra of HYNPs and CONPs.
Due to the use of the same chemicals in both methods the resultant samples provide unique and desirable results. Hence, there is not much appropriate alteration in the occurred peaks and no main significant differences between the spectra of HYNPs and CONPs.
3.3 Grain size and structural studies
X-ray diffraction (XRD) was used to determine the existing crystallite phase of the synthesized \(\hbox {CeO}_{2}\) NPs. XRD patterns of \(\hbox {CeO}_{2}\) NPs which are obtained using hydrothermal and co-precipitation methods are shown in figure 4a and b, respectively. From the XRD pattern of the hydrothermally synthesized \(\hbox {CeO}_{2}\) NPs, we observed a series of characteristic peaks: 28.54 (111), 33.05 (200), 47.47 (220), 56.33 (311), 59.07 (222), 69.40 (400), 76.68 (331) and 79.06 (420) and for the co-precipitation method: 28.46 (111), 32.96 (200), 47.26 (220), 56.10 (311), 58.80 (222), 69.62 (400) and 77.04 (331). These are the Bragg’s angles (2\(\theta \)) and their corresponding Bragg’s reflection plane values (hkl), respectively. These values are in accordance with the Joint Committee on Powder Diffraction Standards (JCPDS file no. 81-0792), which indexed that the synthesized \(\hbox {CeO}_{2}\) NPs are with a face-centred-cubic structure. No other peaks of impurity are observed, suggesting that high-purity \(\hbox {CeO}_{2}\) was obtained. Also the reflection peaks are sharp, so by the broadening of the X-ray line of reflections the average crystallite size of the sample was calculated. XRD data can be utilized to evaluate the average particle size of the \(\hbox {CeO}_{2}\) NPs. Herein, we calculated the crystalline size by Scherrer analysis and the Williamson–Hall (W–H) method using the broadening of peaks in the XRD pattern.
3.3a Crystallite size using Scherrer’s method: The particle size of the \(\hbox {CeO}_{2}\) NPs was determined using the XRD peak. We have calculated average particle sizes of \(\hbox {CeO}_{2}\) NPs using Scherrer’s equation:
where D is an average particle size, \(\lambda \) is the wavelength of Cu-K\(\upalpha \) radiation (0.15406 nm), k is the shape factor (k = 0.9), \(\beta \) is the full-width at half-maximum (FWHM) and \(\theta \) is Bragg’s angle of the peak. The crystallite size was estimated from the line broadening of the maximum intense peak at the (111) plane. The estimated average crystallite size of HYNPs and CONPs is 9.53 and 5.4 nm, respectively.
3.3b Crystallite size by W–H method: In addition to the X-ray peak broadening, the crystallite size and lattice strain are the two independent parameters that contribute to the total peak broadening. The W–H method will assume that the strain is present in the material and it is uniformly distributed. The strain-induced line broadening \({\beta }_\mathrm{s}\) is given by the relation \({\beta }_\mathrm{s}\) = 4\(\varepsilon \) tan \({\theta }_{hkl}\). The total peak broadening is the sum of the contributions of the crystallite size and the strain present in the material. The W–H equation for the uniform determination model is given by [34,35,36,37]
where \({\beta }_\mathrm{s}\) is the FWHM, \(\theta \) is the Bragg’s diffraction angle, k is the shape factor, \(\lambda \) is the wavelength of radiation, D is the crystallite size and \(\varepsilon \) is the lattice strain. Fitting a plot by taking 4 sin \(\theta \) vs. \({\beta }_\mathrm{s}\) cos \(\theta \) is shown in figure 4c and d for the hydrothermal and co-precipitation methods. The crystalline size is obtained from the intercept value of the plot. The estimated size of the HYNPs and CONPs was 9.66 and 7.02 nm, respectively.

XRD pattern of: (a) HYNPs and (b) CONPs and W–H plot for: (c) HYNPs and (d) CONPs.
3.4 Surface morphological studies
The morphology and the nano-nature of the synthesized \(\hbox {CeO}_{2}\) NPs were further examined by TEM. The TEM overview image of HYNPs is shown in figure 5a along with the selected area electron diffraction (SAED) pattern in figure 5b and it is confirmed that NPs are regular and uniform in morphology with a cubic structure. The average size of the \(\hbox {CeO}_{2}\) NPs is found to be 10.5 nm. In figure 5b, we observed the bright spot and intense rings which indicate that the sample is well crystallized in nature. Figure 5c shows the TEM overview image of CONPs. In this figure, we observed that CONPs are narrow in size distribution, which is randomly oriented. It may indicate that \(\hbox {CeO}_{2}\) NPs are well crystallized. But most of the NPs were aggregated, due to the non-dispersion of the particles in a solvent before the sample preparation for TEM. The average particle size of \(\hbox {CeO}_{2}\) NPs is found to be 5.78 nm. Figure 5d presents the SAED pattern of CONPs, representing the rings which could be attributed to reflections of the structure of \(\hbox {CeO}_{2}\). The broadening of the diffraction rings which are less intense than the rings shown in figure 5d suggests that the particles are small and/or are of low crystalline in nature [8, 37].

(a) TEM images of HYNPs, (b) SAED pattern of HYNPs, (c) TEM images of CONPs and (d) SAED pattern of CONPs.

TGA/DSC curves for: (a) HYNPs and (b) CONPs.
3.5 Thermal and stability analysis
To study the thermal characteristics of \(\hbox {CeO}_{2}\) NPs, we carried out TGA and DSC. The sample was allowed to heat at a constant rate of \(10^{\circ }\hbox {C}\) min\(^{-1}\). To avoid any premature heating, it is important to use an inert gas for performing this study. For TGA of HYNPs and CONPs, about 5.5 mg of NPs was placed in a pan and heated from room temperature to \(700^{\circ }\hbox {C}\). In the temperature region between room temperature and \(50^{\circ }\hbox {C}\), 5.76% of weight loss was observed for HYNPs. For CONPs there is 7% weight loss observed in the temperature range of room temperature to \(70^{\circ }\hbox {C}\). This is due to the evaporation of absorbed moisture. Next for both cases, up to \(320^{\circ }\hbox {C}\) 3.8 and 6.8% loss is observed for hydrothermal and co-precipitation NPs, respectively, which indicate the removal of water molecules adsorbed by the NPs. Finally, at 320–700\(^{\circ }\hbox {C}\) there is a weight loss of 2.6% for HYNPs and 1.4% loss for CONPs. This minor weight loss is due to the breakdown of agglomerated organic surface impurities present in the NPs or combustion of organic residues. Totally 12.16 and 15.4% of weight loss was observed for the NPs synthesized by hydrothermal and co-precipitation methods, respectively. Based on the DSC thermograph, each sample shows endothermic peaks. For HYNPs there are two broad endothermic peaks observed at \(\sim \)200 and \({\sim }340^{\circ }\hbox {C}\). For CONPs there are two broad endothermic peaks observed at \(\sim \)67 and \({\sim }315^{\circ }\hbox {C}\) [30, 32, 37].
These endothermic peaks were attributed to the evaporation of observed water and dehydration of dried organic matter. Figure 6a and b shows the TGA and DSC curves of the corresponding HYNPs and CONPs, respectively. There is a remarkable difference in the thermal properties of the NPs synthesized by hydrothermal and co-precipitation methods. That is, up to \({\sim }400^{\circ }\hbox {C}\) HYNPs are more stable than CONPs.

(a) Antibacterial activity of HYNPs with B. subtilis, (b) antifungal activity of HYNPs with C. albicans, (c) antibacterial activity of CONPs with B. subtilis and (d) antifungal activity of CONPs with C. albicans.
3.6 Biological activities
3.6a Antimicrobial assessment: The antimicrobial assay of the synthesized \(\hbox {CeO}_{2}\) NPs was evaluated against the four pathogenic bacteria. Gram-positive bacteria S. aureus and B. subtilis, Gram-negative bacteria E. coli and P. aeruginosa and fungi C. albicans showed zones of inhibition on the culture plates which indicate that the NPs are having potent antimicrobial strength.
The microbial efficiency of \(\hbox {CeO}_{2}\) NPs normally depends on their surface area, size, shape, etc. This interaction between NPs and microbial will observe the inhibition of microbial growth and also the reactive oxygen species generation, which leads to cell death. Our findings are in accordance with Thill et al [38], who suggested three types of interactions between bacteria and Ce NPs: (1) adsorption, (2) oxi-reduction and (3) toxicity. Herein, we have shown that HYNPs have higher antibacterial activities compared to CONPs. As discussed by Lian Wang and Sang-Hocha et al, the shape of the NPs plays a very crucial role. They have been reported that the antibacterial strength is not only dependent on the NPs, instead it is also dependent on the shape of the synthesized NPs. As we have shown in the TEM images of HYNPs and CONPs in figure 5a and c, respectively, there are morphological differences between them. The HYNPs are well dispersed and uniform in nature with a cubic structure looking like nanocubes. CONPs are randomly oriented, aggregated and they look like NPs or spheres. Among these two, we observed the potent bacterial activity for hydrothermally synthesized nanocubes compared to the co-precipitation synthesized NPs. Based on the surface morphology, the intermolecular interactions vary with bacteria [39]. Therefore, based on the shape, we may predict that HYNPs (nanocubes) will get much exposed stable reactive planes. As reported, to form oxygen vacancies very less energy is required for the stable planes on the surface. Therefore, HYNPs will have good interaction with the bacteria; and CONPs have less exposed planes on the surface [40] and less interaction with bacteria. This indicates that the HYNPs exhibit a potent antimicrobial activity to kill microorganisms as compared to the CONPs. Also, the compounds showed comparatively higher antimicrobial activity for the Gram-positive bacteria as compared to that of the Gram-negative ones and very less active for the yeast and hence can be stated that these NPs are more active as the antibacterial agents. Further, we also observed the concentration-dependent antimicrobial activities. Herein, we observed a maximum zone of inhibition for the higher concentration which is shown in table 2. The antimicrobial activities of HYNPs for (A) B. subtilis and (B) C. albicans, and of CONPs for (C) B. subtilis and (D) C. albicans images are shown in figure 7a–d.
3.6b Antioxidant assessment: We have studied the radical scavenging assessments for ascorbic acid, HYNPs and CONPs using the DPPH for five different concentrations (100, 200, 300, 400 and 500 \(\upmu \)g ml\(^{-1})\). The residual DPPH could be quantified by comparing absorbance at 517 nm against the blank, and the percent inhibition could be calculated. RSA assessment of ascorbic acid, HYNPs and CONPs is shown in figure 8 with an error bar and in the same graph the statistics bar shows the significance of that. We found a significant difference among the respective materials. The DPPH value increases as the concentration increases. The recorded value for the lowest concentration is 100 \(\upmu \)g ml\(^{-1}\) for HYNPs and CONPs; their scavenging activities were 39.37 ± 0.47 and 31.78 ± 1.43, respectively. Comparatively, HYNPs possessed a higher scavenging activity compared to CONPs. Thus, HYNPs act as potent antioxidants compared to CONPs and they may be useful for the treatment of several diseases.

RSA of standard ascorbic acid, synthesized HYNPs and CONPs with error bars.
4 Conclusion
The present investigation concludes that \(\hbox {CeO}_{2}\) NPs were successfully synthesized through hydrothermal and co-precipitation methods. The results obtained from both methods were analysed and compared. Herein, we noted the following results:
-
Initially the presence of \(\hbox {CeO}_{2}\) NPs is confirmed by UV–Vis spectroscopy and from Tauc’s plots, it is confirmed that HYNPs have higher band gap energy compared to CONPs.
-
FT-IR analysis revealed the presence of Ce–O bands, O–H bands, C–H bands, nitrates, and noted that there are no significant differences in both the synthesized \(\hbox {CeO}_{2}\) NPs from hydrothermal and co-precipitation methods.
-
XRD data determined the size of the synthesized \(\hbox {CeO}_{2}\) NPs, both are having the face-centred-cubic structure. Further, using XRD data the crystallite size was calculated by Scherrer’s method, W–H method and compared by TEM analysis.
-
Hydrothermally synthesized NPs have almost the same particle size by Scherrer’s method, W–H method and TEM analysis, which strongly concludes that hydrothermally synthesized NPs are well crystallized in nature and thus no agglomeration of particles.
-
TGA/DSC studies found that the HYNPs are more stable than CONPs.
-
The HYNPs showed more potent antimicrobial activities for the four pathogenic bacteria compared to the CONPs.
-
Comparatively, the antioxidant properties of HYNPs are more potent than those of CONPs.
Based on the above findings, we suggest that the hydrothermal method is a very simple, eco-friendly, cost-effective and one-step synthesizing method with all potent results. Further, due to their antimicrobial activities against bacteria we can say that the synthesized \(\hbox {CeO}_{2}\) NPs can be used in medicine as therapeutic agents and fungi-evaluated in vitro studies due to their potent antioxidant activities.
References
Auffan M, Rose J and Bottero J Y 2009 Nat. Nanotechnol. 4 634
Wu Z, Zhang J and Benfield R E 2002 J. Phys. Chem. B 106 4569
Trovarelli A, De Leitenburg C, Boaro M and Dolcetti G 1999 Catal. Today 50 353
Graziani M, Bekyarova E, Fornasiero P and Kas J 1998 Catal. Today 45 179
Tsunekawa S, Sahara R, Kawazoe Y and Kasuya A 2000 Mater. Trans. 41 1104
Tsai M S 2004 Mater. Sci. Eng. B 110 132
Mori T, Wang Y and Drennan J 2004 Solid State Ion. 175 641
Li H, Wang G and Zhang F 2012 RSC Adv. 2 12413
Arul N S, Mangalaraj D and Han J I N 2015 Bull. Mater. Sci. 38 1135
Tok A I Y, Boey F Y C, Dong Z and Sun X L 2007 J. Mater. Process. Technol. 190 217
Pujar M S, Hunagund S M and Desai V R 2018 62nd AIP Conference Proceedings of DAE SSPS 2017 p 50026
Balavi H, Samadanian-Isfahani S, Mehrabani-Zeinabad M and Edrissi M 2013 Powder Technol. 249 549
Masui T, Fujiwara K and Machida K I 1997 Chem. Mater. 9 2197
Sharma V, Eberhardt K M and Sharma R 2010 Chem. Phys. Lett. 495 280
Singh V, Karakoti A and Kumar A 2010 J. Am. Ceram. Soc. 93 3700
Nagy K and Dékány I 2009 Colloids Surf. A 345 31
Bumajdad A, Eastoe J and Mathew A 2009 Adv. Colloid Interface Sci. 147 56
Qian L, Zhu J, Du W and Qian X 2009 Mater. Chem. Phys. 115 835
Gabal M A, Elroby S A K and Obaid A Y 2012 Powder Technol. 229 112
Njagi J, Ispas C and Andreescu S 2008 Anal. Chem. 80 7266
Ispas C, Njagi J, Cates M and Andreescu S 2008 J. Electrochem. Soc. 155 169
Charbgoo F, Ramezani M and Darroudi M 2017 Biosens. Bioelectron. 96 33
Wang D, Kang Y and Doan-Nguyen V 2011 Angew. Chem. 50 4378
Deng W, Chen D, Hu J and Chen L 2015 RSC Adv. 5 80158
He L, Su Y, Lanhong J and Shi S 2015 J. Rare Earths 33 791
Walkey C, Das S and Seal S 2015 Environ. Sci. Nano 2 33
Caputo F, Mameli M and Sienkiewicz A 2017 Sci. Rep. 7 1
Hunagund S M, Desai V R and Barretto D A 2017 J. Photochem. Photobiol. A 346 159
Sujana M G, Chattopadyay K K and Anand S 2008 Appl. Surf. Sci. 254 7405
Babitha K K, Sreedevi A and Priyanka K P 2015 Indian J. Pure Appl. Phys. 53 596
John C 2000 In Encyclopedia of analytical chemistry R A Meyers (ed) (Chichester, UK: John Wiley & Sons Ltd) p 10815
Kumar K S and Jaya N V 2013 Asian J. Chem. 25 6095
Kannan S K and Sundrarajan M 2014 Int. J. Nanosci. 13 1450018
Khorsand Zak A, Abd Majid W H, Abrishami M E and Yousefi R 2011 Solid State Sci. 13 251
Venkateswarlu K, Chandra Bose A and Rameshbabu N 2010 Physica B 405 4256
Hunagund S M, Desai V R and Barretto D A 2017 Adv. Sci. Eng. Med. 9 453
Chandradass J, Nam B and Kim K H 2009 Colloids Surf. A 348 130
Antonie T, Ophelie Z, Olivier S, Franck C, Jernone R, Melaine A et al 2006 Environ. Sci. Technol. 40 6151
Lian W, Hong H, Yunbo Y, Li S, Sijin L, Changbin Z et al 2014 J. Inorg. Biochem. 135 45
Sang-Ho C, Jin H, Matt M, Bongjun Y, Scott J V and Nicholas A K 2015 ACS Nano 9 9097
Acknowledgements
The authors are thankful to the director and technical staff of USIC, Karnatak University, Dharwad for providing UV–Vis and FT-IR instrumentation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pujar, M.S., Hunagund, S.M., Barretto, D.A. et al. Synthesis of cerium-oxide NPs and their surface morphology effect on biological activities. Bull Mater Sci 43, 24 (2020). https://doi.org/10.1007/s12034-019-1962-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12034-019-1962-6