
Abstract
Chalcogenide Ge 20Se80 glass was prepared using the melt-quench technique. The radial distribution function is obtained from X-ray diffraction data in the scattering vector interval 0⋅28 ≤ K ≤ 6⋅87 Å −1. Reverse Monte Carlo (RMC) simulations are useful to compute the partial pair distribution functions, g ij (r), partial structure factors, S ij (K), and total structure factor. Values of r 1/r 2 ratio and bond angle (Θ) indicate that Ge (Se1/2)4 tetrahedra units connected by chains of the chalcogen atoms are present. The partial structure factors have shown that homopolar Ge–Ge and Se–Se bonds are behind the appearance of the first sharp diffraction peak (FSDP) in the total structure factor. Tetrahedral Ge (Se1/2)4 structural units connected by Se–Se chains have been confirmed by the simulated values of the partial coordination numbers and bond angle distributions. Finally, Raman spectra measurements have strongly supported the conclusions obtained either from the calculated Fourier data or from RMC simulations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Chalcogenide glasses present a great potential for application in technological devices, such as optical fibers, memory materials and switching devices, but their use is limited due to several factors. One of them is the difficulty in obtaining information about atomic structures. The structure of chalcogenide glasses in the short-range order (SRO) or intermediate-range order (IRO) is an important and controversial subject. The appearance of the first sharp diffraction peak (FSDP) in the total structure factor indicates the presence of IRO. Germanium selenides have been intensively studied by several methods like X-ray diffraction,[1–4,22,26] neutron diffraction,[5,6] Raman scattering,[7] anomalous X-ray scattering[8] and extended X-ray absorption fine structure.[9] The structure unit in these glasses is Ge(Se 1/2)4 tetrahedra connected through Se chains.
As mentioned earlier,[10] the addition of germanium into the polymeric Se matrix produces a cross-linking of selenium chains, mediated by the formation of Ge(Se 1/2)4 tetrahedra. At low doping (x <15 at%), the tetrahedra are sparsely distributed in the background matrix, with rather flexible interconnections. The feeble Ge–Ge correlations are inadequate to give any detectable FSDP, as it was previously concluded from the partial pair structure studies on GeSe 2.[11] When germanium content reaches 15 at%, the amount of Ge atoms becomes sufficient to join some of the tetrahedral pairs by corner sharing.[12] X-ray diffraction (XRD) of the glassy Ge x Se1−x (with 0 ≤ x ≤ 0⋅33) systems[1,8] have demonstrated that besides the well-established SRO information, a pre-peak appeared in the total structure factor, S(K), at a scattering vector K of about 1⋅1 Å −1. The pre-peak, being a clear evidence for the existence of the IRO, showed a systematic decrease in intensity and shift towards higher K values with the decrease in Ge concentration. A similar result was observed using the neutron diffraction measurement.[5] In recent times,[13] the sorption ability of Ge 20Se 80 thin films, applied as active layers of quartz crystal microbalance for NO 2 gas sensing, has been investigated. It was found that the introduced gas molecules interact electrostatically with the chalcogen atoms of the host material and initiate some degree of structural changes in it.
Reverse Monte Carlo (RMC) simulation[14,15] represents, when used carefully, a powerful tool to extract some information of intermediate and extended-range scale in glassy materials. It assembles three-dimensional atomic configurations using the experimental diffraction data implicitly in the simulation. The intimate connection between computational and experimental processes means that the better quality and higher resolution of the experimental data, the more reliable RMC model of a network structure for vitreous materials. RMC method is an inverse problem in which the experimental data are enforced to build atomic configurations that have the desired structural and electronic properties. The main point is to set up a generalized function containing as much information as possible, and then optimize the function for generating configurations toward exact agreement with the experimental data.
The present paper aims to get the structural correlations of the investigated Ge 20Se80 glass either from Fourier transformation or from RMC simulations. It is intended to confirm the coincidence between the obtained results of both methods. Finally, Raman spectroscopy analysis could strengthen the obtained conclusions.
2 Theoretical background
2.1 Conventional (Fourier) method
According to Faber and Ziman,[16] the total structure factor, S(K), is obtained from the normalized coherent scattered intensity, I c(K), through
where \(K=4\pi \,(\sin \,\theta /\lambda )\) is the transferred momentum, \(\langle f\rangle ^{2}=\sum \nolimits _{i} {(c_{i} f_{i} )^{2}} \) and \(\langle f^{2}\rangle =\sum \nolimits _{i} {c_{i} f_{i}^{2},} \) where c i is the atomic fraction of element i and f(K) the atomic scattering factor.
Fourier transformation of the S(K) data into real space[17] gives the reduced distribution function, G(r), as follows:
where ρ(r) is the local atomic density at a distance r, ρ o the bulk atomic density and M(K) is called the damping factor.[17,18] At short distances (r ≤ 2 Å), see equation (2), G(r) should follow the density line (−4πrρ o) which is used as a quality check of the data. The radial distribution function, defined as the number of atoms lying at distances between r, r+ d r from center of an arbitrary origin atom, is given by
The positions of the first and the second peak in the RDF(r) represent the average values of the first- and second-nearest-neighbor distances r 1 and r 2, respectively. A knowledge of both distances yields a value for the bond angle \({\Theta } =2\sin ^{-1}(r_{2} /2r_{1})\).[5] The area under each peak gives the corresponding coordination number.
2.2 Reverse Monte Carlo method
In the structural analysis using Fourier transformations,[17,19] a modification factor was suggested to reduce the effect of termination data at a finite \(K_{\max }\). This factor in turn, while reduces the spurious oscillations, leads to a broadening of the genuine peaks in g(r). The broadening is wide enough to cause an overlap between the first and second peaks, and consequently introduces significant errors in the obtained structural parameters. One of the main difficulties in the study of glasses and other disordered materials is the production of structural models that agree quantitatively with diffraction data. In normal Monte Carlo simulation, an initial structure is allowed to rearrange in such a way that its energy is minimized. The RMC does not need the inter-atomic potentials and the structural configuration is adjusted so as to minimize instead the difference between the calculated diffraction pattern and that measured experimentally.[20]
Three-dimensional arrangement of N atoms is placed into a cubic cell with periodic boundary conditions. The atomic number density (ρ) should be the same as the experimental value. The positions of the atoms are chosen randomly. The partial pair distribution function[15,21] can be calculated from the initial configuration by
where the superscripts C and o mean ‘calculated’ and ‘old’, respectively, c i is the concentration of atoms type i and n ij (r) is the average number of atoms type j located at distance between r and r+ dr from a central atom of type i. Fourier transform of \(g_{ij}^{\text {Co}} (r)\) to reciprocal space yields the partial static structure factor
where K (= 4 πsin 𝜃/ λ) is the momentum transfer. The total structure factor is calculated as follows:
where f i (K) is the atomic scattering factor of atom type i. The difference between the experimental total structure factor, S E(K), and that calculated from the configuration is given by
where the sum is taken over the m experimental points and σ represents the experimental error. One atom moves at random but if it approaches another atom closer than the cut-off distance, the move is rejected. Otherwise, a new atom is chosen with acceptable move. Then, the new values of the partial pair distribution functions, partial structure factors and the total structure factor can be calculated. The new value of S Cn(K) gives a new difference
where n means ‘new’. If \(\chi _{\text {n}}^{2} <\chi _{\text {o}}^{2}\), the move is accepted and the new configuration becomes the old one. If \(\chi _{\text {n}}^{2} >\chi _{\text {o}}^{2}\), it is accepted with probability \(\exp \left ({{-\left ({\chi _{\text {n}}^{2} -\chi _{\text {o}}^{2}} \right )} / 2} \right )\). Otherwise it is rejected. As the number of accepted atom moves increases, χ 2 will initially decrease until it reaches an equilibrium value. Thus, the atomic configuration corresponding to the equilibrium should be consistent with the experimental total structure factor within the experimental error. From the equilibrium values of the partial pair distribution function, one can calculate the partial coordination number, partial inter-atomic distance and the bond angle distribution.
3 Experimental
Bulk Ge 20Se80 chalcogenide was prepared using the melt-quench technique. High-purity Ge and Se elements (99⋅999%) were, weighed according to their atomic percentages, charged into chemically cleaned silica tube and then sealed under vacuum of ≈1⋅33 ×10−3 Pa. The ampoule was inserted into a furnace where the temperature was raised gradually by rate of 3–4 K/min up to 1300 K and kept constant for 24 h. To get homogeneous melt, the ampoule was frequently rocked inside the furnace at the highest temperature. The quenching was made in ice cold water. The glassy state of the quenched alloy was checked using a Philips (PW-1710) X-ray diffractometer. XRD patterns are recorded at scanning speed of 2⋅4 deg/min using CuK α line (λ=1⋅5418 Å). The experiment was carried out in the scattering angle range 4° ≤ 2 𝜃 ≤ 115° in steps of 0⋅1°, which corresponds to K-range 0.284 ≤ K ≤ 6.874 Å −1. Raman spectra was carried out using the 532 nm line of a diode pumped solid state laser. The scattered light is analyzed with a spectrometer equipped with holographic grating and detected with a Andor Newton CCD camera.
4 Results and discussion
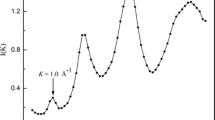
As a starting point, the observed X-ray intensities have been corrected through background subtraction followed by absorption and polarization corrections. The corrected X-ray data are used to calculate the total structure factor[18] as a function of the scattering vector (K=4π sin 𝜃/λ). As shown in figure 1, a FSDP which is commonly observed in covalently bonded materials implies the presence of IRO caused by connecting some of the structural units. In order to determine the position and intensity of the pre-peaks, the S(K) spectra have been analyzed using two pseudo-Voigt functions[1] for the pre- and first-peaks. The pseudo-Voigt function is linear summation of Lorentzian and Gaussian components [c.Gauss.+(1−c)Lorent.],where the Gaussian fraction has a value in the range 0 ≤ c ≤ 1⋅0. The values of the characteristic length (R= 2 π/ K o, where K o is the peak position) and the coherence length (L= 2 π/ ΔK, where ΔK is the half-width at half-maximum of the peak), as calculated from the well-resolved FSDP following the procedures published by, Johnson et al[22] are 5⋅45±0⋅05 and 17⋅44±0⋅52 Å, respectively.

The total structure factor of the Ge 20Se 80 glass as a function of the scattering vector (K). The spectra was analyzed using two pseudo-Voigt functions for the pre- and first-peaks.
The reduced distribution function, G(r), calculated after one damping correction followed by several Kaplow iterations,[23] is shown in figure 2. At small values of r ≤ 1⋅5 Å, see Equation (2), G(r) function shows a straight line with slope is equal to − 4πρ o. The bulk density obtained from the straight portion of G(r), equals (3⋅85± 0.04)×10−2 atom/Å −3. The bulk density can also be calculated from the relation \(\rho _{\text {o}} =N_{\text {A}} \sum \nolimits _{i} {x_{i} d_{i} /A_{i}} , \)where d i , A i and N A are the density, atomic weight of the element i and Avogadro number, respectively.[24] The excellent coincidence between the former value of the bulk density and the calculated one \((\rho _{\text {o}} =3{\cdot }8505\times 10^{-2}~\text {atom}/\text {{\AA }}^{3})\) confirms the high quality of the present glass. The radial distribution function, RDF(r), is shown also in figure 2. At high values of r, the curves should fit the parabola, RDF(r)=4πr 2 ρ o. The broadening appeared in the RDF(r) peaks is expected to cause significant errors in determining the SRO parameters. So instead of RDF(r), the total distribution function, T (r)= RDF(r)/r, is commonly used to get the SRO parameters.[18] The sharpness of the T(r) peaks shown in figure 3 is much better than that appeared in the RDF(r) curve. Gaussian fit of T(r) curve shown in figure 3 has resulted in two well-resolved peaks, where the positions of the first and second peaks are r 1= 2⋅38 Å and r 2= 3⋅82 Å, respectively. These values are in good agreement with those previously obtained by. Rao et al[5]

The reduced distribution function, G(r), and the radial distribution function, RDF (r), versus r of the investigated Ge 20Se 80 glass.

The total distribution function, T(r), versusr of the studied Ge 20Se 80 glass.
The starting point in RMC simulation[21] is to randomly generate the configuration distribution of N=4000 atoms inside a cubic box. The length of the cubic configuration is 23⋅6 Å. According to their atomic percentages, the numbers of Ge and Se atoms inside the cube are 800 and 3200 atoms, respectively. RMC simulation runs for 20 h using the total structure factor fit under the coordination constraints and a minimum approach distance of 1⋅96 Å for any atoms pair. When χ 2 oscillates around an equilibrium value, a three-dimensional molecular image of a disordered structure can be obtained. In order to get an accurate image, the average of five simulation trials was taken. Figure 4 shows the partial pair distribution g Ge−Ge(r), g Ge−Se(r), g Se−Se(r) functions. In fact, most of the important structural parameters such as the coordination number, inter-atomic distance and bond angle distribution can be obtained from the partial pair distribution functions. In the first coordination sphere, the near-zero value of g Ge−Ge(r) indicates that only homopolar Se–Se bonds exist in addition to heteropolar Ge–Se bonds. The average Ge–Ge, Ge–Se and Se–Se bond lengths are, as obtained from the refined RMC model, 2⋅52± 0⋅065, 2⋅43±0⋅065 and 2⋅47±0⋅065 Å, respectively. The average partial coordination numbers, listed in table 1, are close to some extent to those reported by other references.[6,7] Based on the chemical order network model (CONM), the partial coordination numbers are also calculated and listed in table 1. The presence of Se–Se bridges between the tetrahedral units is a possible reason for high partial Se–Se coordination number as compared with that reported previously or computed from the chemically ordered network model. Pan et al[25] have characterized the structural units of the Ge–Se films prepared by pulsed laser deposition using Raman spectroscopy. They concluded that in addition to the basic structural units of edge-sharing GeSe 4/2 tetrahedra, there are Se–Se homopolar bonds in Se-rich GeSe 4 films.

The partial pair distribution g Ge−Ge(r), g Ge−Se(r), g Se−Se(r) functions vs.r of the investigated Ge 20Se 80 glass.
Bond angle distribution functions, Θ(𝜃), are obtained from the final configuration of the investigated glass using triplets program.[7] In which, the bond angle for any reference atom can be calculated from the Cartesian coordinates of the final positions of the surrounding atoms. The Θ(𝜃) functions of the Ge 20Se 80 glass are shown in figure 5. ΘSe−Ge−Se(𝜃) function presents a main peak around 104⋅2°, which is close to the ideal tetrahedral angle of 109°. A small peak appears at 60° can be attributed to the existence of what we can call it as wrong (homo) bonds. During the melt-quench process, the freeze of Ge atoms in some corner positions of the tetrahedral units instead of Se atoms could be responsible for the above small peak. A reverse behavior is given by ΘSe−Se−Se(𝜃) function, where a main peak is located at 60° and a small one at 104⋅2°. Selenium atoms occupying face of a perfect tetrahedron should exhibit internal angles of 60°. The presence of a ΘSe−Se−Se(𝜃) peak at 104⋅2° can be attributed to inter-tetrahedral units. The above bond angle distributions have strongly confirmed that the distorted tetrahedral units in addition to the ideal units are formed inside the cube. These structural units seem to be connected by Se–Se bridges, forming small chains and rings, as previously reported.[24] In the same figure, ΘGe−Ge−Ge(𝜃) function shows a broad distribution from 50° to 150°, which indicates that pairs of tetrahedral units can be connected either by corner or edge share.

Bond angle distribution functions, Θ(𝜃), obtained from RMC simulations.
Fourier transformation of the partial pair distribution gives the corresponding partial scattering factor. The dependence of the partial scattering factors on the scattering vector (K) are shown in figure 6. The partial S(K) are important especially in regions where the FSDP is located. Because of its insignificant contribution to the pre-peak, S Ge−Se(K) function is not shown. It was previously mentioned[26] that the pre-peak is originated by Ge–Ge correlation, which bridging GeSe 4/2 tetrahedra. Recently, Machado et al[7] have used the RMC simulations and noticed the existence of large number of Se–Se pairs in the first coordination shell suggesting that the tetrahedral units are linked by Se–Se bridges. The FSDP appeared in the partial S Ge−Ge(K) and S Se−Se(K) functions indicates that the intermediate range order is not only attributed to Ge–Ge bonds but also to the presence of Se–Se bonds. Summation of the partial scattering factors gives the total RMC scattering factor as a function of the scattering vector. Figure 7 shows a very good coincidence between RMC simulation and the experimental scattering factor to the point no one can distinguish between their values. In the above figure, the RMC curve is shifted downward by 0⋅05 in order to differentiate between their values.

The partial scattering factors, S ij (K), for the studied Ge20Se80 glass.

Experimental scattering factor together with RMC simulation of the studied Ge20Se80 glass. RMC data are shifted downward (–0⋅05) to clarify the coincidence.
Based on the present IRO and SRO parameters, some conclusions about the structural correlations inside the glass matrix are assumed. To confirm such conclusions, Raman spectra measurements of the investigated glass is made and shown in figure 8. Two broad as well as one side (shoulder) peaks have appeared. One of the main peaks located at 267 cm −1 is related to Se–Se pairs. The second main peak located at 200 cm is assigned to the stretching mode of the corner-sharing (CS) GeSe4/2 tetrahedra. The latter peak is accompanied by a shoulder at 215 cm −1, which rises from the vibrations of Se atoms in the four member rings composed of two edge-sharing (ES) tetrahedra.[27] The intensities difference between the peak and its shoulder clarify that the studied glass has a lot of CS tetrahedra and few ES tetrahedra.

Raman spectrum vs. Raman frequency of the investigated Ge20Se80 glass.
5 Conclusions
The FSDP appeared in the structure factor curve implies the presence of IRO caused by connecting some of the structural units. The values of r 1/ r 2 ratio and the corresponding bond angle (Θ), obtained from the conventional (Fourier) method, indicate that the structural units inside the present alloy are Ge(Se1/2)4 tetrahedra connected by chains of the chalcogen atoms. RMC simulations of the X-ray scattering data are useful to compute the partial pair distribution functions, \(g_{_{ij}} (r)\), the partial structure factors, S ij (K), and consequently the total structure factor. The partial structure factors have shown that not only the homopolar Ge–Ge bonds, but also Se–Se bonds are behind the appearance of the FSDP in the total structure factor. The presence of the tetrahedral Ge(Se1/2)4 structural units which connected by Se–Se chains have been confirmed by the simulated values of the partial coordination numbers and the bond angle distributions. Finally, Raman spectra measurements have strongly supported the conclusions obtained either from the calculated Fourier data or from RMC simulations.
References
Wang Y, Ohata E, Hosokawa S, Sakurai M and Matsubara E 2004 J. Non-Cryst. Solids 337 54
Sharma D, Sampath S, Lalla N P and Awasthi A M 2005 Physica B 357 290
Inam F, Shatnawi M T, Tafen D, Billinge S J L, Chen P and Drabold D A 2007 Am. J. Phys.: Condens. Matter 19 455206
Chen G, Inam F and Drabold D A 2010 Appl. Phys. Lett. 97 131901
Rao N R, Krishna P S R, Basu S, Dasannacharya B A, Sangunni K S and Gopal E S R 1998 J. Non-Cryst. Solids 240 221
Tafen D and Drabold D A 2005 Phys. Rev. B 71 54206
Machado K D, de Lima J C, Campos C E M, Gasperini A A M, de Souza S M, Maurmann C E, Grandi T A and Pizani P S 2005 Solid State Commun. 133 411
Hosokawa S, Wang Y, Sakurai M, Bérar J F, Pilgrim W C and Murase K 2003 Nucl. Instrum. Methods Phys. Res. B199 165
Gulbrandsen E, Johnsen H B, Endregaard M, Grande T and Stølen S 1999 J. Solid State Chem. 145 253
Susman S, Price D L, Volin K J, Dejus R J and Montague D G 1988 J. Non-Cryst. Solids 106 26
Salmon P S and Petri I 2003 J. Phys.: Condens. Matter 15 S1509
Warren B E, Krutter H and Morningstar O 1936 J. Am. Ceram. Soc. 19 2025
Chen P, Mitkova M, Tenne D A, Wolf K, Georgieva V and Vergov L 2012 Thin Solid Films 525 141
McGreevy R L and Pusztai L 1988 Mol. Simul. 1 359
McGreevy R L 2001 J. Phys.: Condens. Matter 13 R877
Faber T E and Ziman J M 1965 Philos. Mag. 11 153
Elliott S R 1990 Physics of amorphous materials (New York: Longman) 2nd ed
Moharram A H and Abdel-Basit A M 2005 Physica B 358 279
Szczygielska A, Burian A, Dore J C, Honkimaki V and Duber S 2004 J. Alloys Compd. 362 307
Keen D A and McGreevy R L 1990 Nature 344 423
Johnson R W, Price D L, Susman S, Arai M, Morrison T I and Shenoy G K 1986 J. Non-Cryst. Solids 83 251
Kaplow R, Strong S L and Averbach B L 1965 Phys. Rev. 138 A1336
Machado M D, de Lima J C, Campos C E M, Grandi T A and Pizani P S 2004 J. Chem. Phys. 120 329
Pan R K, Tao H Z, Wang J Z, Wang J Y, Chu H F, Zhang T J, Wang D F and Zhao X J 2013 Optik 124 4943
Fuoss P H, Eisenberger P, Warburton W K and Bienestock A 1981 Phys. Rev. Lett. 46 1537
Dwivedi P K, Tripathi S K, Pradhan A, Kulkarni V N and Agarwal S C 2000 J. Non-Cryst. Solids 266 924
Acknowledgement
This work was supported by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under Grant No. (662-4-D1432). The author, therefore, acknowledges with thanks DSR technical and financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
MOHARRAM, A.H. Short-range order of germanium selenide glass. Bull Mater Sci 38, 111–117 (2015). https://doi.org/10.1007/s12034-014-0824-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12034-014-0824-5