
Abstract
The morbidity of oral squamous cell carcinoma (OSCC) has been rising year after year, making it a major global health issue. But the molecular pathogenesis of OSCC is currently unclear. To study the potential pathogenesis of OSCC, the differentially expressed genes (DEGs) were screened, and multiple databases were used to perform the tumor stage, expression, prognosis, protein–protein interaction (PPI) networks, modules, and the functional enrichment analysis. Moreover, we have identified SP110 as the key candidate gene and conducted various analyses on it using multiple databases. The research indicated that there were 211 common DEGs, and they were enriched in various GO terms and pathways. Meanwhile, one DEG is significantly related to short disease-free survival, four are associated with overall survival, and 12 DEGs have close ties with tumor staging. Additionally, the SP110 is significantly associated with methylation level, HPV status, tumor staging, gender, race, tumor grade, age, and overall/disease-free survival of oral cancer patients, as well as the immune process. The copy number variation of SP110 significantly affected the abundance of immune infiltration. Therefore, we speculate that SP110 could be used as the diagnostic and therapeutic biomarker for OSCC, and can help to further understand oral carcinogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Head and neck squamous cell carcinoma (HNSC) has become an important global public health problem due to its high mortality and poor prognosis [1]. OSCC is the most common aggressive subtype of HNSC [2]. Despite the progress in surgical procedures and chemo-radiotherapy approaches, the 5-year survival rate and overall prognosis of most OSCC patients are still unsatisfactory [3]. Meanwhile, the occurrence and progression of OSCC are thought to be multi-stage complex processes involving intricate regulatory networks. Currently, with the development of sequencing technology, researchers find that DEGs may lead to the transformation of normal oral cells into OSCC [4]. Therefore, it is urgent and necessary to screen for and identify DEGs, differentially expressed signaling pathways, and potential molecular mechanisms in carcinogenesis to aid in the exploration of novel treatment strategies for OSCC.
The SP110 is one of the chromatin “readers” in humans and a member of the speckled protein family. Related research found that the “readers” can also be used as cancer therapeutic targets [5]. A number of studies have illustrated that SP110 is associated with immunity, advanced liver disease without veno-occlusive disease (VOD), tuberculosis infection, and transcriptional regulation [6, 7]. Recently, emerging evidence indicates that the function of SP110 is correlated with multiple cancers [8,9,10]. However, there are few studies on the mechanisms of SP110 in oral cancer.
In the present research, multiple databases were selected to screen and validate the DEGs. SP110 was found to be not only significantly up-regulated in multiple databases, human OSCC tissues, and cell lines but also related to stage and overall/disease-free survival. Our results indicate that SP110 may be involved in the molecular regulation mechanism of OSCC and could be used as a novel potential diagnostic, prognostic, and therapeutic biomarker of OSCC. More importantly, the present research is allowing us to screen potential biomarkers and contribute to a better understanding of oral carcinogenesis.
Materials and Methods
Datasets Acquisition
Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) is a public genomics database that is freely available online [11]. After review, three datasets were selected: The GSE138206 dataset contains six oral cancer tissues and six contralateral normal tissues. All patients were diagnosed with OSCC, including five males and one female. And the dataset is based on microarray data obtained from the “GPL570 ([HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array)” platform. The GSE23558 dataset includes 27 oral cancer samples, four independent controls, and one pooled control. All control group samples are oral cavity tissue from healthy donors. All 27 patients were diagnosed with oral cancer, including 20 males and 7 females. Among the five healthy controls, there are three males, one female, and one pooled sample. This dataset was obtained from the GPL6480 (Agilent-014850 Whole Human Genome Microarray 4x44K G4112F) platform. The GSE37991 dataset consists of oral cancer samples obtained from 40 male OSCC patients with a history of regular alcohol consumption, betel chewing, and smoking. Simultaneously, the adjacent non-tumor epithelium of these 40 patients was taken as a normal control. This dataset was obtained from the GPL6883 (Illumina HumanRef-8 v3.0 expression beadchip) platform.
Data Processing
The DEGs and the P value, adjusted P value, and |logFC| were obtained by GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/), and the adjusted P value < 0.01 and |logFC| ≥ 1 were used as screening thresholds. Meanwhile, the DEGs common to the three datasets were identified for further function analysis.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
In the present study, the GO and KEGG pathway enrichment analyses were performed by ClueGO (v2.5.7) and CluePedia (v1.5.7). The adjusted P < 0.05 was chosen as the significance cutoff point.
PPI Network Construction
The Search Tool for the Retrieval of Interacting Genes (STRING, https://string-db.org/) [12] was used to detect and construct the potential PPI networks of the DEGs using the default parameters. And the Cytoscape (v3.7.1) was used to perform further analysis, identify, and visualize the PPI networks.
Hub Gene Screening and Module Analysis
CytoHubba (v0.1) was used to further investigate the DEGs in the PPI network using the default parameters, and the top 20 node genes were identified as hub genes for next-step analysis. In addition, another plugin for Cytoscape, Molecular Complex Detection (MCODE, v1.5.1), was used to screen the most significant modules in the PPI network with default parameters.
Expression, Stage and Survival Analysis
To explore the role of identified hub genes in oral carcinogenesis, the Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer-pku.cn/#index) [13] was used to perform expression, stage, and survival analyses of the top 20 hub genes using the default parameters.
Analysis of Oncomine, ENCORI and UALCAN Database
Oncomine (https://www.oncomine.org/) is an integrated tumor sequencing database [14], and ENCORI (https://rnasysu.com/encori/) is an open-source database including multiple modules [15]. They were used to further analyze the transcription level of SP110 in different tumors and detect the expression level of SP110 with default parameters. Furthermore, to increase the credibility of our results, the UALCAN (http://ualcan.path.uab.edu/) database [16] was used to detect the relative expression level, methylation level, HPV status, patient’s gender, tumor grade, patient’s race, and patient’s age in the SP110 using the default parameters.
OSCC Sample Collection
The OSCC samples with pathologically definitive diagnoses were provided by Professor Sun from Shanxi Provincial People’s Hospital, which included five OSCC samples and their three normal counterparts. The written informed consent for tissue donation for research was obtained, and the relevant research was approved by the ethics committees of the Shanxi Provincial People’s Hospital (2019-05).
Cell Line Culture
Human OSCC cell line SCC-9 was purchased from FuHeng Biology Co., Ltd. (Shanghai, China); CAL-33 and SCC-25 were provided by Henan Provincial People’s Hospital. And the HOK (human normal oral keratinocyte cell line) and HN6 cell lines were provided by the School and Hospital of Stomatology, Shanxi Medical University. All cell lines were cultured for fewer than six months after resuscitation and evaluated for mycoplasma on a regular basis. Additionally, OSCC cells were grown in Dulbecco’s modified Eagle medium (DMEM, BOSTER, China), and HOK cells were grown in Hyclone 1640 medium (Hyclone, USA). Meanwhile, the medium contains 10% fetal bovine serum (Gibco, USA) and 1% penicillin–streptomycin (Solarbio, China). All cells were maintained in a 37 °C incubator with 5% CO2.
Expression Validation of Candidate Gene
Total RNA from human OSCC tissue and cell lines was extracted by TRIzol (Invitrogen, USA). Hereafter, they were reverse transcribed to cDNA by the Prime Script™ RT Master Mix kit (Takara, Japan). And then qPCR was performed on StepOne Plus (ABI, USA). The reaction conditions were: 95 °C for 30 s, 40 cycles of 95 °C for 5 s, and 60 °C for 30 s; following, a melting curve analysis was performed. Primers were synthesized by Sangon Biotech. The 2−ΔΔCt method was used to analyze the results, which used GAPDH as an endogenous control.
The following primers were used: GAPDH forward 5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse 5′-TGGTGAAGACGCCAGTGGA-3′; SP110 forward 5′-GGAACGCAAAGAACTGGAAAC-3′ and reverse 5′-CATGGAAGACTCGTGGACAAG-3′.
Co-expression Analysis of SP110
The top 100 co-expressed genes significantly associated with SP110 were screened from the Oncomine database using the default parameters. Meanwhile, the PPI network of the screened genes was constructed by the STRING database with default parameters and visualized by Cytoscape.
Functional Analysis of Co-expressed Genes
GO and KEGG enrichment analyses were performed by DAVID [17] (https://david.ncifcrf.gov/), and adjusted P < 0.05 is the threshold. The association between SP110 expression and the abundance of immune infiltrates, including various immune cells, was analyzed by the TIMER (https://cistrome.shinyapps.io/timer/) database with default parameters. Moreover, the relationship between the level of tumor immune infiltration and the Copy Number Variation (CNV) of SP110 was also evaluated by the TIMER database using the default parameters, and P < 0.05 is the threshold.
Statistical Analysis
The quantitative results of qPCR were analyzed using the IBM SPSS Statistics 24 software. And the LSD t-test was used to compare groups. The values are expressed as mean ± standard error of the mean, and P < 0.05 was used to indicate statistical significance. In the differential expression and functional enrichment analysis, the Benjamin and Hochberg false discovery rate (FDR) method was used to correct the adjusted P value and the occurrence of false positive results. The cutoff standard was defined as an adjusted P value < 0.01 and |logFC| ≥ 1. The significance level of GO and KEGG enrichment was calculated using a threshold of an adjusted P value < 0.05. And the co-expression analysis of SP110 was evaluated in the oncomine database using Spearman’s correlation analysis, and a P value < 0.05 was considered statistically significant. Other analyses are performed according to the statistical analysis methods and default parameters provided by the corresponding software.
Brief Description of Bioinformatics Analysis
Initially, we identified three oral cancer datasets from the GEO database and subsequently utilized GEO2R to set an adjusted P value < 0.01 and |logFC| ≥ 1 as the threshold for screening DEGs. Perform GO and KEGG enrichment analysis on the selected DEGs using ClueGO (v2.5.7) and CluePedia (v1.5.7), with adjusted P < 0.05 as the threshold for significant differences. The PPI network of DEGs was constructed using STRING with default parameters and further analyzed and visualized using Cytoscape (v3.7.1); the plugin MCODE (v1.5.1) with default settings was used to filter key modules in the PPI network; Another plugin, CytoHubba (v0.1) with default parameters, was employed to investigate key node genes in the PPI network, and the top 20 nodes were identified as hub genes for subsequent analysis. In order to further validate the expression of hub genes and explore their impact on oral cancer staging and survival, we analyzed the expression, staging, overall survival, and disease-free survival of the top 20 hub genes using GEPIA with default parameters. Select the gene (SP110) that is significantly differentially expressed in oral cancer and has a significant impact on its staging, overall survival, and disease-free survival as the key candidate gene. SP110, which is significantly differentially expressed in oral cancer and has a significant impact on its staging, overall survival, and disease-free survival, has been selected as a key candidate.
Oncomine and ENCORI databases (with their default parameters) were utilized to analyze the expression levels of SP110 in different types of tumors. Meanwhile, the UALCAN database was employed to investigate the expression level, methylation level, and HPV status of SP110 in oral cancer, as well as the relationship between SP110 and factors such as the patient’s gender, tumor grade, race, and age in oral cancer. To ensure the reliability of the screening results, the expression of SP110 was examined in human oral cancer tissues and human oral cancer cell lines. Furthermore, the co-expression genes of SP110 were identified using the Oncomine database with its default parameters, and the top 100 co-expressed genes were screened for further research. Subsequently, the PPI network for the top 100 co-expressed genes was constructed using the aforementioned method and visualized. The top 100 co-expressed genes were subjected to GO and KEGG enrichment analyses using DAVID with default parameters. Finally, the TIMER database was used to analyze the relationship between SP110 expression and the abundance of immune infiltrates (including various immune cells), as well as the association between tumor immune infiltration levels and the CNV of SP110.
Results
Identification of DEGs
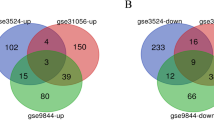
Among the datasets, GSE138206 has six OSCC tissues, six tissues adjacent to cancer, and six contralateral normal tissues. The 27 OSCC tissues in GSE23558 were compared with five control oral cavity tissues. GSE37991 contains 40 OSCC tissues and 40 non-tumor tissues. Figure 1A showed that there were 1041 DEGs in the GSE138206, including 450 upregulated differentially expressed genes (uDEGs) and 591 downregulated differentially expressed genes (dDEGs). And 2080 DEGs were identified from GSE23558, of which 830 are uDEGs and 1250 are dDEGs (Fig. 1B). From GSE37991, 2092 DEGs were identified, consisting of 887 uDEGs and 1205 dDEGs (Fig. 1C). Subsequently, the intersection of the DEGs in three datasets was obtained by Venn analysis. In the intersection, a total of 211 DEGs (Fig. 1D) were detected, including 135 dDEGs (Fig. 1E) and 76 uDEGs (Fig. 1F). Finally, a heat map of the 211 DEGs in the three datasets is shown in Fig. 2A–C.

Analysis of DEGs. Volcano plots of DEGs in GSE138206 (A), GSE23558 (B), and GSE37991 (C) datasets. The red represent up-regulated DEGs, and the green represent down-regulated DEGs. D–F Venn graph of DEGs in gene expression profiling datasets. D The total DEGs in the three datasets. E Down-regulated DEGs in the three datasets. F Up-regulated DEGs in the three datasets

Heat map of the DEGs. A GSE138206. B GSE23558. C GSE37991
GO Enrichment Analyses
From the dDEGs, 69 significant differentially GO terms, including 49 biological processes, 15 molecular functions, and five cellular components, are identified (Fig. 3A). Meanwhile, for the uDEGs, there are 35 significant differentially GO terms containing 32 biological processes, one molecular function, and two cellular components (Fig. 3B). Moreover, the dDEGs are mainly enriched in the inflammatory response to antigenic stimulus, scavenger receptor activity, negative regulation of cell junction assembly, the lateral plasma membrane, and NAD binding. And the uDEGs are mainly contained in response to vitamin D, the collagen catabolic process, the basement membrane, double-stranded RNA binding, and the negative regulation of type I interferon production.

GO enrichment of the DEGs. A GO histogram of the intersecting down-regulated DEGs in three datasets. B GO histogram of the intersecting up-regulated DEGs in three datasets. C Pie chart for GO categories of the intersecting down-regulated DEGs in three datasets. D Pie chart for GO categories of the intersecting up-regulated DEGs in three datasets. E The GO term interaction network in three datasets with intersecting down-regulated DEGs. F The interaction network of GO terms in the intersecting up-regulated DEGs in three datasets
Furthermore, based on the analysis of the plugin of Cytoscape, the enriched GO terms of the dDEGs were mainly divided into 14 groups, among which the long-chain fatty acid metabolic process is the largest group (Fig. 3C); the enriched GO terms of the uDEGs were mainly divided into 12 groups, and the type I interferon signal is the largest group (Fig. 3D). At the same time, the networks of the GO terms indicate that, in the GO terms of dDEGs, the long-chain fatty acid metabolic process has the most connections with other terms (Fig. 3E), and for the GO terms of uDEGs, type I interferon signal has the most connections with other terms (Fig. 3F).
KEGG Enrichment Analysis
We analyzed all the DEGs together in KEGG enrichment, and they are significantly enriched in the retinol metabolism, hepatitis C, Extracellular Matrix (ECM) receptor interaction, small cell lung cancer, drug metabolism, and chemical carcinogenesis signaling pathways (Fig. 4A). In addition, we performed interactive analysis on these significantly enriched pathways and visualized them with cytoscape. Figure 4B not only reveals the interaction between the significantly enriched pathways but also shows the DEGs contained in the pathways. In the KEGG network, we could learn that many of the genes are common along different pathways; in other words, they overlap among pathways.

KEGG enrichment of the DEGs. A KEGG histogram of the intersecting DEGs in three datasets. B KEGG pathway interaction network of intersecting DEGs
PPI Network Analysis and Hub Gene Screening
There are 157 nodes and 288 edges in the PPI network (Fig. 5A). To further explore the interactions, the topological features were analyzed, and the Number of nodes, Topological coefficient, Neighborhood connectivity, and Clustering coefficient were analyzed. The results revealed that, in the network, most of the nodes had a low score and a few nodes were highly connected to the others (Fig. 5B). Moreover, cluster analysis was performed by MCODE APP, and we found that the network mainly has nine clusters, of which the top three are presented in Fig. 5C, D. And the top cluster contained 15 nodes with 85 edges and had the highest score (Fig. 5C). The top two clusters possessed six nodes and 42 edges and also had a close connection (Fig. 5D). Meanwhile, the third cluster included four nodes and six edges (Fig. 5E). Furthermore, we screened the 20 most significant hub genes using the CytoHubba plugin. And Table 1 provided more information about them.

PPI network, topological features, and significant modules. A PPI network. Red nodes represent up-regulated DEGs, and green nodes indicate down-regulated DEGs. B Analysis results of the degrees, topological coefficients, neighborhood connectivity, and clustering coefficients of the PPI network. C–E The top three significant modules in the PPI network
Expression, Stage, and Survival Analysis of the Hub Genes
The GEPIA database was used to perform expression analysis on the top 20 hub genes. At the same time, we also analyzed their role in the stage and prognosis of the OSCC. The results showed that the expression of all 20 hub genes in the GEPIA database was consistent with our previous analysis (Fig. 6). Meanwhile, we also found that, in the top 20 genes, SP110, IFIT3, OAS3, OAS2, DDX58, UBE2L6, CXCL10, CXCL11, GBP5, EPSTI1, MMP3, and SPP1 are closely related to tumor staging (Fig. 7A), and high expression of SP110, MMP1, SERPINE1, and SPP1 is associated with short overall survival (Fig. 7B). Through the multiple screenings mentioned above and by considering various relevant factors comprehensively, we found that among the top 20 key candidate genes, SP110, MMP1, SERPINE1, and SPP1 are significantly associated with the overall survival rate of oral cancer patients. To further explore the potential association between candidate genes and oral cancer, we conducted an analysis of disease-free survival for these four genes. The results indicated that out of the four genes, only SP110 was significantly associated with the disease-free survival rate of oral cancer patients, whereas MMP1, SERPINE1, and SPP1 showed no significant correlation with the disease-free survival rate (Fig. 7C). The high expression of SP110 leads to a significant shortening of the disease-free survival period in oral cancer patients (Fig. 7C). Based on these findings, we discovered that SP110 is significantly differentially expressed and is associated with tumor stage, overall survival, and disease-free survival. Therefore, we selected the SP110 for further verification and analysis.

Expression verification in the GEPIA database of the top 20 significant DEGs. The left column (red) indicates cancer tissues; the other (black) indicates normal tissues

Stage analysis and overall/disease-free survival analysis of the top 20 significant DEGs. A Stage analysis. B Overall survival analysis. C Disease-free survival analysis
SP110 Expression and Function Analysis
In order to better verify the expression and prognostic value of SP110, we further performed analysis using the Oncomine, ENCORI, and UALCAN databases. We compared the mRNA expression of SP110 in cancer and normal samples in the Oncomine database (Fig. 8A). Moreover, the SP110 was significantly overexpressed in the cancer group in all three datasets (the Ginos OSCC, Peng OSCC, and Toruner OSCC) (Fig. 8B).The ENCORI pan-cancer analysis showed that the SP110 was significantly up-regulated in human HNSC samples (Fig. 8C). In the UALCAN database, we also found that the SP110 was significantly up-regulated in oral tumor tissues (Fig. 8D). Meanwhile, to further validate the findings of the analysis described above, we used qPCR to further validate SP110 expression in human OSCC tissues and human OSCC cells. We also observed high expression of SP110 in oral cancer specimens (Fig. 8E). And compared to the HOK cell line, SP110 was significantly up-regulated in the SCC-9, CAL-33, SCC-25, and HN6 cell lines, and it has the highest expression level in the HN6 cell line (Fig. 8F).

Pan-cancer analysis of SP110 expression. A Red represents up-regulated and blue represents down-regulated expression in tumor specimens. The darker shadow indicates a higher significance. The numbers indicate the number of datasets. B SP110 expression in OSCC specimens in three oncomine datasets. C SP110 expression in OSCC specimens in the ENCORI database. D The expression of SP110 in OSCC samples in the UALCAN database. E SP110 expression in human oral cancer specimens. F SP110 expression in the human oral cancer cell lines
Further analysis of the UALCAN database indicated that the promoter methylation level of SP110 was significantly elevated in the primary tumor (Fig. 9A). Significant differences in HPV status were found not only between the normal and tumor groups, but also between the HPV + ve and HPV − ve tumor groups. Furthermore, the results show that the expression of SP110 was significantly up-regulated in both the HPV + and HPV − groups. Meanwhile, SP110 expression was lower in the HPV + ve group than in the HPV − ve group (Fig. 9B).The SP110 was significantly high expressed in female patients (Fig. 9C), indicating that the differential expression of the SP110 was closely related to the patient’s gender. At the same time, SP110 expression was also related to tumor grade (Fig. 9D). Furthermore, the patient’s race analysis indicates that the expression of SP110 in African-Americans was lower than that in Asians, and there were no differences between the other races (Fig. 9E). Furthermore, the patient’s age analysis results revealed that there were significantly differences between the Age (41–60 years)-vs-Age (81–100 years) and Age (61–80 years)-vs-Age (81–100 years) (Fig. 9F).

Analysis of the clinical role of SP110 in HNSC by the UALCAN database. Differential expression of the SP110 is significantly correlated with promoter methylation level (A), HPV status (B), the patient’s gender (C), tumor grade (D), race (E), and the age of the patient (F)
Analysis of Co-expression, Enrichment, Infiltrated Immunity, and CNV
Based on the above, we in-depth screened the top 100 genes co-expressed with SP110 in oral cancer from the Peng Oral Cavity Squamous Cell Carcinoma dataset of the Oncomine database (Fig. 10A). And their PPI network was generated in STRING and imputed into the Cytoscape for visualization and further analysis (Fig. 10B). Furthermore, the GO functional analysis revealed that the co-expressed genes were significantly enriched in defense response, TAP complex, immune response, ATPase activity, peptide antigen binding, innate immune response, peptide antigen-transporting, chemokine activity, and type I interferon signaling pathways (Fig. 10C). KEGG enrichment analysis suggested that the co-expressed genes were mainly enriched in the Toll-like receptor, herpes simplex infection, antigen processing and presentation, and Chemokine and Cytokine–cytokine receptor interaction signaling pathways (Fig. 10D).

Bioinformatics analysis of the genes co-expressed with SP110. A The top 100 genes co-expressed with SP110. B PPI network of the top 100 genes. GO (C) and KEGG pathway (D) enrichment analysis of the top 100 genes. E Correlation between SP110 expression and immune infiltration level. F Correlation of SP110 expression with somatic CNV and immune infiltration levels of six immune cells in OSCC
Based on the functional enrichment analysis, we speculate that differential expression of SP110 may play an important role in the oral tumor immune response. The results indicated that the different infiltrated immune cells were all significantly related to SP110 expression levels in oral cancer (Fig. 10E). Besides, the relevance between immune infiltration and somatic CNV was also determined by TIMER. The results indicated that arm-level gain was related to the infiltration of B cells, macrophages, CD8+ T cells, CD4+ T cells, neutrophils, and dendritic cells, while arm-level deletion was only associated with B cell infiltration. And the deep deletion was also related to CD4+ T cells (Fig. 10F).
Discussion
The International Classification of Diseases defines oral cancer as “the cancer of the oral cavity and pharynx,” and the most common form is oral squamous cell carcinoma. More than 330,000 deaths from oral cancer are reported annually [18]. Although the diagnostic and therapeutic methods for OSCC have increased markedly, the overall survival rate of this life-threatening disease is still at a low level [19]. OSCC occurs and develops in a complicated and multi-step sequential process that includes multiple gene and multi-stage changes. Oral cancer is mainly treated by surgery, radiotherapy, and chemotherapy technologies such as platinum, doxorubicin, five-fluorouracil, etc. However, it has significant toxic and side effects, and the mechanism of the occurrence and malignant transformation of oral cancer is still unknown [2]. So it is of key importance in identifying new potential diagnostic, treatment, and prognostic biomarkers to improve targeted therapy for oral cancer.
As research progressed, it was discovered that the pathogenesis of oral cancer was strongly linked to genetic mutations [20]. Nowadays, with the rapid development of DEG screening technology such as microarrays and high-throughput sequencing, it is easier for researchers to study oral cancer at the genetic level. At the same time, the development of various tumor-related databases has also made it easier to obtain tumor-related DEGs. The GEO database has been widely used to explore the DEGs involved in the diagnosis, prognosis, and therapeutics of various tumors. Similarly, we also use GEO, STRING, GEPIA, Oncomine, UALCAN, ENCORI, and TIMER databases to screen and verify DEGs and perform functional analysis and immune-related analysis.
Analysis of the differential expression in the three datasets in the GEO database shows that there were 211 genes that were significantly differentially expressed in all three datasets. Then, GO and KEGG functional and PPI network interaction analyses for DEGs were performed. At the same time, we characterized the network and obtained the sub-networks and hub genes. Moreover, we selected the top 20 prominent hub genes to verify their expression in other databases (GEPIA) and analyzed their roles in the stage and overall/disease-free survival of oral cancer. The analysis shows that all 20 hub genes have a consistent expression trend in the GEPIA and GEO databases. In addition, in the top 20 genes, there were 12 hub genes (SP110, IFIT3, OAS3, OAS2, DDX58, UBE2L6, CXCL10, CXCL11, GBP5, EPSTI1, MMP3, and SPP1) that were significantly related to tumor staging, four (SP110, MMP1, SERPINE1, and SPP1) that were closely related to overall survival, and one (SP110) that was associated with disease-free survival.
Furthermore, we also found that the up-regulation of SPP1 is associated with both an advanced tumor stage and a worse prognosis. Similarly, Hu et al. pointed out that lipid metabolism-related genes SPP1 and SERPINE1 have the potential to be used to predict the prognosis of oral cancer [21]. Yang et al. discovered that the mRNA and protein levels of the cancer-related gene SPP1 are overexpressed in OSCC samples and are associated with a poor prognosis. They hypothesized that SPP1 could be a potential therapeutic target for inhibiting metastasis in OSCC [22]. Zou et al. identified the key candidate, SPP1, from the complex PPI network, and they confirmed that SPP1 is closely related to OSCC survival. They also found that SPP1 could regulate oral cancer proliferation, migration, and invasion. Meanwhile, SPP1 is considered a prognostic or therapeutic target for OSCC [23, 24].
Simultaneously, the research also demonstrated that up-regulation of SP110 is not only significantly related to the advanced pathological stage but also closely correlated to a worse overall/disease-free survival rate. Inspired by these discoveries, we infer that SP110 could be considered a promising therapeutic biomarker with future clinical significance. Therefore, we decided to select SP110 for in-depth analysis and detection. The results of our subsequent analysis and verification show that the expression level of SP110 in a variety of tumor databases, including Oncomine, UALCAN, and ENCORI, is consistent with the GEO and GEPIA databases. The qPCR verification illustrated that SP110 was significantly overexpressed in human oral tumor tissues and multiple cell lines. Moreover, we also found that the expression of SP110 in the HN6 cell line is the highest. Therefore, in subsequent experiments, the HN6 cell line can be used as a potential candidate cell line and combined with siRNA technology to verify the impact of reduced SP110 expression on oral cancer cell function after interference. Further bioinformatics analysis of SP110 found that the differential expression of SP110 was markedly correlated with the methylation level, HPV status, gender, tumor grade, race, and age of oral cancer patients, which indicates that SP110 has the potential to be a pre-cancer diagnosis and pathological grading for oral cancer, and it may also affect the occurrence and development of oral cancer by regulating methylation. Next, the interaction analysis of SP110 and enrichment analysis of its interaction genes demonstrated that SP110 is significantly associated with immune-related genes and significantly enriched in immune-related pathways and GO terms. Thus, we speculate that SP110 could be regarded as a potential therapeutic target for OSCC. In subsequent research, efforts could be focused on reducing the expression of SP110 in oral cancer to inhibit its progression. In addition, the above results indicate that SP110 may also play an important role in the immune regulation of oral tumors. So it is also possible to combine SP110 with tumor immunity in order to explore a novel treatment method for oral cancer. Ultimately, immune-related analysis of SP110 suggests that dysregulation of SP110 is related to the infiltration levels of six immune cells (CD4C T cells, CD8C T cells, B cells, macrophages, neutrophils, and DCs); at the same time, the differential expression of SP110 is also associated with their copy number variation. The results indicate that the dysregulation of SP110 can affect the occurrence and progression of oral cancer by affecting copy number variation and the infiltration of related immune cells. Based on the above analysis, we believe that SP110 could act as a potential immunotherapeutic target with future clinical significance.
There are only a few studies on SP110 in cancer, but some related reports were found to be consistent with our findings. According to the literature, the transcription regulator SP110 is one of the most frequently up-regulated oncogenes in pregnancy-associated breast cancer. And the research emphasized that the SP110 might be the potential target for breast cancer, which controls the development of breast cancer and could improve fetal implantation [25]. Hu et al. found that SP110 was overexpressed in both mouse and human tumors by establishing an animal model system of mammary cancer [26]. A study of a gene signature based on B cells illustrated that SP110, as one of the B cell-specific genes, was an independent risk factor for overall survival in lung adenocarcinoma [8]. Through literature consulting, it was found that currently only one article has reported the relationship between SP110 and oral cancer. It pointed out that SP110 was significantly overexpressed in oral cancer. Furthermore, SP110 exhibits a high propensity for mutation in oral cancer cell lines, and these mutations may be associated with the disease. Additionally, the mutation profiles of SP110 and SP140 are complex, and these genes may mediate immune cell transcriptional regulation and cell apoptosis through epigenetic regulation [27].
Human SP110 were first observed to exhibit elevated levels in both peripheral blood leukocytes and spleen [5]. And relevant studies show that SP110 could play an indispensable role in the field of other biological functions. Wang et al. pointed out that SP110 is included in remodeling and formation of the chromatin, and upregulation of SP110 leads to fetal liver veno-occlusive disease with immunodeficiency [28]. SP110 also has the potential to be a targeted drug for the treatment of infantile influenza, according to a comprehensive analysis of genes involved in the immune system and virus defense modules [29]. Chang et al. indicated that the polymorphism in SP110 plays a role in controlling the genetic susceptibility of humans to latent and active tuberculosis infections [30]. Sengupta et al. pointed out that SP110 is recruited on the FBP1 promoter rich in H3K18Ac and promotes the recruitment of the acetylase SIRT2 at this site in the presence of HBV. SIRT2, in turn, brings its interactor and transcriptional activator into HNF4αit to the promoter, ultimately leading to the loss of DNA methylation near the homologous site. And the regulation of FBP1 driven by SP110 can promote the progression of hepatitis mediated hepatocellular carcinoma [31]. The research on veno occlusive disease shows that, the mutation of SP110 is one of the root causes of veno-occlusive diseases with immunodeficiency (VODI) [32]. Leu et al. suggested that SP110b regulates NF-κB activity, leading to the production of TNF-α and the concomitant upregulation of NF-κB-induced anti-apoptotic gene expression, thereby inhibiting IFN-γ-mediated monocyte and/or macrophage death. Therefore, SP110b can serve as a potential target for regulating the body’s immunity [33]. Xiaogang et al. found that the rs722555 SNP in the SP110 gene may be a risk factor for tuberculosis in the Mongolian population in the study of genotyping detection [34]. A study combining immunodeficiency and advanced liver disease has shown that functional deletion mutations in the SP110 gene can cause familial venous occlusive disease with immunodeficiency [7]. The analysis of SP110 deletion mutants showed that the interaction between the N-terminal fragment of SP110 (amino acids 1–276) and the NF-κB subunit p50 in the cytoplasm plays a crucial role in the downregulation of TNF-α promoter activity driven by p50 in the nucleus [35]. Knocking down SP110 significantly reduced the viral DNA load in the culture medium supernatant by activating the type I interferon response pathway. Moreover, SP110 can differentially regulate several direct target genes of hepatitis B virus protein X (HBx, a viral cofactor) and is a novel interacting agent of HBx [36].
To our knowledge, our study reported a relationship between significant up-regulation of SP110 and tumor stage, overall/disease-free survival, HPV status, tumor grade, patients’ age and patients’ gender of oral cancer for the first time. Additionally, we discovered that SP110 not only interacts with a variety of genes but is also significantly associated with immune infiltration. Our study indicates that the occurrence and development of OSCC may be regulated by SP110 and its related genes, and SP110 has the capacity to act as a new target for diagnosis, treatment, and prognosis of oral cancer. And the findings expanded current knowledge about the role of SP110 in oral cancer and may help increase treatment options and improve diagnostic accuracy. Furthermore, research on the cellular, in vivo, and protein expression levels during OSCC tumorigenesis and development should be performed to further validate our analysis results and explore the potential of the key pathways as biomarkers.
There are still some potential limitations and addressing methods for this study: (i) The relevant sequencing data was obtained from the GEO database rather than generated by the authors. Since bioinformatics analysis relies on data from public databases, systematic errors in the selected data sources may lead to bias. To overcome this limitation, we validated the screening results using multiple databases to increase the reliability and consistency of the results. (ii) During the bioinformatics analysis process, steps such as data cleaning, screening, and integration may result in information loss or bias. Therefore, when selecting datasets, we ensure that the raw data used has undergone rigorous quality control to avoid biases resulting from poor data quality. And in the analysis process, standardized and normalized data processing and analysis were adopted to reduce the impact of human factors on the results. (iii) The focus of this study is to screen and predict key candidate genes for oral cancer based on bioinformatics methods. In order to improve the reliability of screening and prediction results, we further validated its expression in human oral cancer tissues and cell lines using qPCR experiments. (iv) Different functional analysis methods have different advantages and disadvantages. In this study, the limitations of the DAVID tool include that it only uses the number of genes without considering gene expression levels or differential expression values. Meanwhile, an artificial threshold is required to obtain the genes of interest or differential expression. Moreover, it usually focuses on the most significant genes while ignoring those without significant differences, which may lead to the loss of genes with lower significance but more crucial roles, resulting in reduced detection sensitivity. Furthermore, pathway analysis may be limited by known pathway data and might fail to accurately identify newly discovered pathways or those not yet widely recognized. In order to overcome the abovementioned shortcomings and better screen and predict key candidate genes for oral cancer, we comprehensively utilized various analysis methods besides pathway analysis, such as stage analysis, overall and disease-free survival analysis, immune infiltration analysis, etc., to obtain more comprehensive and accurate results regarding the candidate genes. In addition, functional validation experiments can be conducted in subsequent studies to determine the potential regulatory effect of differentially expressed SP110 on oral cancer. And the specific avenues for future research and putative translational therapy scenarios are as follows:
The specific avenues for future research and putative translational therapy scenarios The present research found that SP110 is significantly overexpressed in oral cancer, and its overexpression is markedly correlated with shortened overall/disease-free survival rates and advanced tumor staging in patients with oral cancer. Meanwhile, SP110 is also significantly associated with the infiltration of immune cells. Therefore, we speculate that the inhibition of SP110 expression may have a significant inhibitory effect on oral cancer. The validation of SP110 expression in oral cancer cells has demonstrated that its expression is higher in HN6 and CAL-33 cell lines than in other cell lines. Thus, when using siRNA technology for further in vitro and in vivo in-depth research and validation, performing related experiments with these two cell lines may yield more pronounced results. In subsequent studies, siRNA technology can be used to reduce the expression of SP110 in oral cancer to investigate its specific mechanism in the occurrence and development of oral cancer. At the same time, it can also be studied whether a reduction in SP110 expression in oral cancer can regulate the tumor immune microenvironment of oral cancer by regulating the infiltration of immune cells, thereby inhibiting the malignant progression of oral tumors.
The novel nano-materials have not only been proven to have good therapeutic effects in various diseases but can also carry relevant anti-cancer genes or inhibitors of oncogenes into cancer cells to achieve a synergistic suppression of tumors with gene therapy [37]. Meantime, the protection provided by nanocarriers can also overcome the shortcomings of genetic formulations being easily degraded and difficult to treat in vivo. Therefore, after clarifying the primary anticancer mechanisms of SP110, a nano anticancer drug that can effectively carry and deliver the siRNA of SP110 should be designed. Integrating SP110 with nanomedicine allows for a multifaceted approach to treating oral cancer from both gene therapy and nanotherapy perspectives. In addition, not only can SP110 regulate the infiltration of related immune cells, but the nanomedicine itself may also exert certain effects on the immune microenvironment of tumors [38]. Based on this, after using nanodrugs to load SP110 siRNA, we can further explore their regulation of the immune microenvironment of oral tumors. Thereby, by combining nanomedicine therapy, gene therapy, and immunotherapy, we can form an innovative, more comprehensive, and safer strategy for the treatment of oral cancer.
Conclusion
SP110 is significantly overexpressed in both oral cancer tissues and cells, and its overexpression is markedly correlated with shortened overall/disease-free survival rates and advanced tumor staging in patients with oral cancer. Meanwhile, SP110 is also significantly associated with the infiltration of immune cells. SP110 has the potential to serve as a biomarker for the treatment and diagnosis of oral cancer.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Chi, H., Peng, G., Song, G., Zhang, J., Xie, X., Yang, J., & Yang, G. (2024). Deciphering a prognostic signature based on soluble mediators defines the immune landscape and predicts prognosis in HNSCC. Frontiers in Bioscience (Landmark Edition), 29(3), 130. https://doi.org/10.31083/j.fbl2903130
Chang, H. C., Yang, C. C., Loi, L. K., Hung, C. H., Wu, C. H., & Lin, Y. C. (2024). Interplay of p62-mTORC1 and EGFR signaling promotes cisplatin resistance in oral cancer. Heliyon, 10(6), e28406. https://doi.org/10.1016/j.heliyon.2024.e28406
Stojanović, M. Z., Krasić, D., Radović, P., Trajkovic, M., Ćosić, A., Petrović, V., & Pešić, P. (2024). Nutritional status and quality of life in patients with oral squamous cell carcinoma before and after surgical oncological treatment: A single-center retrospective study. Medical Science Monitor, 30, e943844. https://doi.org/10.12659/msm.943844
Xu, G., Li, L., Wei, J., Xiao, L., Wang, X., Pang, W., & Song, G. (2019). Identification and profiling of microRNAs expressed in oral buccal mucosa squamous cell carcinoma of Chinese hamster. Scientific Reports, 9(1), 15616. https://doi.org/10.1038/s41598-019-52197-3
Fraschilla, I., & Jeffrey, K. L. (2020). The Speckled Protein (SP) family: Immunity’s chromatin readers. Trends in Immunology, 41(7), 572–585. https://doi.org/10.1016/j.it.2020.04.007
Cai, L., Li, Z., Guan, X., Cai, K., Wang, L., Liu, J., & Tong, Y. (2019). The research progress of host genes and tuberculosis susceptibility. Oxidative Medicine and Cellular Longevity, 2019, 9273056. https://doi.org/10.1155/2019/9273056
Delmonte, O. M., Baldin, F., Ovchinsky, N., Marquardsen, F., Recher, M., Notarangelo, L. D., & Kosinski, S. M. (2020). Novel missense mutation in SP110 associated with combined immunodeficiency and advanced liver disease without VOD. Journal of Clinical Immunology, 40(1), 236–239. https://doi.org/10.1007/s10875-019-00715-3
Han, L., Shi, H., Luo, Y., Sun, W., Li, S., Zhang, N., & Xie, C. (2020). Gene signature based on B cell predicts clinical outcome of radiotherapy and immunotherapy for patients with lung adenocarcinoma. Cancer Medicine, 9(24), 9581–9594. https://doi.org/10.1002/cam4.3561
Korakiti, A. M., Moutafi, M., Zografos, E., Dimopoulos, M. A., & Zagouri, F. (2020). The genomic profile of pregnancy-associated breast cancer: A systematic review. Frontiers in Oncology, 10, 1773. https://doi.org/10.3389/fonc.2020.01773
Pryor, J. G., Brown-Kipphut, B. A., Iqbal, A., & Scott, G. A. (2011). Microarray comparative genomic hybridization detection of copy number changes in desmoplastic melanoma and malignant peripheral nerve sheath tumor. American Journal of Dermatopathology, 33(8), 780–785. https://doi.org/10.1097/DAD.0b013e31820dfcbf
Clough, E., Barrett, T., Wilhite, S. E., Ledoux, P., Evangelista, C., Kim, I. F., & Soboleva, A. (2024). NCBI GEO: Archive for gene expression and epigenomics data sets: 23-year update. Nucleic Acids Research, 52(D1), D138–D144. https://doi.org/10.1093/nar/gkad965
Szklarczyk, D., Kirsch, R., Koutrouli, M., Nastou, K., Mehryary, F., Hachilif, R., & Von Mering, C. (2023). The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Research, 51(D1), D638–D646. https://doi.org/10.1093/nar/gkac1000
Li, C., Tang, Z., Zhang, W., Ye, Z., & Liu, F. (2021). GEPIA2021: Integrating multiple deconvolution-based analysis into GEPIA. Nucleic Acids Research, 49(W1), W242–W246. https://doi.org/10.1093/nar/gkab418
Rhodes, D. R., Kalyana-Sundaram, S., Mahavisno, V., Varambally, R., Yu, J., Briggs, B. B., & Chinnaiyan, A. M. (2007). Oncomine 3.0: Genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia, 9(2), 166–180. https://doi.org/10.1593/neo.07112
Li, J. H., Liu, S., Zhou, H., Qu, L. H., & Yang, J. H. (2014). starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Research, 42(1), D92–D97. https://doi.org/10.1093/nar/gkt1248
Chandrashekar, D. S., Karthikeyan, S. K., Korla, P. K., Patel, H., Shovon, A. R., Athar, M., & Varambally, S. (2022). UALCAN: An update to the integrated cancer data analysis platform. Neoplasia, 25, 18–27. https://doi.org/10.1016/j.neo.2022.01.001
Sherman, B. T., Hao, M., Qiu, J., Jiao, X., Baseler, M. W., Lane, H. C., & Chang, W. (2022). DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Research, 50(W1), W216–W221. https://doi.org/10.1093/nar/gkac194
Mughees, M., Sengupta, A., Khowal, S., & Wajid, S. (2021). Mechanism of tumour microenvironment in the progression and development of oral cancer. Molecular Biology Reports, 48(2), 1773–1786. https://doi.org/10.1007/s11033-020-06054-6
Xu, G., Yang, Y., Yang, J., Xiao, L., Wang, X., Qin, L., & Song, G. (2023). Screening and identification of miR-181a-5p in oral squamous cell carcinoma and functional verification in vivo and in vitro. BMC Cancer, 23(1), 162. https://doi.org/10.1186/s12885-023-10600-3
Ichimura, N., Urata, Y., Kobayashi, T., & Hibi, H. (2024). Mutational landscape of oral mucosal melanoma based on comprehensive cancer genomic profiling tests in a Japanese cohort. Oral Oncology, 152, 106807. https://doi.org/10.1016/j.oraloncology.2024.106807
Hu, Q., Peng, J., Chen, X., Li, H., Song, M., Cheng, B., & Wu, T. (2019). Obesity and genes related to lipid metabolism predict poor survival in oral squamous cell carcinoma. Oral Oncology, 89, 14–22. https://doi.org/10.1016/j.oraloncology.2018.12.006
Yang, W., Zhou, W., Zhao, X., Wang, X., Duan, L., Li, Y., & Hong, L. (2021). Prognostic biomarkers and therapeutic targets in oral squamous cell carcinoma: A study based on cross-database analysis. Hereditas, 158(1), 15. https://doi.org/10.1186/s41065-021-00181-1
Thakore, V. P., Patel, K. D., Vora, H. H., Patel, P. S., & Jain, N. K. (2024). Up-regulation of extracellular-matrix and inflammation related genes in oral squamous cell carcinoma. Archives of Oral Biology, 161, 105925. https://doi.org/10.1016/j.archoralbio.2024.105925
Xie, W., Sun, G., Xia, J., Chen, H., Wang, C., Lin, J., & Wang, P. (2023). Identification of novel tumor-associated antigens and evaluation of a panel of autoantibodies in detecting oral cancer. BMC Cancer, 23(1), 802. https://doi.org/10.1186/s12885-023-11247-w
Thanmalagan, R. R., Naorem, L. D., & Venkatesan, A. (2017). Expression data analysis for the identification of potential biomarker of pregnancy associated breast cancer. Pathology Oncology Research, 23(3), 537–544. https://doi.org/10.1007/s12253-016-0133-y
Hu, Y., Sun, H., Drake, J., Kittrell, F., Abba, M. C., Deng, L., & Aldaz, C. M. (2004). From mice to humans: Identification of commonly deregulated genes in mammary cancer via comparative SAGE studies. Cancer Research, 64(21), 7748–7755. https://doi.org/10.1158/0008-5472.can-04-1827
Chang, K. W., Lin, C. E., Tu, H. F., Chung, H. Y., Chen, Y. F., & Lin, S. C. (2020). Establishment of a p53 null murine oral carcinoma cell line and the identification of genetic alterations associated with this carcinoma. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms21249354
Wang, T., Ong, P., Roscioli, T., Cliffe, S. T., & Church, J. A. (2012). Hepatic veno-occlusive disease with immunodeficiency (VODI): First reported case in the U.S. and identification of a unique mutation in Sp110. Clinical Immunology, 145(2), 102–107. https://doi.org/10.1016/j.clim.2012.07.016
Zarei Ghobadi, M., Mozhgani, S. H., Farzanehpour, M., & Behzadian, F. (2019). Identifying novel biomarkers of the pediatric influenza infection by weighted co-expression network analysis. Virology Journal, 16(1), 124. https://doi.org/10.1186/s12985-019-1231-8
Chang, S. Y., Chen, M. L., Lee, M. R., Liang, Y. C., Lu, T. P., Wang, J. Y., & Yan, B. S. (2018). SP110 polymorphisms are genetic markers for vulnerability to latent and active tuberculosis infection in Taiwan. Disease Markers, 2018, 4687380. https://doi.org/10.1155/2018/4687380
Sengupta, I., Mondal, P., Sengupta, A., Mondal, A., Singh, V., Adhikari, S., & Das, C. (2022). Epigenetic regulation of fructose-1,6-bisphosphatase 1 by host transcription factor Speckled 110 kDa during hepatitis B virus infection. FEBS Journal, 289(21), 6694–6713. https://doi.org/10.1111/febs.16544
Marquardsen, F. A., Baldin, F., Wunderer, F., Al-Herz, W., Mikhael, R., Lefranc, G., & Recher, M. (2017). Detection of Sp110 by flow cytometry and application to screening patients for veno-occlusive disease with immunodeficiency. Journal of Clinical Immunology, 37(7), 707–714. https://doi.org/10.1007/s10875-017-0431-5
Leu, J. S., Chen, M. L., Chang, S. Y., Yu, S. L., Lin, C. W., Wang, H., & Yan, B. S. (2017). SP110b controls host immunity and susceptibility to tuberculosis. American Journal of Respiratory and Critical Care Medicine, 195(3), 369–382. https://doi.org/10.1164/rccm.201601-0103OC
Cui, X., Yuan, T., Ning, P., Han, J., Liu, Y., Feng, J., & Wu, C. (2022). Polymorphisms in the ASAP1 and SP110 genes and its association with the susceptibility to pulmonary tuberculosis in a Mongolian population. Journal of Immunology Research, 2022, 2713869. https://doi.org/10.1155/2022/2713869
Leu, J. S., Chang, S. Y., Mu, C. Y., Chen, M. L., & Yan, B. S. (2018). Functional domains of SP110 that modulate its transcriptional regulatory function and cellular translocation. Journal of Biomedical Science, 25(1), 34. https://doi.org/10.1186/s12929-018-0434-4
Sengupta, I., Das, D., Singh, S. P., Chakravarty, R., & Das, C. (2017). Host transcription factor Speckled 110 kDa (Sp110), a nuclear body protein, is hijacked by hepatitis B virus protein X for viral persistence. Journal of Biological Chemistry, 292(50), 20379–20393. https://doi.org/10.1074/jbc.M117.796839
Drabczyk, A., Kudłacik-Kramarczyk, S., Jamroży, M., & Krzan, M. (2024). Biomaterials in drug delivery: Advancements in cancer and diverse therapies-review. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms25063126
Wu, X., Li, Y., Wen, M., Xie, Y., Zeng, K., Liu, Y. N., & Zhao, Y. (2024). Nanocatalysts for modulating antitumor immunity: Fabrication, mechanisms and applications. Chemical Society Reviews. https://doi.org/10.1039/d3cs00673e
Funding
This work was supported by the National Natural Science Foundation of China (31772551 and 31970513), Shanxi Scholarship Council of China (2021-086), the special found for Science and Technology Innovation Teams of Shanxi Province (202204051002032), the Central Government’s Guide to Local Science and Technology Development Fund (YDZJSX2022A060), and the Natural Science Foundation of Shanxi Province (20210302124093).
Author information
Authors and Affiliations
Contributions
Conception and design: Guoqiang Xu and Guohua Song. Administrative support: Guohua Song and Jiping Gao. Provision of study materials or patients: Guoqiang Xu, Litao Qin, Jiping Gao, Xiaotang Wang and Guohua Song. Collection and assembly of data: Guoqiang Xu, Xiaotang Wang and Litao Qin. Data analysis and interpretation: Guoqiang Xu. Manuscript writing: Guoqiang Xu and Xiaotang Wang. Final approval of manuscript: All authors.
Corresponding author
Ethics declarations
Competing Interests
The authors have no competing interests to declare that are relevant to the content of this article.
Ethical Approval
This study was performed in line with the principles of the Declaration of Helsinki. The written informed consent for tissue donation for research was obtained, and the relevant research was approved by the ethics committees of the Shanxi Provincial People’s Hospital (2019-05).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xu, G., Wang, X., Qin, L. et al. SP110 Could be Used as a Potential Predictive and Therapeutic Biomarker for Oral Cancer. Mol Biotechnol (2024). https://doi.org/10.1007/s12033-024-01212-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12033-024-01212-8