
Abstract
Neutrophil extracellular traps (NETs) play a central role in chronic airway diseases. However, the precise genetic basis linking NETs to the development of severe asthma remains elusive. This study aims to unravel the molecular characterization of NET-related genes (NRGs) in severe asthma and to reliably identify relevant molecular clusters and biomarkers. We analyzed RNA-seq data from the Gene Expression Omnibus database. Interaction analysis revealed fifty differentially expressed NRGs (DE-NRGs). Subsequently, the non-negative matrix factorization algorithm categorized samples from severe asthma patients. A machine learning algorithm then identified core NRGs that were highly associated with severe asthma. DE-NRGs were correlated and subjected to protein–protein interaction analysis. Unsupervised consensus clustering of the core gene expression profiles delineated two distinct clusters (C1 and C2) characterizing severe asthma. Functional enrichment highlighted immune-related pathways in the C2 cluster. Core gene selection included the Boruta algorithm, support vector machine, and least absolute contraction and selection operator algorithms. Diagnostic performance was assessed by receiver operating characteristic curves. This study addresses the molecular characterization of NRGs in adult severe asthma, revealing distinct clusters based on DE-NRGs. Potential biomarkers (TIMP1 and NFIL3) were identified that may be important for early diagnosis and treatment of severe asthma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Asthma, a heterogeneous inflammatory disease, is driven by diverse immune mechanisms [1]. Although the introduction of inhaled glucocorticoids has proven effective in reducing asthma-related deaths, the global prevalence of asthma has continued to rise in many countries over the past decade. This trend underscores the critical role of gene-environment interactions, particularly in genetically predisposed populations. Phenotypes, complex traits shaped by a mixture of genetic and environmental influences, serve as critical determinants in asthma diagnosis, treatment strategies, and prognosis of future risk events [2,3,4,5]. In particular, the majority of asthmatics have type 2 eosinophilic inflammation. However, in severe asthma, a significant proportion of patients have neutrophil-predominant lung inflammation [6]. Despite the clinical significance of severe asthma, our understanding of the contribution of non-type 2 immune mechanisms to asthma remains limited. Whether neutrophils are active participants or mere "bystanders" in the asthmatic context remains a mystery [7]. In addition, there is a lack of effective and feasible biomarkers to differentiate non-Th2 asthmatics and their response to treatment [2].
The unraveling of neutrophil biology reached new depths with the revelation of neutrophil extracellular traps (NETs) intricate reticular structures released into the extracellular milieu upon neutrophil activation. These structures consist of DNA backbone complexes embedded within multi-protein DNA–protein complexes. Initially characterized as a defense mechanism designed to engulf and neutralize invading microorganisms, the evolving research landscape has fostered a growing body of evidence highlighting the pivotal role of NETs in chronic inflammatory airway diseases, including asthma and chronic obstructive pulmonary disease [8,9,10]. In the context of persistent inflammatory responses, NETs not only exert direct cytotoxic effects on lung epithelial and endothelial cells but also exacerbate damage by releasing proteases into the adjacent extracellular environment. In addition, NETs exert deleterious effects by exposing self-antigens and immunostimulatory proteins and by activating plasma cell-like dendritic cells to release interferon [11]. The discovery of NETs brings us to a new cellular and molecular mechanism, and their unique biological activity may provide a potential new therapeutic target for patients with non-type 2 asthma. An in-depth understanding of the key pathways driven by NETs in shaping asthma pathology holds the promise of refining patient stratification into more precise subgroups, thereby improving clinical outcomes. However, the current detailed and comprehensive studies of NET-related genes (NRGs) in severe asthma still require further in-depth exploration.
This study aimed to comprehensively analyze the association between NRGs and severe asthma and to identify reliable molecular clusters and biomarkers based on NRGs to provide more accurate guidance for the diagnosis and treatment of severe asthma. We first searched for differentially expressed NRGs (DE-NRGs) in patients with severe asthma using the Gene Expression Omnibus (GEO) database and NRGs summarized in previous literature and databases. The Non-negative matrix factorization (NMF) based on DE-NRGs classified patients with severe asthma into two clusters with different molecular profiles. We have also identified core genes associated with severe asthma through various machine learning methods and experimental validation to provide more accurate information for early diagnosis of severe asthma.
Materials and Methods
Data Collection and Processing
GSE74986 is a dataset focused on moderate-to-severe asthma, sourced from the GEO database (https://www.ncbi.nlm.nih.gov/gds/). The experimental platform employed for this dataset is Agilent's GPL6480 platform. The GSE74986 dataset consists of RNA samples derived from bronchoalveolar lavage cells obtained from 86 subjects. Among these, 74 samples were collected from individuals with asthma, including 28 classified as moderate asthmatics and 46 as severe asthmatics. The remaining 12 samples were obtained from healthy subjects serving as controls. In this study, we used gene expression profiling data from 46 patients diagnosed with severe asthma, along with 12 healthy controls. In addition, we chose the GSE64913 and GSE76262 datasets as independent validation sets because both datasets were designed to compare the differences in gene expression profiles between severe asthma and healthy subjects, the sample size and data quality control were relatively better, the sampling sites were derived from airways and bronchi, and both datasets have been reported and used in the literature. To acquire gene symbols for each probe matrix, matching platform files were utilized, and subsequent analysis involved the normalization of all microarray datasets using the "limma" R package [12].
For the identification of NRGs in this study, we retrieved information from various sources, including the GeneCards database [13] (http://www.genecards.org), the OMIM database [14](https://www.omim.org/), the NCBI gene database [15] (https://www.ncbi.nlm.nih.gov/), as well as pertinent literature on the subject [16, 17]. We assembled a total of 403 genes associated with NETs, and further details can be found in supplementary Table 1.
Identification of DE-NRGs
Differential expression analyses were performed using the "limma" R package [12] to compare two sample types: patients with severe asthma and healthy subjects. A difference threshold of |logFC|> 1 and an adjusted p-value of < 0.05 were used to identify significantly differentially expressed genes (DEGs). By intersecting the DEGs with the NRGs, we obtained the DE-NRGs specific to the samples from severe asthma patients and the samples from healthy controls.
Functional Enrichment Analysis
To explore the role of DEGs in severe asthma, we used the "clusterprofiler" package [18] in R for gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. We set a q-value < 0.05 as the threshold value.
Non-Negative Matrix Factorization Algorithm
Asthma phenotypes are influenced by both genetic and environmental factors, resulting in variable responses to different treatments. To investigate the variation among patients with severe asthma, we used the R package "NMF" [19] to perform cluster analysis based on the expression data of DE-NRGs to identify potential molecular subtypes. For cluster analysis, we applied the "Brunet" criterion and performed 30 iterations. The average contour width of the common membership matrix was calculated using the "consensusmap" function within the "NMF" R package. The resulting consensus matrix was visualized and evaluated for clustering stability, using the average contour width as a measure of stability. Cluster stability was assessed by calculating the cophenetic correlation coefficient, a commonly used measure of clustering stability. The cophenetic correlation coefficient reflects the similarity between the original dissimilarity matrix and the matrix obtained from the clustering result. Higher values of the cophenetic correlation coefficient indicate greater clustering stability. The clustering performance of the model was evaluated by calculating the residual sum of squares (RSS). The optimal k was determined by calculating the differences between the cophenetic correlation coefficients and selecting the index position with the highest difference. Based on the above algorithm and the optimal k, we categorized the samples from patients with severe asthma into distinct molecular clusters.
Gene Set Variation Analysis
The mRNA expression profiles of the two molecular clusters in severe asthma were analyzed using the gene set variation analysis (GSVA) [20], a non-parametric unsupervised analysis method primarily used to evaluate the enrichment characteristics of gene sets in microarray and transcriptome data. GSVA allows the identification of gene sets associated with specific biological processes, pathways, or functions. By calculating the expression activity score of a gene set, we can assess functional differences between samples and determine whether different pathways are enriched. In this GSVA analysis, we used Hallmarker, KEGG, and GO-related gene sets as reference sets. The GSVA score for each gene set was quantified to assess its enrichment level.
Calculation of Immune Cell Infiltration
To assess immune infiltration between patients with severe asthma and healthy controls, we used ssGSEA [21] to quantify the relative abundance of immune cells and pod-plot to compare immune cell infiltration.
Identification of NETs-Related Hub Gene Based on the Machine Learning Algorithm
Three machine learning algorithms, Boruta [22], Support Vector Machine (SVM) [23], and Least Absolute Shrinkage and Selection Operator (LASSO) [24] were used to screen for key genes associated with DE-NRGs. The Boruta algorithm is a machine learning algorithm used for feature selection, which is mainly used to extract the most informative features in a dataset, thus improving the performance and generalization of the model, which can be implemented using the "Boruta" R package [22]. SVM is a feature selection algorithm that finds the most discriminative features for a task by combining the classification or regression capabilities of SVM with the feature culling strategy of random forest to identify the subset of features that are most informative for a classification or regression task, which can be implemented using the "e1070" R package [25]. This algorithm can be implemented using the "e1070" R package. In LASSO regression, we used 10 resampling iterations based on tenfold cross validation to select the best lambda values. Area under the curve (AUC) values were calculated using receiver operating characteristic (ROC) curves to identify potential candidate biomarkers with diagnostic significance for severe asthma.
Donor Consent
Blood samples were collected in accordance with the Declaration of Helsinki, and written informed consent was obtained from study participants. All samples were collected with the approval of the Ethics Committee of the China-Japan Friendship Hospital (No:2021-GZR-70). 10 ml of peripheral blood was collected from three patients with severe asthma and three healthy subjects.
Quantitative Real-Time PCR
Total RNA was isolated and extracted using Tritol reagent (#9109, TaKaRa Bio), and reverse transcription was performed using a PrimeScript™ RT Reagent Kit (#RR036A, TaKaRa Bio). Quantitative real-time PCR was then performed according to the Hieff® qPCR SYBR Green Master Mix (Low Rox Plus; Yeasen) protocol using specific primers. GAPDH was used as an internal control and the 2−△△Ct method was used to calculate relative mRNA levels. The primer sequences for TIMP1 and NFIL3 in this study were as follows Forward: 5'-CCTCTGGCATCCTGTTGTTG-3', Reverse: 5'- GGTATAAGGTGGTCTGGTTG-3'; Forward: 5'-CCGAGAACGTCGGAAACTGA-3', Reverse: 5'-TTGGCTTTGATCCGGAGCTT-3'.
Neutrophil Isolation and NET Induction
Human peripheral blood neutrophils were isolated using the Polymorphprep isolation solution method as described previously [26]. Experiments were performed in RPMI-1640 (without phenol red) supplemented with 10 mM HEPES and 5% heat-inactivated FBS. For NET experiments, cells were seeded at 5*10^5 cells/well (24-well plate) and stimulated with or without 100 nm PMA (#HY-18739, Medchemexpress) for 3 h. Neutrophils inoculated onto PDL-coated glass coverslips were stained as previously described [27]. Briefly, after neutrophil induction, cells were fixed with 4% paraformaldehyde and samples were then stained with Sytoxgreen (#KGA260, Keygen). Images were captured using a Leica orthogonal fluorescence microscope.
Results
Identification of DEGs
Figure 1 shows the flow chart of our study. Differential expression analysis was performed on the gene expression profiles of 12 healthy control samples and 46 severe asthma samples. A total of 1681 severe asthma-related DEGs were identified, consisting of 568 up-regulated genes and 1113 down-regulated genes, based on the conditions described in the Materials and Methods section. Volcano and heat maps of the DEGs are shown in Fig. 2A and Fig. 2B, respectively.

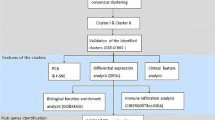
Flowchart of this study. NETs, Neutrophil Extracellular Traps; NRGs, NET-associated genes; SA, Severe Asthma; NMF, Non-Negative Matrix Factorization; GSVA, Gene Set Variance Analysis; SVM-RFE, Support Vector Machine Recursive Feature Elimination; LASSO, Least Absolute Shrinkage and Selection Operator; ROC, Receiver Operating Characteristic Curves

Identification of DEGs. A Volcano plots: Volcano plots were constructed based on fold change values > 1 and adjusted P values < 0.05. B The Heatmap of DEG shows the trend of gene expression in different samples and is represented by different colors. The top 100 genes were ranked according to the adjusted P value, as shown in the figure. DEGs: differentially expressed genes
Identification of DE-NRGs and Their Functional Enrichment Analysis, Gene Expression Patterns, and Construction of PPI Networks
By integrating 1681 DEGs with our collection of 403 NRGs, 50 DE-NRGs were finally obtained (Fig. 3A). To visualize their expression patterns in different samples, we generated a heat map of the DE-NRGs (Fig. 3B). Furthermore, the gene relationship network graph (Fig. 3C) depicted the correlation among these DE-NRGs. In addition, we constructed a network of DE-NRGs that included 20 other genes, namely DSTN, BNIP3L, AGER, CFL1, TLR3, NCF2, AMOTL2, PDLIM1, CFL2, S100A9, SYNE4, CYBA, TIRAP, AGGF1, IRAK4, C1QB, CCR6, IL1B, LY96, and FCER1G (Fig. 3D). To gain insight into the potential role of DE-NRGs in severe asthma, we performed a functional enrichment analysis. The results of GO analysis revealed that DE-NRGs were associated with various biological processes, including granule lumen secretion, cellular response to bacterial-derived molecules, positive regulation of leukocyte activation, cytokine receptor binding, and signaling receptor activation activity (Fig. 3E). In addition, KEGG enrichment analysis revealed significant enrichment in pathways such as the NOD-like receptor signaling pathway, Toll-like signaling pathway, TNF signaling pathway, NF-κB signaling pathway, HIF-1 signaling pathway, and IL-17 signaling pathway (Fig. 3F).

Identification of DE-NRGs and their gene expression patterns, PPI networks, and functional enrichment analysis. A Gene overlaps between DEGs and NRGs. B Heat map of DE-NRGs expression. C PPI network of DE-NRGs and their interacting proteins. (D) Correlation matrix of DE-NRGs. E GO enrichment analysis of DE-NRGs in terms of biological processes, cellular components, and molecular functions. F KEGG pathway analysis of DE-NRGs. DEGs, differentially expressed genes; NRGs, NET-associated genes; DE-NRGs, differentially expressed NRGs; PPI, protein–protein interaction; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes
Stratification of Patients with Severe Asthma based on DE-NRGs
To differentiate between severe asthma patients with varying degrees of severity, a cluster analysis was conducted using the NMF algorithm. This analysis was based on the expression profiles of the 50 DE-NRGs across all severe asthma patient samples. By assessing co-expression, dispersion, and profile metrics, the optimal number of clusters was determined to be k = 2 (Fig. 4A, B).

Non-negative matrix factorization (NMF) analysis of samples from patients with severe asthma. A The consensus plot for NMF clustering at k = 2 is presented (A). B The distributions of co-expression, residuals, residual sum of squares (RSS), and profile metrics are visualized on a scale ranging from 2 to 10 (B). C A heatmap illustrates the expression patterns of DE-NRGs (C). D A box plot displays the expression levels of 16 DE-NRGs between the two clusters (D). RSS, residual sum of squares; DE-NRGs, differentially expressed NET-related genes. *P < 0.05, **P < 0.01, ***P < 0.001
Accordingly, all samples from severe asthma patients were categorized into two clusters using the NMF algorithm: C1 cluster (n = 23) and C2 cluster (n = 23). The expression patterns of DE-NRGs in these two severe asthma clusters are illustrated in Fig. 4C. Notably, the C1 cluster exhibited upregulated expression of DEFA3, DEFB1, DNASE1L3, DYSF, HTRA1, JAK3, MMP9, ROMO1, S100A12, S100A8, SLPI, TECPR2, TICAM1, and TNFSF12. Conversely, the C2 cluster displayed upregulated expression of ACTB, ACTG1, APEX1, CASP1, CLEC7A, CTSC, CYBB, FCGR3B, FN1, HAVCR2, IL1A, ITGB1, LYZ, MME, MSR1, MYD88, OLR1, PIK3CA, PLAUR, PTPN22, RIPK3, SUCNR1, TLR4, and VDAC1 (Fig. 4D).
Enrichment Analysis of GSVA in Different Clusters of Severe Asthma
To elucidate the different biological features between the two severe asthma clusters, we performed GSVA enrichment analysis using the Hallmarks gene set (h.all.v7.2.symbols.gmt) based on the MSigDB database. By histogram presentation, we could observe that the C1 cluster was up-regulated in the following biological processes compared to the C2 cluster: apical junctions, KRAS signaling, allograft rejection, and coagulation. In contrast, the C1 cluster was downregulated in apoptosis, angiogenesis, P53 signaling, TNFA signaling via NFκB, inflammatory response, interferon-gamma response, PI3K-AKT-MTOR signaling, IL6-JAK-STAT3 signaling, MTORC1 signaling, and protein secretion (Fig. 5).

Plot of GSVA results. Histogram of GSVA enrichment results in the Hallmarker reference set. GSVA, Gene Set Variation Analysis
Differences in Immune Infiltration Characteristics Between Subclusters
The heatmap (Fig. 6A) showing immune cell correlations revealed significant positive correlations between neutrophils and resting or activated dendritic cells, follicular helper T cells, and naive B cells. Conversely, neutrophils showed significant negative correlations with regulatory T cells, γδ T cells, resting NK cells, and mast cells. The results of the ANOVA analysis (Fig. 6B) showed variations in the number of immune cells in different subclusters, indicating changes in the immune microenvironment among these subclusters. Simultaneously, box plot results (Fig. 6C) showed distinct trends in immune cell abundance between the C1 and C2 clusters. The C2 cluster showed a higher abundance of neutrophils, eosinophils, naive B cells, follicular helper T cells, and resting and activated dendritic cells compared to the C1 cluster. In contrast, the C1 cluster showed a higher abundance of γδ T cells, resting NK cells, plasma cells, memory B cells, naive CD4 T cells, monocytes, M0 phase macrophages, and resting mast cells compared to the C2 cluster.

Analysis of immune infiltration in subclusters of severe asthma. A Correlation heat map of 22 infiltrating immune cells. B Heat map of differentially expressed immune cells between two clusters. C Box plots of differentially expressed immune cells between two clusters. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Correlation Analysis of DE-NRGs with Immune Cell Infiltration
We investigated the relationship between immune cell ratios and the expression of the 50 DE-NRGs in individuals with severe asthma. This investigation aimed to uncover potential biomarkers associated with immune cell ratios. The correlation heatmap (Fig. 7) visually depicted these associations. Notably, the heatmap revealed that the expression levels of certain genes, including ACTN4, CCL5, DNASE1L3, JAK3, TECPR2, TNFSF12, DEFB1, DEFA3, S100A12, S100A8, SLPI, DYSF, HTRA1, TICAM1, ROMO1, and MMP9, were negatively correlated with the neutrophil ratio. Conversely, the expression of the remaining 34 genes showed a positive correlation with the neutrophil ratio.

Results of correlation analysis between DE-NRGs and immune cells. Correlation heatmap showing the correlation between DE-NRGs in mRNA expression profiles and immune cells. *P < 0.05, **P < 0.01, ***P < 0.001
Machine Learning Algorithm Identifies NETs Related Central Genes in Severe Asthma
In LASSO logistic regression, a selection of 16 genes emerged as potential central candidates, as determined by the optimal lambda value of 0.01134282 (Fig. 8A, B). Among the different models, the SVM model showed the lowest classification error when considering four candidate genes: SLC44A2, TIMP1, S100A12, and NFIL3 (Fig. 8C). Using the Boruta feature selection method to predict feature attributes, the changing Z-scores are shown in Fig. 8D. Finally, the Boruta algorithm identified a set of 26 genes (Fig. 8E). Finally, the three algorithms together selected the genes SLC44A2, TIMP1, and NFIL3, which were identified as central genes (Fig. 8F). We then entered the validation phase by testing the three identified candidate genes in two different datasets—GSE64913 and GSE76262. The results, as shown by the ROC curves, underline a promising diagnostic potential for TIMP1 (with AUC values of 0.683 and 0.71) and NFIL3 (with AUC values of 0.737 and 0.671). Unfortunately, SLC44A2 showed less favorable diagnostic accuracy with AUC values of 0.417 and 0.514, respectively (Fig. 9). In addition, we performed further validation using the chronic lung disease without asthma dataset (GSE47460), which again showed specific expression of TIMP1 and NFIL3 in the neutrophil-associated severe asthma differential gene (Supplementary Table 2 and supplementary Fig. 1). These results highlight the prospective applicability of TIMP1 and NFIL3 as predictive biomarkers for severe asthma.

Identification of core genes in severe asthma. A LASSO coefficient profiles of 16 selected genes and generation of coefficient distribution plots for log sequences. B Selection of the best-penalized coefficients lambda by a tenfold cross-validation process with 10 replicates. lambda values generate the minimum average binomial deviation used for feature selection. C RMSE of candidate central gene combinations for the SVM algorithm. Minimum classification error obtained with four candidate genes considered. (D) Z-score variation plot. E Boruta's algorithm selected 26 feature genes and ranked them in order of importance. F Venn diagram showing the overlap of candidate genes in the above three algorithms. LASSO, least absolute shrinkage and selection operator; SVM, support vector machine; RMSE, root mean square error

Verification of the performance of centralized center genes. (A–C) GSE64913 (D–F) GSE76262
Validation of TIMP1 and NFIL3 Expression and Extent of NETs Release in Severe Asthma
Both independent data sets suggested that TIMP1 and NFIL3 might be available as predictive markers for severe asthma, and to further validate the reliability of the results, we performed an experimental verification. We isolated peripheral blood neutrophils from healthy individuals and patients with severe asthma, respectively, and verified the mRNA expression of TIMP1 and NFIL3 in neutrophils from healthy individuals and patients with severe asthma by qPCR, which showed that the expression of TIMP1 and NFIL3 was significantly upregulated in severe asthma group compared to healthy control group (p < 0.05; Fig. 10A). In addition, we treated neutrophils with Phorbol 12-myristate 13-acetate, a stimulant widely used to understand the neutrophil pathway and NETs formation[28], and the results showed that patients in the severe asthma group had a significantly higher level of NET release than healthy control group (Fig. 10B).

Experimental validation procedure. A mRNA expression of TIMP1 and NFIL3 in peripheral blood neutrophils of control and severe asthma groups. B Fluorescence microscopy experiments showing the level of extracellular DNA release after stimulation with 100 nm PMA for 180 min (Images represent 3 independent experiments with neutrophils from different donors). PMA, Phorbol Myristate Acetate. **p < 0.01
Discussion
In this investigation, we used the GEO database to shed light on gene expression levels in both healthy controls and patients with severe asthma. To provide a molecular characterization of NRGs in severe asthma, further analysis of 50 DE-NRGs showed that the vast majority of them had a strong tendency to co-express. This observation suggests a potential concerted action of these genes in the intricate pathogenesis of severe asthma. Protein–protein interaction (PPI) analysis revealed that the genes that interact with the DE-NRGs are predominantly involved in responding to stimuli from biogenic molecules such as bacteria and their constituents. In addition, these genes play a key role in orchestrating the production and release of interleukin-6 (IL-6). It has been found that the origin, efficacy in microbial eradication, or potential deleterious effects of NETs may adopt different profiles depending on the characteristics inherent to different pathogenic microorganisms, including factors such as the presence of virulence traits, microbial load, and microorganism dimensions, among others [29]. Previous study has found that IL-6 exerts a robust influence on the initiation of NETs formation [30]. In the context of critically ill COVID-19 patients, it is noteworthy that the early surge in IL-6 levels showed a positive correlation with the extent of NETosis and subsequent respiratory impairment [31]. Interestingly, the interplay doesn't just flow in one direction, as NETs are capable of reciprocally modulating IL-6 receptor expression. An insightful investigation by Winslow et al. revealed that Haemophilus influenza-induced NETosis may be a mechanism for increasing soluble IL-6 receptor levels. This intricate interaction also holds significant sway over the phenotypic attributes of individuals struggling with chronic obstructive pulmonary disease [32].
Subsequent functional enrichment analysis unveiled their pronounced enrichment in the NOD signaling pathway, Toll-like signaling pathway, and TNF signaling pathway. Previous studies have shown that the above pathways play an important role in NETs formation. NOD-like receptors constitute a family of intracellular innate immune sensors that are triggered by the recognition of microbe-associated and damage-associated molecular patterns. Notably, NOD2 and NLRP3 are significantly expressed in neutrophils, and their activation promotes neutrophil IL-8 and IL-1β secretion and migration toward inflammatory stimuli [33]. In a mouse model lacking NLRP3, both the density, and rate of NET formation were significantly reduced compared to the wild type, highlighting the important role of NLRP3 in NETosis [34]. Toll-like receptors (TLRs) serve as recognition sensors for foreign compounds such as viral and bacterial products, thanks to their diverse extracellular structural domains rich in leucine repeats. Investigations by Tadie et al. revealed that the hazard-associated molecular pattern protein, high mobility group protein 1 (HMGB1), induces NET generation both in vitro and in vivo through a TLR4-dependent mechanism [35]. Building on this, Wang et al. extended the understanding by showing that HMGB1 mediates NETs formation through its engagement with TLR2 and TLR4. This interaction with TLR2 and TLR4 contributes to the generation of NETs by HMGB1 and consequently triggers the recruitment of neutrophils [36]. TNF-α, a widely studied pleiotropic cytokine of the TNF superfamily, plays a prominent role as an initiator of the TNF signaling pathway. TNF-α and NF-κB levels are significantly elevated in individuals with severe asthma [37, 38]. A recent investigation highlighted the critical role of TNF signaling in NET formation during Staphylococcus aureus skin infections [39].
Using the expression profiles of the 50 different DE-NRGs, we then performed cluster analysis using NMF to effectively classify patients with severe asthma into distinct C1 and C2 clusters. Within the C2 cluster, genes with elevated expression levels were observed to be positively correlated with the degree of neutrophil infiltration. These highly expressed genes within the C2 cluster exhibited significant enrichment in pathways such as TNFA, which showed substantial dissimilarity compared to the C1 cluster. This revealing finding suggests that individuals within the C2 cluster of severe asthma samples may exhibit heightened immune-inflammatory responses and a propensity for NET formation, potentially indicating a more challenging clinical prognosis. Asthma is widely recognized for its clinical and therapeutic diversity. This complexity underscores the importance of tailored management strategies to optimize therapeutic efficacy. Consequently, the findings from this study hold promise for yielding valuable biological perspectives into distinct clinical phenotypes and facilitating the stratification of patients for more targeted and effective interventions.
With the rapid development of artificial intelligence, machine learning algorithms, as an important branch of artificial intelligence, have been widely used in the field of identification and screening of key genes due to their excellent feature differentiation ability and applicability to high-dimensional feature data [40,41,42,43]. In this study, we used three specifically selected machine learning classifiers, namely Boruta, SVM and LASSO, to merge the predictive performance anchored in DE-NRG expression profiles to identify SLC44A2, TIMP1 and NFIL3 as alternative key core genes. We then further validated the alternative key core genes using two independent validation sets, and our results showed that TIMP1 and NFIL3 showed diagnostic utility for severe asthma in the context of both validation sets. The results of our in vitro experiments also supported this conclusion, as TIMP1 and NFIL3 were highly expressed in patients with severe asthma, and neutrophils in patients with severe asthma produced and released NETs at a significantly higher rate than in healthy controls, suggesting that the degree of neutrophil stress in patients with severe asthma may be more pronounced and may contribute to the exacerbation of symptoms in patients with asthma.
TIMP1 is emerging as a novel soluble matrix metalloproteinase inhibitor that is rapidly expressed on the cell surface when human neutrophils are activated [44]. The work of Wang et al. revealed for the first time that TIMP1 is present on NETs released by activated neutrophils. Similarly, Schoeps et al. found in a mouse model genetically engineered to carry pancreatic ductal adenocarcinoma that TIMP1 significantly promoted the formation of NETs within the tumor. Remarkably, the removal of either TIMP1 or NETs resulted in prolonged survival in these mouse subjects [45]. NFIL3, also known as E4BP4, is a basic leucine zipper-type transcription factor originally identified as a transcriptional repressor and activator [46,47,48]. NFIL3 has been implicated in immune-mediated diseases, with a growing body of literature suggesting that NFIL3 is associated with IgE class switching and resistance to glucocorticoid therapy[49, 50]. A study by Ke et al. found that NFIL3 may be involved in neutrophil-mediated cellular injury during myocardial infarction and is a core gene for NETs-associated endothelial injury. The role of TIMP1 and NFIL3 in asthma is not yet fully understood, and in particular their potential mechanism with NET formation is intriguing and warrants further study in the future.
However, it is important to acknowledge several limitations of this study. First, the results were derived from a relatively small sample of patients with severe asthma, so a larger cohort is needed to obtain more robust and reliable results. Second, given the evolving understanding of NETs, there is room for refinement in the construction of the NET-related gene set. Furthermore, a more comprehensive exploration of the cellular and molecular mechanisms underlying NRGs is imperative to gain a deeper insight into their role in the context of asthma pathogenesis. Finally, this study focused primarily on gene expression data; future investigations should expand their scope to include broader facets involved in severe asthma pathogenesis, including epigenetics, proteomics, and metabolomics, to gain a more comprehensive understanding and insight. Such expanded research efforts will undoubtedly contribute to a more nuanced understanding of the intricate mechanisms involved in severe asthma.
In conclusion, based on our current understanding, this study provides the first in-depth exploration of the molecular characterization of NRGs in adult patients with severe asthma. Through an intricate analysis of DE-NRGs, we have effectively categorized individuals affected by severe asthma into two distinct clusters. In addition, our exploration has yielded two potential biomarkers—TIMP1 and NFIL3—that hold promise for opening up new avenues for the diagnosis and treatment of severe asthma. As we look to the future, future investigations may delve deeper into the functional attributes of these markers and their potential role in refining severe asthma management and therapeutic strategies, thereby making more substantial contributions to the well-being of patients.
Data Availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Abbreviations
- NETs:
-
Neutrophil extracellular traps
- NRGs:
-
NET-related genes
- DE-NRGs:
-
Differentially expressed NRGs
- DEGs:
-
Differentially expressed genes
- GEO:
-
Gene expression omnibus
- NMF:
-
Non-negative matrix factorization
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- RSS:
-
Residual sum of squares
- GSVA:
-
Gene set variation analysis
- SVM:
-
Support vector machine
- LASSO:
-
Least absolute shrinkage and selection operator
- AUC:
-
Area under the curve
- ROC:
-
Receiver operating characteristic
- NETosis:
-
Neutrophil extracellular trap cell death
- PPI:
-
Protein–protein interaction
- IL-6:
-
Interleukin-6
- TLR:
-
Toll-like receptor
- HMGB1:
-
High mobility group protein 1
- MMP:
-
Matrix metalloproteinase
References
Wills-Karp, M. (2018). Neutrophil ghosts worsen asthma. Science Immunology, 3(26), eaau0112.
Chung, K. F. (2016). Asthma phenotyping: A necessity for improved therapeutic precision and new targeted therapies. Journal of Internal Medicine, 279(2), 192–204.
Froidure, A., Mouthuy, J., Durham, S. R., Chanez, P., Sibille, Y., & Pilette, C. (2016). Asthma phenotypes and IgE responses. European Respiratory Journal, 47(1), 304–319.
Bønnelykke, K., & Ober, C. (2016). Leveraging gene-environment interactions and endotypes for asthma gene discovery. The Journal of Allergy and Clinical Immunology, 137(3), 667–679.
DeVries, A., & Vercelli, D. (2016). Epigenetic mechanisms in asthma. Annals of the American Thoracic Society, 13(Suppl 1), S48–S50.
Bird, L. (2018). Asthma exacerbated by neutrophil ghosts. Nature Reviews Immunology, 18(10), 602–603.
Machida, K., Aw, M., Salter, B. M. A., Ju, X., Mukherjee, M., Gauvreau, G. M., et al. (2020). The role of the TL1A/DR3 axis in the activation of group 2 innate lymphoid cells in subjects with eosinophilic asthma. American Journal of Respiratory and Critical Care Medicine, 202(8), 1105–1114.
Radermecker, C., Sabatel, C., Vanwinge, C., Ruscitti, C., Maréchal, P., Perin, F., et al. (2019). Locally instructed CXCR4hi neutrophils trigger environment-driven allergic asthma through the release of neutrophil extracellular traps. Nature Immunology, 20(11), 1444–1455.
Papayannopoulos, V. (2018). Neutrophil extracellular traps in immunity and disease. Nature Reviews Immunology, 18(2), 134–147.
Jorch, S. K., & Kubes, P. (2017). An emerging role for neutrophil extracellular traps in noninfectious disease. Nature Medicine, 23(3), 279–287.
Twaddell, S. H., Baines, K. J., Grainge, C., & Gibson, P. G. (2019). The emerging role of neutrophil extracellular traps in respiratory disease. Chest, 156(4), 774–782.
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research, 43(7), e47.
Rebhan, M., Chalifa-Caspi, V., Prilusky, J., & Lancet, D. (1997). GeneCards: Integrating information about genes, proteins and diseases. Trends in Genetics, 13(4), 163.
McKusick, V. A. (2007). Mendelian inheritance in man and its online version, OMIM. American Journal of Human Genetics, 80(4), 588–604.
Edgar, R., Domrachev, M., & Lash, A. E. (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research, 30(1), 207–210.
Wu, J., Zhang, F., Zheng, X., Zhang, J., Cao, P., Sun, Z., et al. (2022). Identification of renal ischemia reperfusion injury subtypes and predictive strategies for delayed graft function and graft survival based on neutrophil extracellular trap-related genes. Frontiers in Immunology, 13, 1047367.
Zhang, Y., Guo, L., Dai, Q., Shang, B., Xiao, T., Di, X., et al. (2022). A signature for pan-cancer prognosis based on neutrophil extracellular traps. Journal for Immunotherapy of Cancer, 10(6), e004210.
Yu, G., Wang, L. G., Han, Y., & He, Q. Y. (2012). clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS: A Journal of Integrative Biology, 16(5), 284–287.
Gaujoux, R., & Seoighe, C. (2010). A flexible R package for nonnegative matrix factorization. BMC Bioinformatics, 11, 367.
Hänzelmann, S., Castelo, R., & Guinney, J. (2013). GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics, 14, 7.
Bindea, G., Mlecnik, B., Tosolini, M., Kirilovsky, A., Waldner, M., Obenauf, A. C., et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity, 39(4), 782–795.
Kursa, M. B., & Rudnicki, W. R. (2010). Feature selection with the boruta package. Journal of Statistical Software, 36, 1–13. https://doi.org/10.18637/jss.v036.i11
Gold, C., & Sollich, P. (2003). Model selection for support vector machine classification. Neurocomputing, 55(1), 221–249.
Tibshirani, R. (1996). Regression Shrinkage and Selection via the Lasso. Journal of the Royal Statistical Society Series B (Methodological), 58(1), 267–288.
Guyon, I., Weston, J., Barnhill, S., & Vapnik, V. (2002). Gene selection for cancer classification using support vector machines. Machine Learning, 46(1), 389–422. https://doi.org/10.1023/A:1012487302797
Hu, Y. (2012). Isolation of human and mouse neutrophils ex vivo and in vitro. In R. B. Ashman (Ed.), Leucocytes: Methods and protocols (Vol. 844, pp. 101–113). Totowa: Humana Press. https://doi.org/10.1007/978-1-61779-527-5_7
de Buhr, N., & von Köckritz-Blickwede, M. (2020). Detection, visualization, and quantification of neutrophil extracellular traps (NETs) and NET markers. Methods in Molecular Biology, 2087, 425–442.
Damascena, H. L., Silveira, W. A. A., Castro, M. S., & Fontes, W. (2022). Neutrophil activated by the famous and potent PMA (Phorbol Myristate Acetate). Cells, 11(18), 2889.
de Jesus, G.-C., & Zarate, X. (2022). Neutrophil extracellular traps: Modulation mechanisms by pathogens. Cellular Immunology, 382, 104640.
Joshi, M. B., Lad, A., Bharath Prasad, A. S., Balakrishnan, A., Ramachandra, L., & Satyamoorthy, K. (2013). High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Letters, 587(14), 2241–2246.
Mukhopadhyay, S., Sinha, S., & Mohapatra, S. K. (2021). Analysis of transcriptomic data sets supports the role of IL-6 in NETosis and immunothrombosis in severe COVID-19. BMC Genomic Data, 22(1), 49.
Winslow, S., Odqvist, L., Diver, S., Riise, R., Abdillahi, S., Wingren, C., et al. (2021). Multi-omics links IL-6 trans-signalling with neutrophil extracellular trap formation and Haemophilus infection in COPD. European Respiratory Journal, 58(4), 2003312.
Ekman, A. K., & Cardell, L. O. (2010). The expression and function of Nod-like receptors in neutrophils. Immunology, 130(1), 55–63.
Burke, R. M., Dale, B. L., & Dholakia, S. (2021). The NLRP3 inflammasome: Relevance in solid organ transplantation. International Journal of Molecular Sciences, 22(19), 10721. https://doi.org/10.3390/ijms221910721
Tadie, J. M., Bae, H. B., Jiang, S., Park, D. W., Bell, C. P., Yang, H., et al. (2013). HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. American Journal of Physiology. Lung Cellular and Molecular Physiology, 304(5), L342-349.
Wang, Y., Du, F., Hawez, A., Mörgelin, M., & Thorlacius, H. (2019). Neutrophil extracellular trap-microparticle complexes trigger neutrophil recruitment via high-mobility group protein 1 (HMGB1)-toll-like receptors(TLR2)/TLR4 signalling. British Journal of Pharmacology, 176(17), 3350–3363.
Berry, M. A., Hargadon, B., Shelley, M., Parker, D., Shaw, D. E., Green, R. H., et al. (2006). Evidence of a role of tumor necrosis factor alpha in refractory asthma. New England Journal of Medicine, 354(7), 697–708.
Gagliardo, R., Chanez, P., Profita, M., Bonanno, A., Albano, G. D., Montalbano, A. M., et al. (2011). IκB kinase-driven nuclear factor-κB activation in patients with asthma and chronic obstructive pulmonary disease. The Journal of Allergy and Clinical Immunology, 128(3), 635–645.
Youn, C., Pontaza, C., Wang, Y., Dikeman, D. A., Joyce, D. P., Alphonse, M. P., et al. (2023). Neutrophil-intrinsic TNF receptor signaling orchestrates host defense against Staphylococcus aureus. Science Advance, 9(24), eadf8748.
Ray, A., Das, J., & Wenzel, S. E. (2022). Determining asthma endotypes and outcomes: Complementing existing clinical practice with modern machine learning. Cell Reports Medicine, 3(12), 100857.
Shamji, M. H., Ollert, M., Adcock, I. M., Bennett, O., Favaro, A., Sarama, R., et al. (2023). EAACI guidelines on environmental science in allergic diseases and asthma—Leveraging artificial intelligence and machine learning to develop a causality model in exposomics. Allergy, 78(7), 1742–1757.
Ding, X., Qin, J., Huang, F., Feng, F., & Luo, L. (2023). The combination of machine learning and untargeted metabolomics identifies the lipid metabolism -related gene CH25H as a potential biomarker in asthma. Inflammation Research, 72(5), 1099–1119.
Gaudillo, J., Rodriguez, J. J. R., Nazareno, A., Baltazar, L. R., Vilela, J., Bulalacao, R., et al. (2019). Machine learning approach to single nucleotide polymorphism-based asthma prediction. PLoS ONE, 14(12), e0225574.
Price, B., Dennison, C., Tschesche, H., & Elliott, E. (2000). Neutrophil tissue inhibitor of matrix metalloproteinases-1 occurs in novel vesicles that do not fuse with the phagosome. Journal of Biological Chemistry, 275(36), 28308–28315. https://doi.org/10.1074/jbc.M002263200
Schoeps, B., Eckfeld, C., Prokopchuk, O., Böttcher, J., Häußler, D., Steiger, K., et al. (2021). TIMP1 triggers neutrophil extracellular trap formation in pancreatic cancer. Cancer Research, 81(13), 3568–3579.
Zeng, L., Chen, D., Xue, Y., Zhang, M., Wu, Y., & Yang, W. (2023). A new border for circadian rhythm gene NFIL3 in diverse fields of cancer. Clinical and Translational Oncology, 25(7), 1940–1948.
Zhang, W., Zhang, J., Kornuc, M., Kwan, K., Frank, R., & Nimer, S. D. (1995). Molecular cloning and characterization of NF-IL3A, a transcriptional activator of the human interleukin-3 promoter. Molecular and Cellular Biology, 15(11), 6055–6063.
Cowell, I. G. (2002). E4BP4/NFIL3, a PAR-related bZIP factor with many roles. BioEssays, 24(11), 1023–1029.
Carey, K. T., Tan, K. H., Ng, J., Liddicoat, D. R., Godfrey, D. I., & Cole, T. J. (2013). Nfil3 is a glucocorticoid-regulated gene required for glucocorticoid-induced apoptosis in male murine T cells. Endocrinology, 154(4), 1540–1552. https://doi.org/10.1210/en.2012-1820
Kashiwada, M., Levy, D. M., McKeag, L., Murray, K., Schröder, A. J., Canfield, S. M., et al. (2010). IL-4-induced transcription factor NFIL3/E4BP4 controls IgE class switching. Proceedings of the National Academy of Sciences, 107(2), 821–826.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
J Lin had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. K Shen contributed to the study conception, design, and material preparation. The first draft of the manuscript was written by K Shen, and J Lin commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
The study was approved by the Ethics Committee of the China-Japan Friendship Hospital (No:2021-GZR-70). All subjects gave written informed consent to participate in the study.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shen, K., Lin, J. Unraveling the Molecular Landscape of Neutrophil Extracellular Traps in Severe Asthma: Identification of Biomarkers and Molecular Clusters. Mol Biotechnol (2024). https://doi.org/10.1007/s12033-024-01164-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12033-024-01164-z