
Abstract
The application of repertoire selection technologies for the study and characterization of tumor heterogeneity is an area of great interest in the field of tumor biology and immunotherapy. Among the most promising approaches, phage display has been successfully used to select peptides and antibody fragments to a variety of different targets, including cancer cells, immune cells and cytokines. Peptides selected from phage display have been used to guide the delivery of lytic peptides, cytotoxic drugs, and nanoparticles to cancer cells with the aim to obtain more efficient and less toxic therapeutics. Additionally, antibodies developed through phage display are being used in the treatment of autoimmune diseases and in some cases metastatic cancers. This review provides a short description of how phage libraries are designed, and highlights the conversion of the isolated binders into human therapeutics and use in targeted therapies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Phage display is a molecular technique in which phage DNA is genetically modified to express on the phage surface a peptide or protein fused to one of the phage coat proteins. This strategy is fundamentally different from other bacterial expression systems in that the displayed peptides or proteins and the DNA encoding them are physically linked. This physical linkage allows for screening large numbers of phage clones in fluid phase and enrichment of high-affinity binding clones whose coding sequences are then identified through DNA sequencing. The strategy was first described by George Smith in 1985 [1]. Since then, the technology has gained attention leading to the construction of phage libraries for both biotechnological and medical applications [2,3,4,5]. Proteins expressed on the phage surface may be peptides or antibody fragments, such as single-chain variable fragments (scFvs). Despite other in vitro methods, like yeast surface display [6], ribosomal display [7], or puromycin display [8], phage display is the most used method for the selection of human antibodies and cancer cell-binding peptides. Indeed, several studies have successfully applied the phage display technology to isolate peptides specifically targeting surface antigens on tumor cells or tumor-associated vasculature in mice [9,10,11]. Targeted delivery of compounds to tumor vessels and tumor cells can enhance tumor detection and therapy. In addition to tumor-targeting peptides, phage display opened up a whole new realm of possibilities in antibody discovery and engineering. By using this technology, human monoclonal antibodies are now produced without immunization [4, 11]. In 2018, the Nobel Prize in chemistry was awarded to George P. Smith and Gregory P. Winter for the phage display of peptides and antibodies. And to Frances H. Arnold for the directed evolution of enzymes. All three laureates have used the evolution principles to create proteins/antibodies that can be used as pharmaceuticals. This review provides a short description of the different peptide and antibody libraries, and discusses how they can be used to isolate cell-targeting peptides and human antibodies that can counteract autoimmune diseases, and potentially cure cancers.
Phage Vectors and Display Formats
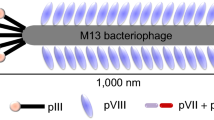
The most common bacteriophages used in phage display are M13 and fd filamentous phages though T4, and T7 phage have also been used [2]. The M13 phage has high capacity for replication and is able to accommodate large foreign DNA, making it the most used phage display vector. It is a non-lytic phage consisting of a limited number of structural proteins assembled around a single-stranded circular genome DNA (ssDNA) encoding the phage proteins (Fig. 1a). The infection of host bacteria is carried out by the attachment of phage pIII to the F′-pilus of E. coli (Fig. 1b). The ssDNA enters the bacteria where it is converted by the host enzymes into double-stranded DNA that generates ssDNA and phage proteins. Phage assembly occurs in or near the inner membrane of host bacteria and in the periplasm [12]. Each M13 particle has 3–5 copies of the pIII protein, as opposed to the most abundant coat protein pVIII with 2700 copies. These two coat proteins have often been used for expression of peptides, and high-affinity binders have been selected from both systems. Antibody fragments are usually fused to N-terminus of pIII protein.

Filamentous phage M13. a Schematic representation of filamentous phage. The different coat proteins are indicated. b Life cycle of the filamentous phages. The phage binds to the tip of F′-pilus of E. coli via the N-terminus of pIII protein. Subsequently, the phage coat proteins are removed by the host TolA protein and the ssDNA is injected into the bacterial cell where it is converted into a double-stranded form, called the replicative form [12]. After replication and expression of phage coast proteins by host enzymes, ssDNA molecules are then coated with pV protein dimers to prevent their conversion into the replicative form. The proteins pI and pXI interact with pIV to form a channel to facilitate phage secretion. Within the channel, pV is replaced by pVIII and then mature phage particles are assembled and released. Thioredoxin in the inner membrane facilitates the removal of pV from the DNA and addition of coat proteins. Note that after gene expression and protein synthesis, phage coat proteins are exported to the periplasm
With respect to display formats, two main types of phage-derived vectors have been used [13,14,15]: the one-gene system where all copies of one coat protein are modified and the two-gene system where the phage genome carries both wild type and a recombinant version of the coat protein (Fig. 2a, b). In the one-gene system, the peptide or antibody fragment is displayed in a multivalent format where all copies of the coat protein are expressed as fusion proteins. Larger polypeptides affect the function of pVIII protein, whereas for pIII larger fusion proteins are better tolerated although some fusions may interfere with the interaction between pIII and F′ pilus, which can negatively affect both phage infectivity and library diversity [12]. The two-gene system uses either a tandem repeat of the gene coding for the coat protein in the phage genome, in which one of the copies is modified, or a phagemid system (Fig. 2b). The relatively small size of phagemids allows the cloning of larger fusion protein gene fragments. Phagemids have higher transformation efficiencies than phage vectors, hence facilitating the construction of large repertoires of peptide or antibody libraries [13]. Additionally, phagemids are genetically more stable than recombinant phages under multiple propagations. Unlike the phage vector, which contains all genes coding for phage coat proteins, the phagemid vector contains only the gene coding for the pIII fusion protein along with an antibiotic resistance gene. Thus, co-infection with a helper phage is required in phagemid system to provide the proteins necessary for the assembly of intact phagemid virions. A helper phage such as M13KO7 or VCSM13 is usually used [15]. In most phagemid systems, the majority of copies are derived from the helper phage, resulting in monovalent display. In general, the phagemids are best suited for the display of antibodies such as scFv and Fab fragments, but can also be used to display peptides at a low valency [13].

Different formats of phage display. In the one-gene system (a), all copies of the coat protein are modified, and in the two-gene system (b), there are both wild type and recombinant versions of the coat protein on the phage. In the two-gene system one version of the coat protein is modified (used for cloning) and the second version (wild type gene) is provided by the helper phage or the M13 phage
With respect to expression, the incorporation of an amber stop codon between the displayed protein and the phage coat protein permits fusion protein expression in suppressor strains of E. coli such as TG1. However, in non-suppressor strains such as HB2151, the soluble form of the recombinant protein will be produced because such strains will not incorporate a glutamine at the amber codon [15]. As mentioned above, the less commonly used T7 lytic phage that lyses its bacterial host has also been used for the construction of peptide libraries [16, 17]. In T7 phage system, peptide sequences are typically fused to the C-terminal of the 10B capsid protein. T7 libraries exhibit less bias than filamentous phage libraries and longer peptide libraries (12–20 mer) are most often used for the discovery of cell-binding peptides [16]. In contrast, short peptide libraries (7–12 mer) are more popular in M13 phage display.
Peptide Libraries
Construction of Random Peptide Libraries
In general, combinatorial peptide libraries are generated by inserting DNA with random sequences into a phage vector. Such DNA is generated in a PCR reaction using chemically made degenerative DNA oligonucleotide containing, for example, the NNK design rule [2, 18]. N stands for an equal mixture of the deoxyribonucleotides G, A, T and C, and K stands for an equal mixture of G and T (Fig. 3). All amino acids are encoded by the 32 different codons represented by the NNK design rule. Moreover, this design leads to the elimination of two stop codons, except the amber stop codon (UAG) that can be translated into glutamine in a SupE + E. coli strain such as TG1 cells. Subsequent to PCR amplification, purification, and digestion with the appropriate restriction enzymes, the DNA encoding the random peptides is ligated into pIII or pVIII gene, encoding either the minor or major coat protein of M13 phage, respectively. After ligation, DNA is electroporated into appropriate E. coli cells. Although the electroporation efficiency of the competent cells is high, usually the yield is much lower for ligation products. Hence, several electroporations are needed to obtain larger libraries (> 109 phages). After bacterial growth and phage preparation, the complexity of each library should be analyzed by sequencing random individual clones. All amino acids should be present in the library and the frequency of each amino acid should be in accordance with the frequency predicted from the number of possible codons for each amino acid. Apparently, some amino acids are overrepresented and some under-represented in all libraries, but there was no marked deviation from a random distribution of residues in any of the libraries [18, 19]. In principle, the lower copy number of pIII allows the selection of high-affinity binders, whereas the high copy number of pVIII rather selects for low-affinity binders.

Random peptide libraries. The concept of random peptide phage display is based on insertion of random oligonucleotides at the appropriate location within one of the phage coat proteins such as the N-terminus of pIII or pVIII. Each amino acid residue in the inserted peptides is randomly encoded by the degenerated codon (NNK). Prior to cloning, the random oligonucleotides are then converted into double-stranded DNAs using an annealing primer and klenow DNA polymerase. Finally, the double-stranded DNAs are digested with the appropriate restriction enzymes to produce DNA fragments that can be cloned directly into a phage vector. The letter n represents the number of inserted amino acid residues. Large peptide inserts up to 38 aa can be inserted into the N-terminus of pIII protein without the loss of phage infectivity or particle assembly [20]. It should be noted that the incorporation of conserved cysteine residues at the desired locations in the degenerated oligonucleotides (e.g., C(NNK)nC) allows the generation of cyclic peptide libraries [21]
In addition to linear (unstructured) peptides, cyclic peptides have been displayed on the surface of M13 or T7 phage [19,20,21,22]. These constrained peptide libraries are constructed by placing two or more invariant cysteines (C) in different positions in the peptide sequences. For example, the CXnC format (where X = any amino acids and n = number of amino acids) has been used. The cysteines in the peptides are automatically linked by oxidation either during the assembly of the bacteriophage in the periplastic space of the bacteria or in the culture medium. Phages displaying the cyclic peptides by an intramolecular disulfide bond tend to exhibit higher target-binding capacity because their rigid structures minimize conformational entropy loss associated with the binding. Moreover, cyclic peptides are far less susceptible to proteolysis and often have increased biological activity because of their conformational rigidity. It should be noted that several phage display peptide libraries are commercially available, e.g., PhD-7, PhD-12, and PhD-C7C (cyclic with a disulfide bond) libraries can be purchased from New England Biolabs. Disulfide-constrained library of the T7 phage is most frequently constructed by using T7Select10-3b or 415-1b vector. Both vectors are commercially available from Novagen.
Affinity Selection
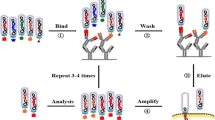
Both peptide and antibody libraries are typically screened by iterative cycles of selection against a target of interest followed by amplification of the bound phages in E. coli cells. This screening process is called biopanning, and in a way mimics the in vivo immune selection process described in “Mimicking the Immune System” section. In brief, the affinity selection method is based on repeated cycles of incubation, washing, elution of bound phages, phage amplification, and re-selection of amplified phages. The target can be an immobilized protein or a whole cell. With respect to live cells, the screening process is usually done by incubating the phage library with target cells, and after washing off non-bound phages, target-bound phages are eluted and amplified in bacteria (Fig. 4a). The process of selection and amplification is normally repeated 3–5 times to ensure the enrichment of highly specific binding phages. The selection can be performed with adherent, non-adherent cells, or in vivo. When using complex targets such as mammalian cells, enhancement of specific binding above the background phage binding is usually needed, as unspecific binding to common membrane receptors is expected to impact the quality of the obtained binders. To circumvent this problem, prior to affinity selection, pre-absorption of the phage library on non-target cells can be used to deplete phages that bind to common receptors. Each biopanning step must also be optimized for the type of cells being used. In cases where ligands for the investigated targets are known, competitive elution can be used.

Affinity selection. In cell-based protocol (a), the phage library is incubated with the desired target cells. After incubation, unbound phages are removed by washing and bound phages are eluted and propagated in E. coli, and then used for further rounds of selection. To enrich for specific binders, usually 3–5 rounds are performed. In in vivo protocol (b), the library is often i.v. injected into the animal and allowed to circulate for few minutes. Subsequently, phages are recovered from the desired organ or tissues, amplified in E. coli and then used for a second round of selection using a second animal. After the desired rounds of selection, phage clones are rescued from tissues, amplified and then sequenced to determine the peptide sequences
In addition to in vitro selection, several groups have successfully identified targeting ligands by using in vivo biopanning [23, 24]. Briefly, following injection of a peptide or antibody library into an animal, the tissues of interest are harvested, washed, and the eluted phages are amplified in E. coli and then used in subsequent rounds of injection and selection (Fig. 4b) [23]. Peptides identified using in vivo biopanning may prove to be of better clinical significance given that they are selected in the disease model of choice. However, most animal models do not accurately mimic the human disease condition. For instance, subcutaneous xenograft models cannot mimic the complexity of the tumor vasculature in humans as the extent of angiogenic heterogeneity in malignant neoplasms is regulated by the organ microenvironment. Regardless of the method used in phage selection, several cancer cell-binding peptides and antibody fragments were identified using phage display technology. The examples listed below illustrate the versatility of phage display with respect to ligand discovery and engineering.
Examples of Cell-Targeting Peptides
Peptide libraries were initially applied to the epitope mapping of monoclonal antibodies and to study the specificity of auto-antibodies present in patient sera [25,26,27]. Shortly after its discovery by Smith, several groups have also implemented the technology to select cell-binding peptides for use in cancer therapy [28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76] (Table 1). Using in vivo biopanning, Ruoslahti et al. [23] have isolated several organ and tumor-homing peptides. Several integrin proteins, the αv integrins in particular, function as receptors for the peptides that contain the Arg-Gly-Asp (RGD) binding site [23, 28]. Early studies found that the binding of the GACRGDCLGA peptide with one disulfide bond was 10 times more efficient than linear RGD-containing peptides. Further studies by the same group identified the RGD-4C peptide (CGCRGDCFC), which contains two disulfide bonds, to be more efficient and selective for the αv integrins [29]. The RGD-4C peptide bound preferentially to integrins at sites of tumor angiogenesis and inflamed synovium in vivo, and can be internalized into targeted cells. The peptide was used to enhance the delivery of chemodrugs to cancer cells and to reduce the toxicity associated with the free drugs [30, 31]. Additionally, the RGD-4C peptide was conjugated to either a pro-apoptotic peptide domain to treat tumors and inflammatory arthritis, or to radioactive labels for imaging of tumor tissues [28].
While the RGD peptides have been shown to be efficient at tumor targeting, they lack selectivity as many cells express the αv integrins. Oyama et al. [37] isolated from phage display a peptide, named H2009.1, (RGDLATRQLAQEDGVVGVR) which bound to the integrin αvβ6 and did not bind other more widely expressed RGD-binding integrins. Integrin αvβ6 is highly expressed in many malignancies, but is usually expressed at low or undetectable levels in normal adult tissues. The authors also established an experimental protocol for selecting peptides that target specific cellular compartments such as the lysosomes [52]. Sugahara et al. [47] used a cyclic peptide library displayed on T7 phage to identify peptides that bind tumor blood vessels in an experimental mouse model of human prostate cancer. One of the selected cyclic peptides iRGD (internalizing RGD) with sequence CRGDKGPDC was internalized by the prostate cancer cell line PPC1. The binding and internalization of the peptide requires both αvβ3 integrin and neuropilin-1 (NRP-1), a membrane-bound receptor that plays an important role in normal neuronal and vascular development. NRP-1 is highly expressed in a variety of solid tumors such as prostate, breast, pancreatic, lung, ovarian, and gastrointestinal carcinomas [53]. Targeted delivery of chemodrugs such as abraxane with the iRGD peptide significantly improved the antitumor potency of the conjugates [47]. Additionally, magnetic resonance imaging with iRGD peptide-coated iron oxide nanoworms in orthotopic xenograft tumor-bearing mice improved the sensitivity of tumor imaging. Interestingly, the cell-penetrating activity of iRGD peptide was far greater than that of conventional RGD peptides [47]. Hence, the iRGD peptide could be developed as vehicles to deliver drug payload or imaging probes to tumor sites. Similarly, Li et al. [54] identified a cyclic peptide (CTPSPFSHC) that bound to endothelial cells present in human colorectal cancer. The peptide was used to guide the delivery of pro-apoptotic peptides and chemodrugs to tumor cells [54]. Pasqualini et al. [32] also isolated the NGR peptide (CNGRCVSGCAGRC), a bicyclic tumor-targeting peptide that bound to aminopeptidase N (CD13), which is highly expressed by endothelial tumor cells such as scirrhous gastric cancer, pancreatic cancer, and non-small cell lung carcinoma. Similar to RGD peptide [55], NGR peptide-doxorubicin conjugates inhibited tumor growth and prolonged mouse survival [56]. Moreover, NGR-liposomal doxorubicin formulations inhibited neuroblastoma tumor growth in an orthotopic xenograft mouse model. Given the role of aminopeptidase N in angiogenesis, the data highlight a potential new target for both inhibiting angiogenesis and targeting tumor cells.
The cyclic LyP-1 peptide (CGNKRTRGC) was the first lymphatic vessel targeting peptide identified by phage display screening using human xenograft models of breast cancer [33]. It recognizes both lymphatic vessels and tumor cells. Conjugation of paclitaxel to the LyP-1 peptide enhanced the antitumor activity of the drug [57, 58]. Similarly, affinity selection of a peptide phage library on bladder tumor cells led to the selection of a cyclic peptide (CSNRDAARRC) that bound to bladder tumor cells [35]. When fused to an apoptotic peptide [(KLAKLAK)2 peptide], the fusion peptide selectively bound to HT1376 bladder tumor cells and efficiently internalized into the cells but not to other tumors or normal cells [59]. Moreover, the fusion peptide inhibited tumor growth. The peptide sequence (KLAKLAK)2 is known to induce apoptosis in mammalian cells by disrupting the mitochondrial membrane, and only when internalized [60].
We have optimized the affinity selection conditions and isolated cancer cell- or immune cell-binding peptides with good affinity and specificity [34, 61]. For example, the LTVSPWY peptide was isolated using an affinity selection protocol designed to enrich for internalized phages [34]. As such, the selected peptides are attractive vehicle to deliver cytotoxic drugs and can also be used in diagnostics. With respect to specificity, the LTVSPWY peptide bound preferentially to HER2/ErbB2-positive cancer cells and was able to deliver therapeutics and imaging agents to cancer cells in vitro and in vivo [62,63,64,65]. Moreover, the LTVSPWY peptide has been used to target lytic peptides to cancer cells. The fusion peptides selectively induced cell death in breast cancer cell lines when compared to free (KLAKLAK)2 lytic peptide [66, 67]. With respect to specificity, only three peptides selected from phage display, LTVSPW, KCCYSL, and MYWGDSHWLQYWYE, have been found to preferentially recognize HER2-positive tumors [10, 48].
The epidermal growth receptor (EGFR) is a cell surface protein that binds to epidermal growth factors and is overexpressed in a variety of human tumors, making it an excellent target for drug delivery [68]. Li et al. [49] screened a phage peptide library and identified a peptide named GE11 (YHWYGYTPQNVI) that showed specific binding to EGFR. The peptide was internalized into cancer cells with high EGFR expression levels. GE-11-lytic peptide conjugates killed cancer cells in culture and inhibited tumor growth in mice [69]. Additionally, doxorubicin-peptide conjugates killed EGFR-positive cells and peptide liposome formulations have been used to image EGFR-positive tumors in mice [70]. In addition to EGFR, vascular endothelium growth factor (VEGF) receptors are also involved in cancers [71]. Qin et al. [50] selected a peptide (CSDSWHYWC) that showed high affinity to VEGF receptor 3. Activation of this receptor by its ligand in several types of solid tumors increased cancer cell mobility and invasion potential, resulting in cancer cell metastasis. Today, anti-angiogenesis agents represent standard-of-care therapies for multiple types of cancers [71].
Matrix metalloproteinases (MMPs) derived from both tumor cells and stromal compartment are regarded as a major player assisting tumor cells during metastasis [72]. Koivunen et al. [51] isolated several cyclic peptides that were found to be selective inhibitors of MMP-2 and MMP-9. These peptides inhibited endothelial and tumor cell migration in vitro and, more important, prevent tumor growth and invasion in mouse models of human breast carcinoma. Moreover, targeting liposomal formulations of doxorubicin with the CTT peptide increased survival of tumor-bearing mice [73, 74].
Glioblastoma multiforme (GBM) remains one of the most lethal primary brain tumors despite surgical and therapeutic advancements [75]. Interleukin 13 receptor α2 (IL-13Rα2) is overexpressed in a majority of patients with GBM, but is not found in normal brain cells. Pandya et al. [36] screened a disulfide-constrained heptapeptide phage display library on GBM cells, and isolated several binding peptides. One of these peptides, CGEMGWVRC bound to IL-13Rα2 with high specificity and homed to both subcutaneous and orthotopic human GBM xenografts expressing IL-13Rα2 when administered intravenously.
Lung cancer is the leading cause of cancer-related mortality in both men and women, accounting for about 27% of all cancer deaths [76]. The most common type is non-small cell lung cancer (NSCLC) which makes up about 80–85% of all cases. The majority of NSCLC patients are diagnosed with advanced-stage disease and almost all will develop incurable metastatic disease. NSCLC-targeting peptides could be used to guide the delivery of drugs, nanoparticles, and toxins. McGuire et al. [38] affinity selected peptide libraries on a large panel of NSCLC cell lines and isolated 11 novel peptides, some of which homed to tumors in vivo and bound to patient tumor samples ex vivo. A significant correlation was found with an epithelial or mesenchymal phenotype and peptide-binding profiles. Similarly, Chang et al. [39] isolated a peptide (TDSILRSYDWTY) that specifically bound to NSCLC cell lines both in vivo and in vivo. Conjugation of this peptide to liposomes containing doxorubicin and vinorelbine increased the therapeutic potency of the conjugates when compared to free drugs [39]. Using large cell carcinoma cell line H460, Chi et al. also selected three peptides (GAMHLPWHMGTL, NPWEEQGYRYSM, NNPWREMMYIEI) that bound to both SCLC and NSCLC cell lines [40]. The peptides enhanced the therapeutic efficacy of liposomal drugs through their targeting and endocytosis abilities. Hence, they can be incorporated into theranostic strategies to enhance drug efficacy and reduce side effects associated with free chemodrugs.
Although several peptides derived from random peptide phage libraries have been used to guide the delivery of chemodrugs and apoptotic peptides to obtain more efficient and less toxic therapeutics, systematic experimental approaches for receptor identification are missing. This is a key challenge as the identification of peptide-binding receptors is necessary for basic and clinical applications. Compared to alterations that may be discovered through genomic and proteomic profiling, aberrant post-translational modifications on proteins or lipids, such as glycosylation, could be targeted by the selected peptides. Moreover, the binding of the receptors to the peptides could be facilitated by the neighboring proteins. Indeed, plasma membrane proteins typically need to be embedded in their natural environment within living cells to exhibit binding properties. Although useful for interaction studies, affinity-based techniques such as immunoprecipitations are usually limited to high-affinity interactions and will usually be ineffective when involving plasma membrane proteins that are often hydrophobic and present in relatively low levels. Hence, specific methods should be developed for cancer cell-binding peptides or antibody like fragments selected from phage display libraries. For example, the use of functional cross-linking reagents such as Sulfo-SBED would allow the capture of the receptors bound to peptides on the surface of live cells followed by identification of the captured receptors by mass spectrometry [77, 78]. We have used the Sulfo-SBED method and identified several peptide- and antibody-binding membrane proteins that are under investigation. The Sulfo-SBED reagent contains an amine reactive group for ligand labeling separated by a disulfide bond from a biotin and a UV-activated aryl azide cross-linking group.
Improving Peptide Stability, Affinity, and Circulation Time
As therapeutics, peptides often suffer from several limitations such as poor oral bioavailability, rapid clearance, short half-life, and sometimes low solubility [79]. Thus, the major challenge currently facing the field is to move peptides selected from phage display into the clinic. Given the significant advances in medicinal chemistry, a set series of optimization techniques may be used to improve the affinity and stability of the selected peptides. With respect to binding, alanine scanning can rank residues in terms of importance to binding affinity, and motifs of two or more key residues can then be identified and optimized. l-Amino acids may also be replaced with their d-amino acid counterparts to improve activity and/or confer increased stability to enzymatic degradation. However, the change in chirality upon incorporation of d-amino acids into cyclic peptides can alter backbone conformation and may reduce binding affinity. The degradation of linear peptides by proteases can be prevented via peptide cyclization, modifications that block the C- and N-terminus, and inclusion of unnatural amino acids [80]. Moreover, tetramerization of the selected peptides can increase their serum stability and binding affinity [81]. Multivalent targeting strategy can provide sufficient avidity to overcome the low affinity of monovalent peptides selected from phage libraries.
Notably, the kidney filtration threshold for proteins is 60 kDa. Proteins between 45 and 60 kDa still show limited filtration; however, those with a size of < 15 kDa are freely filtered out [82]. Therefore, peptides with low molecular weights (< 2–15 kDa) are susceptible to rapid filtration via the kidney. To increase circulation time, usually therapeutic peptides and smaller proteins are fused either to polyethylene glycol, human serum albumin (HSA), or Fc fragment of human IgG [83, 84]. Both HSA and IgG bind to neonatal Fc receptor (FcRn) [85], leading to increased circulation times due to antibody escape from intracellular lysosomal degradation mediated by FcRn [86]. Accordingly, fusing cancer cell-binding peptides selected from peptide libraries to the Fc domain of human IgG offers a new way for the development of new targeted immunotherapies. Like antibodies, the fusion protein forms a homodimer due to the strong interactions of the CH3 domains. Given their small size (52–64 kDa), peptide-Fc fusions are expected to have good tissue penetration as compared to antibodies (150 kDa) [86, 87].
By coupling cancer cell-binding peptides to the Fc domain of human IgG1, we have married the targeting specificities of some peptides with the effector function of antibodies such as the induction of antibody-dependent cellular cytotoxicity (ADCC) [88, 89]. Similarly, Qin and colleagues converted a peptide selected from phage display into a peptide-Fc fusion that depleted myeloid-derived suppressor cells in tumor-bearing mice [90]. The fusion protein eliminated myeloid-derived suppressor cells without affecting other pro-inflammatory cells, including dendritic cells and lymphocytes. Of note, several Fc-fusion proteins have been used as research tools and more significantly as protein therapeutics. For example, etanercept, which is composed of the 75 kDa soluble extracellular domain of the TNF-α receptor II fused to the Fc domain of human IgG1, is the first successful example of using a soluble receptor-Fc fusion protein as a therapeutic drug [91, 92]. A peptide mimic of thrombopoietin selected from phage display was the first peptide-Fc fusion protein (romiplostim, AMG-531) developed for the treatment of chronic immune thrombocytopenia purpura [93]. Trebananib (AMG 386) is also a peptide-Fc fusion with dual specificity to angiopoietin (Ang)-1 and -2 [94]. AMG 386 neutralizes the binding of two soluble ligands, Ang-1 and Ang-2, to the Tie-2 receptor tyrosine kinase, resulting in the inhibition of tumor angiogenesis [95]. Collectively, these data show that peptides derived from phage libraries can be turned into true human therapeutics or delivery agents.
Antibody Libraries
Mimicking the Immune System
Phage antibody libraries are constructed by PCR-based cloning of VH and VL repertoires by random pairing into a phage or phagemid vector system and displayed on the surface of bacteriophage [96]. As such, the technique mimics the assembly and production of antibodies by B cells. Indeed, each B cell is a self-replicating packaged system containing antibody genes that encodes the antibody displayed on its surface. First, in the natural course of B cell development, functional immunoglobulin (Ig) genes are assembled randomly from heavy chain variable (VH) and light chain variable (VL) gene segments, leading to the generation of the naïve repertoire capable of reacting with unlimited number of foreign antigens [97]. Second, during the activation period with antigens, each B cell undergoes a process called somatic hypermutation in which amino acid residues particularly in the complementarity-determining regions (CDRs) are mutated [98]. This step of somatic hypermutation plays an essential role in increasing both the affinity and selectivity of antibodies for their target antigens (Fig. 5). In phage display, this in vivo step can be mimicked by strategies such as site-directed mutagenesis and heavy/light chain shuffling [96]. Subsequent to affinity maturation, B cells expressing antibodies with high affinity for the target antigen are clonally expanded to generate memory B cells and antibody secreting plasma cells. Of note, the repertoire of Igs is selectable because in each B cell both genotype and phenotype are linked as in phage antibody display libraries.

Selection of antigen-binding B cells. Naïve B cell repertoire consists of around 2.3 × 107 B cells, each expressing a unique antibody-binding site on their surface (IgM). Exposure to antigen selects from this repertoire those lymphocytes that produce antigen-reactive antibodies, and induces the process of somatic mutations in the V genes, resulting in generation of antibodies with various affinity to the same antigen. The B cells displaying the best antibody with the highest affinity will be selected and further differentiated into either memory B cells or antibody secreting B cells, known as plasma cells. These processes of somatic mutation and differentiation occur in the germinal center and require T cell help
Challenges of Making Human Antibodies
There has been interest in using antibodies clinically since the beginning of the nineteenth century. However, developing specific antibodies was initially limited to polyclonal antibodies, which required a large numbers of animals immunized with the desired antigen. In 1975, Milstein and Köhler [99] developed the hybridoma technology for the production of monoclonal antibodies (mAbs). The method is based on the fusion of spleen cells (antibody secreting B cells) from an immunized animal with murine myeloma cells, resulting in the generation of hybrid cells (hybridomas) that could be maintained in tissue culture to secrete specific mAbs (Fig. 6). This technology gave rise to the field of therapeutic antibodies, and in 1985 the first mAb, Muromonab-anti-CD3, was approved by the FDA. However, the immunogenicity of murine-derived mAbs precludes their use in chronic or recurrent human diseases such as inflammation or cancer. Mouse mAbs are frequently recognized as foreign by the patients leading to the development of human anti-mouse antibody (HAMA) responses [100]. By replacing mouse sequences with human framework homologous sequences, antibody humanization partially bypasses the HAMA responses [101].

Principle of hybridoma technology. Mouse spleen B cells from an immunized mouse and mouse myeloma cells are fused with the use of the polyethylene glycol (PEG) mediated cell fusion method, and then selected in hypoxanthine aminopterin-thymidine (HAT) medium. The HGPRT gene is required for the generation of nucleosides from nucleic acids via the salvage pathway. The myeloma cells used for hybridoma technology are defective in HGPRT gene and therefore will not grow in HAT medium. The fusion cells will survive because they contain a functional HGPRT gene (from the B cells). Primary B cells will not survive in culture very long and eventually all will die. The selection process in HAT medium will yield healthy hybrid cells that contain the HGPRT gene which can be further screened for specific antibody production using the antigen used for immunization. Positive cultures will be cloned by limiting dilution using 96-well culture plates, resulting in the generation of hybridoma clones that produce monoclonal antibodies [99]
Taking into consideration the challenges mentioned above, and inspired by phage display of peptide libraries and the assembly of Ig genes in B cells, Winter and colleagues demonstrated that large combinatorial antibody libraries created by random combination of genes for the heavy and light chain variable domains can be displayed on the surface of filamentous phage M13 [96, 102]. Notably, advances in recombinant DNA technology through the introduction of the polymerase chain reaction (PCR) technique have permitted the rapid isolation and cloning of various antibody domains and the construction of antibody libraries. Defined sets of primers specific for the different VH and VL gene families facilitated the amplification of all VH and VL gene repertoire that is randomly spliced together to create single-chain variable (scFv) or Fab fragment antibody libraries expressed on the surface of phages, thereby enabling rapid affinity selection from large libraries [102,103,104,105,106].
Origin of Ig V Genes
Antibody gene segments can be obtained either from normal B cell sources or synthetic methods (Fig. 7). In natural libraries, the V genes are derived from non-immune or immune donors. Phage antibody libraries with a natural antibody repertoire from different species than human have also been cloned and displayed on phages [11]. The first naïve phage antibody library was made by amplification of rearranged variable domain sequences from peripheral blood lymphocytes of non-immunized donors [96]. These libraries from non-immune donors permit the isolation of antibodies against a wide range of antigens, including non-immunogenic and toxic antigens [106]. The major drawback of these libraries is that the selected mAbs often have low affinity compared to that of antibodies from primary immune responses. However, once an antibody with desired specificity has been identified, its affinity and binding kinetics can be improved via mutagenesis, which is often done by introducing random or specific mutations in the CDR3 regions or the exchange of the light chains by chain shuffling [107,108,109]. Light chain shuffling combines the heavy chains of a selected antibody with the whole light chain repertoire of the naïve library. Antibody phage libraries can also be constructed from immunized patients or animals [110,111,112]. The use of immune libraries as a source of diversity is attractive and can lead to the selection of therapeutic antibodies with high affinities comparable to those of antibodies produced in secondary immune responses. The selection of such high-affinity antibodies can be achieved even from immune libraries with relatively small diversity [110]. Immune antibody libraries have been constructed from the rearranged V-gene repertoire of patients with microbial infections (viruses, parasites, bacteria), tumor transformation, and autoimmune diseases [111,112,113,114,115]. These type of antibody libraries can be used for selection of antibodies against several antigens of the same disease. However, if a single antigen is used for immunization, a new library would have to be constructed for each antigen.

Generation of antibody libraries. Repertoires of antibody fragments can be generated by PCR from rearranged V genes derived either from naïve or activated B cells subsequent to immunization or infection. Rearranged V-gene segments coding for the heavy and the light chains are PCR amplified from cDNAs, randomly paired to generate diverse repertoires. The assembled scFv repertoires are then cloned into a phagemid vector in order to be expressed on the surface of the phage as single-chain Fv or Fab antibody libraries. Similarly, V-gene repertoires can be assembled from human V-gene segments rearranged in vitro (synthetic repertoires). Mutations within the CDR regions can be introduced in order to mimic immune libraries
Unlike natural libraries, synthetic antibody libraries display artificially made diversity in V-gene segments. Randomized CDR regions are usually inserted into the synthetic VH and VL framework sequences. These libraries allow the isolation of high-affinity antibodies against any antigen, including non-immunogenic, toxic, and self antigens. The HuCal library was the first generated synthetic library consisting of 1010 fully human Fab fragments, which has yielded antibodies with nanomolar (nM) affinities to a number of antigens [116]. The framework diversity is limited to seven heavy and seven light chain consensus sequences. More recently, a large fully synthetic human Fab antibody library (named Ylanthia) has been generated by means of 36 fixed VH/VL framework pairings and contains a diversity of 1011 [117].
Naïve libraries in which the CDR3s of the heavy chains are replaced by random sequences are often called semi-synthetic libraries [118]. Semi-synthetic libraries are a combination of donor-derived Ig sequence with synthetic diversity in all or some of the CDR sequences, particularly the CDR3 domain. The introduction of diversity in the CDRs of germline V-gene segments bypasses the use of immunization [119]. The first semi-synthetic library was constructed by Hoogenboom and Winter [120]. Here, a repertoire of human VH genes from 49 human germline VH gene segments was rearranged in vitro in combination with a synthetic CDR3 of five or eight residues. These in vitro rearranged VH genes were cloned with a human Vλ3 light chain as scFv fragments for phage display. Since then, several synthetic antibody libraries were constructed and used to select high-affinity human antibodies. Generating antibodies against self or toxic antigens is limited if immunization is to be employed. In particular, human autoantigens are highly conserved amongst most routinely used laboratory mammals such as mice or rats.
Improving Antibody Function
Similar to peptides, once an antibody fragment candidate that binds to a specific target is identified, it can be further engineered for enhanced pharmacology and beyond. Engineering strategies aim to improve stability, effector functions, and reduce aggregation. Fc modifications to enhance antibody function or circulation times are common [4]. In the case of neutralizing Abs, the effector function should be reduced or eliminated. Due to their lack of effector functions, IgG4 antibodies represent the preferred IgG subclass for receptor blocking. Recombinant human IgG antibodies completely devoid of immune effector functions can be generated by mutations in the CH2 domain of human IgG1 or IgG2 [4]. Stability strategies include formulation adjustment, specific point mutations, and alteration of the disulfide bond in the Fab domain [121,122,123]. Bioinformatic approaches can also be used to improve antibody affinity [124, 125]. For example, Lippow et al. [126] generated higher affinity variants for three antibody targets by computationally selecting mutations that improved antibody–antigen interaction energy, focusing on binding electrostatics. Therefore, one may search for affinity improving mutations by evaluating for example electrostatics and van der Waals energies involved in antibody–antigen complex formation. Regardless of the modifications, a good balance between affinity, solubility, stability, and activity needs to be maintained in order to maximize the chances of any lead to work in clinical trials. Although outside the scope of this review, phage antibody fragments can be converted into bispecific antibodies or chimeric antigen receptors, with the aim to retarget T cells to a tumor for cancer immunotherapy [127]. Moreover, phage antibodies can be included in a wide variety of tailored theranostic agents. Various nanotechnology strategies are now exploiting antibodies and peptides as targeting ligands and as payloads for diagnostics and/or drug delivery [128].
Examples of Phage-Derived Antibodies in Clinical Use
Given the induction of HAMA by murine antibodies, clinical mAbs should be as human as possible. Antibodies that are fully human are now being produced from phage display and transgenic mice [105, 129]. The number of phage library-derived antibodies currently in clinical use, such as adalimumab and belimumab, demonstrates that the technology is a reliable discovery platform. In addition to therapy, antibody fragments are particularly desirable for tumor imaging, where better tissue penetration and rapid clearance from the body are required. Once a Fab or scFv antibody fragment with the desired affinity and specificity for a target protein has been selected, it may prove useful to convert it into an intact human antibody. Moreover, its affinity may significantly improve due to the bivalent nature of the IgG format. Compared with antibody fragments, whole IgGs have an extended serum half-life and are therefore the preferred format for therapeutic mAbs. The antibodies listed in Table 2 were converted to the IgG format from the antibody fragments initially isolated from phage libraries.
Adalimumab was the first phage display-derived mAb, as well as the first human antibody, approved for therapy [138]. It is a human IgG1 antibody that binds to TNF-α and neutralizes its function. Adalimumab is approved for the treatment of inflammatory diseases such as rheumatoid arthritis, psoriasis, and ulcerative colitis [139]. The B lymphocyte stimulator (BLyS), also known as BAFF, has a unique role in B cell differentiation and is involved in the pathogenesis of autoimmune diseases such as systemic lupus erythematosus (SLE) [140, 141]. Affinity selection of a scFv phage library on recombinant BLyS protein identified some binders, one of which was affinity matured to generate an antibody named belimumab that is approved for the treatment of autoimmune diseases, mainly SLE [130].
Several growth and angiogenic factors, including epidermal growth factor receptor (EGFR), insulin-like growth factor-1 receptor (IGF-1R), transforming growth factor β (TGF-β), and vascular endothelial growth factor A (VEGF-A), play important roles in diverse diseases such as cancer and autoimmunity [142, 143]. Moreover, some of these receptors also showed higher expression levels in tumors that recurred. These findings prompted the development of a number of drugs that targeted these receptors. Bevacizumab is a humanized antibody that binds to VEGF-A and neutralizes its activity [144]. This antibody was affinity matured using phage display in the Fab format to generate ranibizumab that differs from bevacizumab at five residues in the variable domains and one residue in the constant domain [132]. Based on several clinical data, ranibizumab was approved by FDA in 2006 for the treatment of age-related macular edema. And later, it was approved for the treatment of diabetic macular edema, and diabetic retinopathy [132]. Ramucirumab is a fully human IgG1 mAb that binds to the VEGF-R2 and functions as a receptor antagonist [133]. Necitumumab was identified by screening an antibody library on epidermal carcinoma cells (A431), which express high levels of EGFR [134]. By binding to the EGFR, the antibody blocks the binding of EGF, resulting in the inhibition of the receptor activation. Necitumumab was approved in 2015 for combination therapy with gemcitabine and cisplatin for the treatment of squamous NSCLC. Fresolimumab is a human IgG4 mAb that binds and neutralizes all TGF-β isoforms [145]. It is developed for the treatment of idiopathic pulmonary fibrosis, focal segmental glomerulosclerosis, and cancer. Cixutumumab is a human IgG1 mAb that binds to the human IGF-1 receptor and blocks receptor activation [136]. The antibody is still under clinical evaluation.
TNF-related apoptosis inducing ligand (TRAIL), a member of the TNF superfamily, interacts with its functional death receptors and induces apoptosis in a wide range of cancer types [146, 147]. Hence, TRAIL has been considered as an attractive agent for cancer therapy and numerous small agonist molecules were developed. Mapatumumab is phage display-derived human mAb that binds to TRAIL-R1 and induces apoptosis in cancer cells. It demonstrated safety and tolerability in cancer patients with advanced solid tumors or non-Hodgkin’s lymphoma, both as a single agent and in combination with chemotherapy [135]. Adams et al. also used phage display to develop a fully human, affinity-matured IgG1 monoclonal antibody (Apomab) that induced apoptosis upon interaction with human TRAIL-R2 [137]. The antibody showed a potent antitumor activity as a single agent and in combination with chemotherapy in several human xenograft models (colorectal, non-small cell lung, and pancreatic cancer cell lines).
In addition to cancer and autoimmune diseases, phage display can be used to generate antibodies against infectious agents. Anthrax is a zoonotic infection caused by Bacillus anthracis, which produces two protein toxins containing the protective antigen (PA) with either or both edema factor (EF) and lethal factor (LF) [148]. Toxin complexes are internalized by mammalian cells via endocytosis and the LF and EF factors are released into the cytosol where they trigger anthrax pathogenesis. Raxibacumab, a phage library-derived human mAb, binds to PA and neutralizes its function, thereby blocking the interaction of LF and EF factors with the toxin receptor in mammalian cells [131]. The antibody was selected from a naïve human scFv phage library, and in 2012, the FDA approved it to treat inhalational anthrax. Rabies is another infectious disease that spreads from animals to humans. The rabies virus causes the disease by infecting nerve cells. Using an immune scFv library, several neutralizing antibodies were selected. The best were named foravirumab and rafivirumab and both are under clinical development [149]. In addition to the optimized antibodies indicated above, numerous scFv antibody fragments have been selected from antibody libraries and are under preclinical or clinical development [11, 105].
More recently, we have developed a selection protocol to isolate scFv antibodies with “pan-cancer binding abilities” from a semi-synthetic human scFv antibody library [150]. The library was sequentially affinity selected on a panel of human cancer cell lines and specific scFv antibody fragments were selected. One of the selected scFv antibody fragments was converted into human IgG1 antibody. The engineered antibodies (named MS5-Fc) inhibited the growth of three human tumor xenografts, suggesting that it may be useful in treating a diverse array of oncologic indications. After binding to cells, the antibody-receptor complexes showed different distribution patterns depending on the type of cancer cells. As shown in Fig. 8, prostate cancer cell lines internalized the antibody-receptor complexes, while lymphoma and breast cancer cell lines did not. Moreover, the antibody-receptor complexes exhibited distinct membrane distribution profiles in different cells. These results indicate that antibody internalization and membrane distribution is not only dependent on the antibody and the antigen recognized by the antibody, but also on the cell types and neighboring proteins.

Confocal microscopy images showing the distribution of the MS5 antibody-receptor complexes. Cancer cells were stained with fluorescein-labeled antibody at 4 °C and then incubated at 37 °C for 3 h to allow internalization. Nuclei were visualized with Hoechst 33342 staining (blue)
Collectively, phage display technology has opened the way for the generation of human antibodies that recognize any desired antigen/cells with a high specificity. Presently, there are > 60 approved mAbs for human therapy and > 50 more in late-stage clinical trials. Rituximab, trastuzumab, bevacizumab, and the best-selling adalimumab are just some examples of the mAbs currently on the market [151]. It should be noted that in addition to peptides and antibodies, cDNAs generated from tumor cells and fragmented genome DNA from microbes were cloned and expressed in-frame with phage coat proteins. Screening these phage-displayed libraries with patient sera has proven to be a powerful strategy to identify immunogenic proteins and diseases caused or perpetuated by microorganisms [152,153,154,155].
Concluding Remarks
During cancer development, cells acquire a variety of genetic and epigenetic modifications which result in changes in proteomic profiles. In addition to aberrant glycosylations, these genetic alterations can result in a change in the type, number, and arrangement of the cell surface receptors. Although microarrays and RNA-Seq offer genome-wide surveys of the transcriptome, both technologies cannot detect the changes indicated above. Peptide and antibody phage libraries offer the possibility of probing for such changes on cancer cell surface. In contrast to purified protein targets, however, the affinity selection of phage libraries on whole cells is more likely to enrich for peptides or antibody fragments that bind to cell surface receptors in their natural conformation (e.g., correct protein folding, association with neighboring proteins, and expression level). Furthermore, this method of selection requires no prior knowledge about the targeted receptors. However, affinity selection on whole cells or tumor tissues is still a challenging task. The process becomes more difficult if the cell surface antigens are unknown. Presently, there is no effective and reliable screening method that can be applied to all cells. Each screening must be optimized for the type of cells being used. While peptides and scFv antibodies selected from phage libraries can be used for delivery of therapeutics without knowing their cell surface targets, receptor identification is critical for their use in patients. The development of appropriate label transfer reagents such as the Sulfo-SBED reagent should allow the identification of cell surface receptors following ligand binding on live cells. Whether used as research, diagnostic, therapeutic, or drug delivery agents, peptides and phage antibodies will continue to impact modern biotechnologies in the future, further justifying the awarding of the Nobel prize in chemistry.
Abbreviations
- cDNA:
-
Complementary DNA
- CDR:
-
Complementarity-determining region
- EGF:
-
Epidermal growth factor
- Fab:
-
Antigen-binding fragment
- FDA:
-
Food and drug administration
- Ig:
-
Immunoglobulin
- mAb:
-
monoclonal antibody
- NSCLC:
-
Non-small cell lung cancer
- scFv:
-
Single-chain variable fragment
- TNF:
-
Tumor necrosis factor
- VEGF:
-
Vascular endothelial growth factor
- VH:
-
Variable heavy chain
- VL:
-
Variable light chain
References
Smith, G. P. (1985). Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science, 228, 1315–1317.
Smith, G. P., & Petrenko, V. A. (1997). Phage display. Chemical Reviews, 97, 391–410.
Nixon, A. E., Sexton, D. J., & Ladner, R. C. (2014). Drugs derived from phage display: From candidate identification to clinical practice. MAbs, 6, 73–85.
Kennedy, P. J., Oliveira, C., Granda, P. L., & Samento, B. (2018). Monoclonal antibodies: Technologies for early discovery and engineering. Critical Reviews in Biotechnology, 38, 394–408.
Barbas, C. F. 3rd, Kang, A. S., Lerner, R. A., & Benkovic, S. J. (1991). Assembly of combinatorial antibody libraries on phage surfaces: The gene III site. Proceedings of the National Academy of Sciences of the United States of America, 88, 7978–7982.
Cherf, G. M., & Cochran, J. R. (2015). Applications of yeast surface display for protein engineering. Methods in Molecular Biology, 1319, 155–175.
Yan, X., & Xu, Z. (2006). Ribosome-display technology: Applications for directed evolution of functional proteins. Drug Discovery Today, 11, 911–916.
Ohashi, H., Ishizaka, M., Hirai, N., & Miyamoto-Sato, E. (2013). Efficiency of puromycin-based technologies mediated by release factors and a ribosome recycling factor. Protein Engineering, Design and Selection, 26, 533–537.
Gray, B. P., & Brown, K. C. (2014). Combinatorial peptide libraries: Mining for cell-binding peptides. Chemical Reviews, 114, 1020–1081.
Shadidi, M., & Sioud, M. (2003). Selective targeting of cancer cells using synthetic peptides. Drug Resistance Updates, 6, 363–371.
Frenzel, A., Schirrmann, T., & Hust, M. (2016). Phage display-derived human antibodies in clinical development and therapy. MAbs, 8, 1177–1194.
Straus, S. K., & Bo, H. E. (2018). Filamentous bacteriophage proteins and assembly. Subcellular Biochemistry, 88, 261–279.
Qi, H., Lu, H., Qiu, H. J., Petrenko, V., & Liu, A. (2012). Phagemid vectors for phage display: Properties, characteristics and construction. Journal of Molecular Biology, 417, 129–143.
Enshell-Seijffers, D., Smelyanski, L., & Gershoni, J. M. (2001). The rational design of a ‘type 88’ genetically stable peptide display vector in the filamentous bacteriophage fd. Nucleic Acids Research, 29, E50–E50.
Hoogenboom, H. R., Griffiths, A. D., Johnson, K. S., et al. (1991). Multi-subunit proteins on the surface of filamentous phage: Methodologies for displaying antibody (Fab) heavy and light chains. Nucleic Acids Research, 19, 4133–4137.
Krumpe, L. R., Atkinson, A. J., Smythers, G. W., et al. (2006). T7 lytic phage-displayed peptide libraries exhibit less sequence bias than M13 filamentous phage-displayed peptide libraries. Proteomics, 6, 4210–4222.
Matsumoto, Y., Shindo, Y., Takakusagi, Y., et al. (2011). Screening of a library of T7 phage-displayed peptides identifies alphaC helix in 14-3-3 protein as a CBP501-binding site. Bioorganic & Medicinal Chemistry, 19, 7049–7056.
Cwirla, S. E., Peters, E. A., Barrett, R. W., & Dower, W. J. (1990). Peptide on phage: A vast library of peptides for identifying ligands. Proceedings of National Academy of Sciences of the United States of America, 87, 6378–6382.
Sioud, M., Førre, Ø, & Dybwad, A. (1996). Selection of ligands for polyclonal antibodies from random peptide libraries: Potential identification of autoantigens that may trigger B and T cell responses in autoimmune diseases. Clinical Immuology and Immunopathology, 79, 105–114.
Bonnycastle, L. L., Mehroke, J. S., Rashed, M., Gong, X., & Scott, J. K. (1996). Probing the basis of antibody reactivity with a panel of constrained peptide libraries displayed by filamentous phage. Journal of Molecular Biology, 258(5), 747–762.
Deyle, K., Kong, X.-D., & Heinis, C. (2017). Phage selection of cyclic peptides for application in research and drug development. Accounts of Chemical Research, 50, 1866–1874.
Ladner, R. C. (1995). Constrained peptides as binding entities. TIBTECH, 13, 426–430.
Teesalu, T., Sugahara, K. N., & Ruoslahti, E. (2012). Mapping of vascular ZIP codes by phage display. Methods in Enzymology, 503, 35–56.
Deramchia, K., Jacobin-Valat, M. J., Vallet, A., et al. (2012). In vivo phage display to identify new human antibody fragments homing to atherosclerotic endothelial and subendothelial tissues. The American Journal of Pathology, 180(6), 2576–2589.
Felici, F., Castagnoli, L., Musacchio, A., Jappelli, R., & Cesareni, G. (1991). Selection of antibody ligands from a large library of oligopeptides expressed on a multivalent exposition vector. Journal of Molecular Biology, 222(2), 301–310.
Fack, F., Hügle-Dörr, B., Song, D., et al. (1997). Epitope mapping by phage display: Random versus gene-fragment libraries. Journal of Immunological Methods, 206, 43–52.
Dybwad, A., Bogen, B., Natvig, J. B., Førre, O., & Sioud, M. (1995). Peptide phage libraries can be an efficient tool for identifying antibody ligands for polyclonal antisera. Clinical & Experimental Immunology, 102, 438–442.
Sun, Y., Kang, C., Liu, F., et al. (2017). RGD peptide-based target drug delivery of doxorubicin nanomedicine. Drug Development Research, 78, 283–291.
Koivunen, E., Wang, E., & Ruoslahti, E. (1995). Phage libraries displaying cyclic peptides with different ring sizes: Ligands specificities of the RGD-directed integrins. Biotechnology, 13, 265–270.
Arap, W., Pasqualini, R., & Ruoslahti, E. (1998). Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science, 279, 377–380.
Kibria, G., Hatakeyama, H., Ohga, N., Hida, K., & Harashima, H. (2013). The effect of liposomal size on the targeted delivery of doxorubicin to integrin αvβ3-expressing tumor endothelial cells. Biomaterials, 34, 5617–5627.
Pasqualini, R., Koivunen, E., Kain, R., et al. (2000). Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Research, 60(3), 722–727.
Laakkonen, P., Porkka, K., Hoffman, J. A., & Ruoslahti, E. (2002). A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nature Medicine, 8(7), 751–755.
Shadidi, M., & Sioud, M. (2003). Identification of novel carrier peptides for the specific delivery of therapeutics into cancer cells. The FASEB Journal, 17, 256–258.
Lee, S. M., Lee, E. J., Hong, H. Y., et al. (2007). Targeting bladder tumor cells in vivo and in the urine with a peptide identified by phage display. Molecular Cancer Research, 5, 11–19.
Pandya, H., Gibo, D. M., Garg, S., Kridel, S., & Debinski, W. (2012). An interleukin 13 receptor α 2-specific peptide homes to human glioblastoma multiforme xenografts. Neuro-Oncology, 14, 6–18.
Oyama, T., Sykes, K. F., Samli, K. N., et al. (2003). Isolation of lung tumor specific peptides from a random peptide library: Generation of diagnostic and cell-targeting reagents. Cancer Letters, 202(2), 219–230.
McGuire, M. J., Gray, B. P., Li, S., et al. (2014). Identification and characterization of a suite of tumor targeting peptides for non-small cell lung cancer. Scientific Reports, 4, 4480.
Chang, D. K., Lin, C. T., Wu, C. H., & Wu, H. C. (2009). A novel peptide enhances therapeutic efficacy of liposomal anti-cancer drugs in mice models of human lung cancer. PLoS ONE, 4(1), e4171.
Chi, Y.-H., Hsiao, J.-K., Lin, M.-H., et al. (2017). Lung cancer-targeting peptides with multi-subtype indication for combinational drug delivery and molecular imaging. Theranostics, 7, 1612–1632.
Hong, F. D., & Clayman, G. L. (2000). Isolation of a peptide for targeted drug delivery into human head and neck solid tumors. Cancer Research, 60, 6551–6556.
Jiang, Y.-Q., Wang, H.-R., Li, H.-P., et al. (2006). Targeting of hapeatoma cell and suppression of tumor growth by a novel 12mer peptide fused to superantigen TSST-1. Molecular Medicine, 12, 81–87.
Nishimura, S., Takahashi, S., Kamikatahira, H., et al. (2008). Combinatorial targeting of the macropinocytotic pathway in leukemia and lymphoma cells. Journal of Biological Chemistry, 283, 11752–11762.
Kim, Y., Lillo, A. M., Steiniger, S. C. J., et al. (2006). Targeting heat shock proteins on cancer cells: Selection, characterization, and cell-penetrating properties of a peptidic GRP78 ligand. Biochemistry, 45, 9434–9444.
Zhang, J., Spring, H., & Schwab, M. (2001). Neuroblastoma tumor cell-binding peptides identified through random peptide phage display. Cancer Letters, 171, 153–164.
Zitzmann, S., Mier, W., Schad, A., et al. (2005). A new prostate carcinoma binding peptide (DUP-1) for tumor imaging and therapy. Clinical Cancer Research, 1, 139–146.
Sugahara, K. N., Teesalu, T., Karmali, P. P., et al. (2009). Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell, 16, 510–520.
Karasseva, N. G., Glinsky, V. V., Chen, N. X., Komatireddy, R., & Quinn, T. P. (2002). Identification and characterization of peptides that bind human ErbB-2 selected from a bacteriophage display library. Journal of Protein Chemistry, 21, 287–296.
Li, Z., Zhao, R., Wu, X., et al. (2005). Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. The FASEB Journal, 19, 1978–1985.
Qin, X., Wan, Y., Li, M., et al. (2007). Identification of a novel peptide ligand of human vascular endothelia growth factor receptor 3 for targeted tumour diagnosis and therapy. Journal of Biochemistry, 142, 79–85.
Koivunen, E., Arap, W., Valtanen, H., et al. (1999). Tumor targeting with a selective gelatinase inhibitor. Nature Biotechnology, 17(8), 768–774.
Umlauf, B. J., Mercedes, J. S., Chung, C. Y., & Brown, K. C. (2014). Identification of a novel lysosomal trafficking peptide using phage display biopanning coupled with endocytic selection pressure. Bioconjugate Chemistry, 25(10), 1829–1837.
Jubb, A. M., Strickland, L. A., Liu, S. D., et al. (2012). Neuropilin-1 expression in cancer and development. The Journal of Pathology, 226, 50–60.
Li, Z. J., Wu, W. K., Ng, S. S., et al. (2010). A novel peptide specifically targeting the vasculature of orthotopic colorectal cancer for imaging detection and drug delivery. Journal of Controlled Release, 148, 292–302.
Nohara, S., Kato, K., Fujiwara, D., et al. (2016). Aminopeptidase N (APN/CD13) as a target molecule for scirrhous gastric cancer. Clinics and Research in Hepatology and Gastroenterology, 40, 494–503.
Pastorino, F., Brignole, C., Marimpietri, D., et al. (2003). Vascular damage and anti-angiogenic effects of tumor vessel-targeted liposomal chemotherapy. Cancer Research, 63(21), 7400–7409.
Herringson, T. P., & Altin, J. G. (2011). Effective tumor targeting and enhanced anti-tumor effect of liposomes engrafted with peptides specific for tumor lymphatics and vasculature. International Journal of Pharmaceutics, 411(1–2), 206–214.
Karmali, P. P., Kotamraju, V. R., Kastantin, M., et al. (2009). Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomedicine, 5, 73–82.
Jung, H. K., Kim, S., Park, R. W., et al. (2016). Bladder tumor-targeted delivery of pro-apoptotic peptide for cancer therapy. Journal of Controlled Release, 235, 259–267.
Leuschner, C., & Hansel, W. (2004). Membrane disrupting lytic peptides for cancer treatments. Current Pharmaceutical Design, 10, 2299–2310.
Sioud, M., Skorstad, G., Mobergslien, A., & Sæbøe-Larssen, S. (2013). A novel peptide carrier for efficient targeting of antigens and nucleic acids to dendritic cells. The FASEB Journal, 27, 3272–3283.
Neo, S. H., Lew, Q. J., Koh, S. M., et al. (2016). Use of a novel cytotoxic HEXIM1 peptide in the directed breast cancer therapy. Oncotarget, 7, 5483–5494.
Wang, X. F., Birringer, M., Dong, L. F., et al. (2007). A peptide conjugate of vitamin E succinate targets breast cancer cells with high ErbB2 expression. Cancer Research, 67, 3337–3344.
Luo, H., Yang, J., Jin, H., et al. (2011). Tetrameric far-red fluorescent protein as a scaffold to assemble an octavalent peptide nanoprobe for enhanced tumor targeting and intracellular uptake in vivo. The FASEB Journal, 25, 1–9.
Moreno, M., Zurita, E., & Giralt, E. (2014). Delivering wasp venom for cancer therapy. Journal of Controlled Release, 182, 13–21.
Sioud, M., & Mobergslien, A. (2012). Selective killing of cancer cells by peptide-targeted delivery of an anti-microbial peptide. Biochemistry Pharmacology, 84, 1123–1132.
Koller, C. M., Kim, Y., & Schmidt-Wolf, I. G. (2013). Targeting renal cancer with a combination of WNT inhibitors and a bi-functional peptide. Anticancer Research, 33, 2435–2440.
Sigismund, S., Avanzato, D., & Lanzetti, L. (2018). Emerging functions of the EGFR in cancer. Molecular Oncology, 12(1), 3–20.
Song, S., Liu, D., Peng, J., et al. (2008). Peptide ligand-mediated liposome distribution and targeting to EGFR expressing tumor in vivo. International Journal of Pharmaceutics, 363, 155–161.
Kohno, M., Horibe, T., Haramoto, M., et al. (2011). A novel hybrid peptide targeting EGFR-expressing cancers. European Journal of Cancer, 47, 773–783.
Carmeliet, P. (2005). VEGF as a key mediator of angiogenesis in cancer. Oncology, 69(Suppl 3), 4–10.
Shay, G., Lynch, C. C., & Fingleton, B. (2015). Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biology, 44–46, 200–206.
Medina, O. P., Söderlund, T., Laakkonen, L. J., et al. (2001). Binding of novel peptide inhibitors of type IV collagenases to phospholipid membranes and use in liposome targeting to tumor cells in vitro. Cancer Research, 61(10), 3978–3985.
Mäkelä, A. R., Enbäck, J., Laakkonen, J. P., et al. (2008). Tumor targeting of baculovirus displaying a lymphatic homing peptide. The Journal of Gene Medicine, 10, 1019–1031.
Osuka, S., & Van Meir, E. G. (2017). Overcoming therapeutic resistance in glioblastoma: The way forward. The Journal of Clinical Investigation, 127(2), 415–426.
Siegel, R. L., Miller, K. D., & Jemal, A. (2016). Cancer statistics. CA: A Cancer Journal of Clinicians, 66, 7–30.
App, C., Knop, J., Huff, T., et al. (2014). Peptide labeling with photoactivatable trifunctional cadaverine derivative and identification of interacting partners by biotin transfer. Analytical Biochemistry, 456, 14–21.
Bhogal, M. S., Lanyon-Hogg, T., Johnston, K. A., Warriner, S. L., & Baker, A. (2016). Covalent label transfer between peroxisomal importomer components reveals export-driven import interactions. Journal of Biological Chemistry, 291, 2460–2468.
Otvos, L. Jr., & Wade, J. D. (2014). Current challenges in peptide-based drug discovery. Frontiers in Chemistry, 2, 62.
Katsara, T., Selios, T., Deraos, S., et al. (2006). Round and round we go: Cyclic peptides in disease. Current Medicinal Chemistry, 13, 2221–2232.
Li, S., McGuire, M. J., Lin, M., et al. (2009). Synthesis and characterization of a high-affinity αvβ6-specific ligand for in vitro and in vivo applications. Molecular Cancer Therapeutics, 8, 1239–1249.
Erbas, B., & Tuncel, M. (2016). Renal function assessment during peptide receptor radionuclide therapy. Seminars in Nuclear Medicine, 6, 462–478.
Mero, A., Clementi, C., Veronese, F. M., & Pasut, G. (2011). Covalent conjugation of poly(ethylene glycol) to proteins and peptides: Strategies and methods. Methods in Molecular Biology, 751, 95–129.
Ehrlich, G. K., Michel, H., Truitt, T., et al. (2013). Preparation and characterization of albumin conjugates of a truncated peptide YY analogue for half-life extension. Bioconjugate Chemistry, 24, 2015–2024.
Roopenian, D. C., & Akilesh, S. (2007). FcRn: The neonatal Fc receptor comes of age. Nature Reviews Immunology, 7, 715–725.
Unverdorben, F., Richter, F., Hurt, M., et al. (2015). Pharmacokinetic properties of IgG and various Fc fusion proteins in mice. MAbs, 8, 120–128.
Strohl, W. R. (2015). Fusion proteins for half-life extension of biologics as a strategy to make biobeters. BioDrugs, 29, 215–239.
Sioud, M., Westby, P., Olsen, J. K., & Mobergslien, A. (2015). Generation of new peptide-Fc fusion proteins that mediate antibody-dependent cellular cytotoxicity against different types of cancer cells. Molecular Therapy-Methods & Clinical Development, 4, 2:e15043.
Mobergslien, A., Peng, Q., Vasovic, V., & Sioud, M. (2016). Cancer cell-binding peptide fused Fc domain activates immune effector cells and blocks tumor growth. Oncotarget, 15, 75940–75953.
Qin, H., Lerman, B., Sakamaki, I., et al. (2014). Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nature Medicine, 20, 676–681.
Tracy, D., Klareskog, L., Sasso, E. H., Salfeld, J. G., & Tak, P. P. (2008). Tumour necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacology & Therapeutics, 117, 244–279.
Klareskog, L., van der Heijde, D., de Jager, J. P., et al. (2004). Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: Double-blind randomised controlled trial. The Lancet, 363, 675–681.
Molineux, G., & Newland, A. (2010). Development of romiplostim for the treatment of patients with chronic immune thrombocytopenia: From bench to bedside. British Journal of Haematology, 150, 9–20.
Herbst, R. S., Hong, D., Chap, L., et al. (2009). Safety, pharmacokinetics, and antitumor activity of AMG 386, a selective angiopoietin inhibitor, in adult patients with advanced solid tumors. Journal of Clinical Oncology, 27, 3557–3565.
Karlan, B. Y., Oza, A. M., Richardson, G. E., et al. (2012). Randomized, double-blind, placebo-controlled phase II study of AMG 386 combined with weekly paclitaxel in patients with recurrent ovarian cancer. Journal of Clinical Oncology, 30, 362–371.
McCafferty, J., Griffiths, A. D., Winter, G., & Chiswell, D. J. (1990). Phage antibodies: Filamentous phage displaying antibody variable domains. Nature, 348, 552–554.
Alt, F. W., Yancopoulos, G. D., Blackwell, T. K., et al. (1984). Ordered rearrangement of immunoglobulin heavy chain variable region segments. The EMBO Journal, 3, 1209–1219.
Tonegawa, S. (1983). Somatic generation of antibody diversity. Nature, 302, 575–581.
Köhler, G., & Milstein, C. (1975). Continuous cultures of fused cells secreting antibody of predefined specificity. Nature, 256, 495–497.
Tjandra, J. J., Ramadi, L., & McKenzie, I. C. (1990). Development of human anti-murine antibody (HAMA) response in patients. Immunology and Cell Biology, 68, 367–375.
He, X. Y., Xu, Z., Melrose, J., et al. (1998). Humanization and pharmacokinetics of a monoclonal antibody with specificity for both E- and P-selectin. The Journal of Immunology, 160, 1029–1035.
Marks, J. D., Hoogenboom, H. R., Bonnert, T. P., et al. (1991). By-passing immunization: Human antibodies from V-gene libraries displayed on phage. Journal of Molecular Biology, 222, 581–597.
Lerner, R. (2016). Combinatorial antibody libraries: New advances, new immunological insights. Nature Reviews Immunology, 16, 498–508.
Winter, G., Griffiths, A. D., Hawkins, R. E., & Hoogenboom, H. R. (1994). Making antibodies by phage display technology. Annual Review of Immunology, 12, 433–455.
Hammers, C. M., & Stanley, J. R. (2014). Antibody phage display: Technique and applications. The Journal of Investigative Dermatology, 134, 1–5.
Unkauf, T., Miethe, S., Fühner, V., et al. (2016). Generation of recombinant antibodies against toxins and viruses by phage display for diagnostics and therapy. Advances in Experimental Medicine and Biology, 917, 55–76.
Hoogenboom, H. R. (2005). Selecting and screening recombinant antibody libraries. Nature Biotechnology, 23, 1105–1116.
Wu, H., Beuerlein, G., Nie, Y., et al. (1998). Stepwise in vitro affinity maturation of vitaxin, an alpha v beta 3-specific humanized mAb. Proceedings of the National Academy of Sciences of the United States of America, 95, 6037–6042.
Ho, M., Kreitman, R. J., Onda, M., & Pastan, I. (2005). In vitro antibody evolution targeting germline host spots to increase activity of an anti-CD22 immunotoxin. Journal of Biological Chemistry, 280, 607–617.
Beerli, R. R., & Rader, C. (2010). Mining human antibody repertoires. MAbs, 2, 365–378.
Ohlin, M., & Borrebaeck, C. A. (1996). Characterization of human antibody repertoire following active immune responses in vivo. Molecular Immunology, 33, 583–592.
Graus, Y. E., de Baets, M. H., Parren, P. W., et al. (1997). Human anti-nicotinic acelylcholine receptor recombinant Fab fragments isolated from thymus-derived phage display libraries from myasthenia gravis patients reflect predominant specificities in serum and block the action of pathogenic serum antibodies. The Journal of Immunology, 158(4), 1919–1929.
Rahumatullah, A., Ahmad, A., Noordin, R., & Lim, T. S. (2015). Delineation of BmSXP antibody V-gene usage from a lymphatic filariasis based immune scFv antibody library. Molecular Immunology, 67(2 Pt B):512–523. https://doi.org/10.1016/j.molimm.2015.07.040.
Hamidon, N. H., Suraiya, S., Sarmiento, M. E., et al. (2018). Immune TB antibody phage display library as a tool To study B cell immunity in TB infections. Applied Biochemistry and Biotechnology, 184(3), 852–868. https://doi.org/10.1007/s12010-017-2582-5.
Scott, A., Walper, B., Lee, P. S., Anderson, G. P., & Goldman, E. R. (2013). Selection and characterization of single domain antibodies specific for Bacillus anthracis spore proteins. Antibodies, 2, 152–167.
Knappik, A., Ge, L., Honegger, A., et al. (2000). Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. Journal of Molecular Biology, 296(1), 57–86.
Tiller, T., Schuster, I., Deppe, D., et al. (2013). A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. MAbs, 5(3), 445–470.
Barbas, C. F., Bain, J. D., Hoekstra, D. M., & Lerner, R. (1992). Semi synthetic combinatorial libraries: A chemical solution to the diversity problem. Proceedings of National Academy of Sciences of the United States of America, 89, 4457–4461.
Benhar, I. (2007). Design of synthetic antibody libraries. Expert Opinion on Biological Therapy, 7, 763–779.
Hoogenboom, H. R., & Winter, G. (1992). By-passing immunisation. Human antibodies from synthetic repertoires of germline VH gene segments rearranged in vitro. Journal of Molecular Biology, 227, 3–8.
Beck, A., Wurch, T., Bailly, C., & Corvaia, N. (2010). Strategies and challenges for the next generation of therapeutic antibodies. Nature Review Immunology, 10(5), 345–352.
Beck, A., & Reichert, J. M. (2012). Marketing approval of mogamulizumab: A triumph for glycol-engineering. MAbs, 4, 419–425.
Lee, C. C., Perchiacca, J. M., & Tessier, P. M. (2013). Toward aggregation-resistant antibodies by design. Trends in Biotechnology, 31, 612–620.
Roy, A., Nair, S., Sen, N., Soni, N., & Maddhusudhan, M. S. (2017). In silico methods for design of biological therapeutics. Methods, 131, 33–65.
Kiyoshi, M., Caaveiro, J. M., Miura, E., et al. (2014). Affinity improvement of a therapeutic antibody by structure-based computational design: Generation of electrostatic interactions in the transition state stabilizes the antibody-antigen complex. PLoS ONE, 9(1), e87099. https://doi.org/10.1371/journal.pone.0087099.
Lippow, S. M., Wittrup, K. D., & Tidor, B. (2007). Computational design of antibody-affinity improvement beyond in vivo maturation. Nature Biotechnology, 25(10), 1171–1176.
Morse, R. J., & Maus, M. V. (2016). Bispecific antibodies and CARs: Generalized immunotherapeutics harnessing T cell redirection. Current Opinion in Immunology, 40, 24–35.
Alibakhshi, A., Abarghooi Kahaki, F., Ahangarzadeh, S., et al. (2017). Targeted cancer therapy through antibody fragments-decorated nanomedicines. Journal of Controlled Release, 268, 323–334.
Green, L. L. (1999). Antibody engineering via genetic engineering of the mouse: XenoMouse strains are a vehicle for the facile generation of therapeutic human monoclonal antibodies. Journal of Immunological Methods, 231, 11–23.
Halpern, W. G., Lappin, P., Zanardi, T., et al. (2006). Chronic administration of belimumab, a BLyS antagonist, decreases tissue and peripheral blood B-lymphocyte populations in cynomolgus monkeys: Pharmacokinetics, pharmacodynamics, and toxicologic effects. Toxicological Sciences, 91, 586–599.
Mazumdar, S. (2009). Raxibacumab. MAbS 1, 531–538.
Steinbrook, R. (2006). The price of sight—ranibizumab, bevacizumab, and the treatment of macular degeneration. New England Journal of Medicine, 355, 1409–1412.
Spratlin, J. L., Cohen, R. B., Eadens, M., et al. (2010). Phase I pharmacologic and biologic study of ramucirumab (IMC-1121B), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. Journal of Clinical Oncology, 28, 780–787.
de Haard, H. J., van Neer, N., Reurs, A., et al. (1999). A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. Journal of Molecular Chemistry, 274, 18218–18230.
Hotte, S. J., Hirte, H. W., et al. (2008). A phase 1 study of mapatumumab in patients with advanced solid malignacies. Clinical Cancer Research, 14, 3450–3455.
Weickhardt, A., Doebele, R., Oton, A., et al. (2012). A phase I/II study of erlotinib in combination with the anti-insulin-like growth factor-1 receptor monoclonal antibody IMC-A12 (cixutumumab) in patients with advanced non-small cell lung cancer. Journal of Thoracic Oncology, 7, 419–426.
Adams, C., Totpal, K., Lawrence, D., et al. (2008). Structural and functional analysis of the interaction between the agonistic monoclonal antibody Apomab and the proapoptotic receptor DR5. Cell Death and Differentiation, 15(4), 751–761.
Kaymakcalan, Z., Sakorafas, P., Bose, S., et al. (2009). Comparisons of affinities, activities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clinical Immunology, 131, 308–316.
Welch, B. (2008). Adalimumab (Humira) for the treatment of rheumatoid arthritis. American Family Physician, 78, 1406–1408.
Mackay, F., & Browning, J. L. (2002). BAFF: A fundamental survival factor for B cells. Nature Reviews Immunology, 2, 465–475.
Stohl, W. (2017). Inhibition of B cell activating factor (BAFF) in the management of systemic lupus erythematosus (SLE). Expert Review in Clinical Immunology, 13, 623–633.
Witsch, E., Sela, M., & Yarden, Y. (2010). Roles for growth factors in cancer progression. Physiology, 25, 85–101.
McMahon, G. (2000). VEGF receptor signaling in tumor angiogenesis. The Oncologist, 5(Suppl 1), 3–10.
Chen, Y., Wiesmann, C., Fuh, G., et al. (1999). Selection and analysis of an optimized anti VEGF antibody: Crystal structure of an affinity-matured Fab in complex with antigen. Journal of Molecular Biology, 293, 865–881.
Oude Munnink, T. H., Arjaans, M. E., Timmer-Bosscha, H., et al. (2011). PET with the 89Zr-labeled transforming growth factor-beta antibody fresolimumab in tumor models. Journal of Nuclear Medicine, 52, 2001–2008.
Mérino, D., Lalaoui, N., Morizot, A., Solary, E., & Micheau, O. (2007). TRAIL in cancer therapy: Present and future challenges. Expert Opinion on Therapeutic Targets, 11, 1299–1314.
von Karstedt, S., Montinaro, A., & Walczak, H. (2017). Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nature Reviews Cancer, 17, 352–366.
Liu, S., Moayeri, M., & Leppla, S. H. (2014). Anhrax lethal and edema toxins in anthrax pathogenesis. Trends in Microbiology, 22, 317–325.
Kramer, R. A., Marissen, W. E., Goudsmit, J., et al. (2005). The human antibody repertoire specific for rabies virus glycoproteins as selected from immune libraries. European Journal of Immunology, 35, 2131–2145.
Sioud, M., Westby, P., Vasovic, V., Fløisand, Y., & Peng, Q. (2018). Development of a new high-affinity human antibody with antitumor activity against solid and blood malignancies. The FASEB Journal, 32(9), 5063–5077.
Reichert, J. M. (2017). Antibodies to watch in 2017. MAbs, 9,167–181.
Sioud, M., & Hansen, M. H. (2001). Profiling the immune response in patients with breast cancer by phage-displayed cDNA libraries. European Journal of Immunology, 31(3), 716–725.
Sioud, M., Hansen, M., & Dybwad, A. (2000). Profiling the immune responses in patient sera with peptide and cDNA display libraries. International Journal of Molecular Medicine, 6(2), 123–128.
Zantow, J., Just, S., Lagkouvardos, I., et al. (2016). Mining gut microbiome oligopeptides by functional metaproteome display. Scientific Reports, 6, 34337.
Dybwad, A., Førre, O., Natvig, J. B., & Sioud, M. (1995). Structural characterization of peptides that bind synovial fluid antibodies from RA patients: A novel strategy for identification of disease-related epitopes using a random peptide library. Clinical Immunology and Immunopathology, 75, 45–50.
Acknowledgements
This work was supported by the Norwegian Cancer Society (Grant No. 182593).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author has no potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sioud, M. Phage Display Libraries: From Binders to Targeted Drug Delivery and Human Therapeutics. Mol Biotechnol 61, 286–303 (2019). https://doi.org/10.1007/s12033-019-00156-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-019-00156-8