
Abstract
Papillary thyroid carcinoma (PTC) is a common endocrine malignancy. The pathology of PTC is far from clear. As a kinase that can be targeted, the role of TNIK in PTC has not been investigated. This study was focused on the effects and molecular mechanisms of TNIK in PTC. Both public datasets and clinical specimens were used to verify TNIK expression. The effects of TNIK were investigated in both cell lines and mice models. Transcriptome analysis was used to explore the underlying mechanism of TNIK. Immunofluorescence, wound healing, and qRT-PCR assays were used to validate the mechanism of TNIK in PTC. The therapeutic effects of TNIK inhibitor NCB-0846 were evaluated by flow cytometry, western blot, and subcutaneous xenografts mice. TNIK expression was upregulated in PTC tissues. TNIK knockdown could suppress cell proliferation and tumor growth in no matter cell models or nude mice. The transcriptome analysis, GO enrichment analysis, and GSEA analysis results indicated TNIK was highly correlated with cytoskeleton, cell motility, and Wnt pathways. The mechanistic studies demonstrated that TNIK regulated cytoskeleton remodeling and promoted cell migration. NCB-0846 significantly inhibited TNIK kinase activity, induced cell apoptosis, and activated apoptosis-related proteins in a dose-dependent manner. In addition, NCB-0846 inhibited tumor growth in tumor-bearing mice. In summary, we proposed a novel regulatory mechanism in which TNIK-mediated cytoskeleton remodeling and cell migration to regulate tumor progression in PTC. TNIK is a therapeutic target in PTC and NCB-0846 would act as a novel targeted drug for PTC therapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Thyroid cancer is a type of endocrine malignant tumor that has gained widespread attention due to its rapid growth [1]. Over the past three decades, the incidence and morbidity of thyroid cancer were steadily growing around the world [2,3,4,5]. Papillary thyroid carcinoma (PTC) accounts for approximately 84% of all thyroid cancers, making PTC the most common type of pathology [6]. Therefore, it is critical to understand how PTC occurs and its molecular mechanism.
TRAF2 and NCK interacting kinase (TNIK) plays a multifunctional role in cell growth, development, differentiation, and apoptosis [7]. Evidence have showed that TNIK expression is elevated in various tumors, such as gastric cancer, colorectal cancer, and prostate cancer [8,9,10]. TNIK amplification could promote the progression of gastric cancer, while TNIK interference could inhibit cell proliferation and tumor growth in colorectal cancer [8, 9]. As reported in our previous study, TNIK was highly expressed in PTC [11]. However, TNIK has not yet been studied in terms of its function and mechanism in PTC.
Typically, early treatment would lead to a favorable outcome of PTC [12]. Currently, targeted therapy is considered to be a promising treatment for PTC patients. For example, the FDA has approved Vandetanib, Cabozantinib, Sorafenib, and Lenvatinib as the main small-molecule inhibitors for advanced differentiated thyroid cancer [13]. However, these inhibitors are susceptible to drug resistance when used in the later stages of the disease. The small-molecule compound NBC-0846 is an oral TNIK kinase inhibitor that has shown significant anti-tumor effects in gastric and colorectal cancers [9]. However, the anti-tumor efficacy and mechanism of NCB-0864 in PTC are not clear.
In this study, we found that TNIK silencing inhibited PTC cell proliferation and suppressed PTC growth in vivo. Mechanistically, TNIK regulated cytoskeleton remodeling and promoted cell migration. Moreover, we verified that NCB-0846 suppressed cell proliferation and tumor growth, and promoted apoptosis by activating the expression of apoptosis-related genes. This evaluation study to evaluate the TNIK effects and small-molecule inhibitors will provide a new strategy to solve the problem of chemotherapy resistance in the clinical application of PTC.
Materials and methods
Data processing and gene expression analysis
PTC datasets obtained from gene chips and RNA-sequencing were available through the Gene Expression Omnibus (GEO). Formatted family files and Series Matrix files derived from GSE33630, GSE27155, GSE3678, and GSE3467 were downloaded and used to analyze TNIK expression.
Cell lines and cell culture
TPC-1, KTC-1, and BCPAP were purchased from the Cell Resource Center (IBMS, CAMS/PUMC, China). The cells were cultured in DMEM (Corning, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin at 37 °C under 5% CO2. A monolayer of cultured cells was observed. Cell passage was performed using 0.25% trypsin (Keygen Biotech, China).
Lentivirus infection
To generate stably transfected cell lines, the cells were plated at a density of 5 × 105 cells in 6-well plates and infected according to the instructions of the lentivirus. The lentivirus for TNIK knockdown was built by Genechem company in Shanghai, China. After 48-h transfection, cells were cultured in DMEM with 0.5-µg/mL puromycin (LABLEAD, China) for selection. The stably transfected cells were used in the following experiments.
Cell proliferation
In order to measure cell viability, the CCK-8 assay (Beyotime, China) was used. The PTC cells were seeded in 96-well plates at a density of 3,000 cells per well. Then, the cells received continuous incubation for 1, 2, 3, 4, and 5 days and 20 µl of CCK-8 was added to each well at the designed time points. After 3-h incubation, the optical density was measured at 450-nm wavelength on a microplate reader (CLARIO star, Germany).
RNA-sequencing and data processing
Total RNA extract was obtained from TPC-1 cells that had been transfected and sequenced according to the protocols for RNA-Sequencing. In terms of fragments per kilobase per million reads (FPKM) values, transcript expression levels were calculated. We identified differentially expressed genes (DEGs) by adjusting the P-value by 0.05 and the absolute fold change (|FC|) by 1.5 for each gene. Gene Ontology (GO) enrichment analysis was performed and a significantly enriched set of GO terms was determined by correcting P values below 0.05. The Cytoscape (http://www.cytoscape.org/) embedded ClueGO was used to interpret the biological functions of DEGs. All the GO terms and KEGG/Reactome pathways were integrated into ClueGO to create a functionally organized GO/pathway term network. Gene Set Enrichment Analysis (GSEA) software was used to conduct the enrichment analysis.
RNA extraction and qRT-PCR assay
We isolated total RNA using the Direct-zol RNA Miniprep Kits (Zymo Research). A reverse transcription kit (Takara) was used to convert the total RNA into cDNA. SYBR Green Master mix (BIO-RAD) was used for the amplification of the cDNA template. The GAPDH was used as the reference gene and three repeat holes were set. The qPCR was conducted at 95 °C for 10 min; 95 °C for 15 s; 60 °C for 35 s; 72 °C for 30 s; and 40 loops on a ViiA7™ Real-Time PCR System. According to the cycle threshold (CT) value, the relative gene expression was determined using a 2−ΔΔCt approach. Here are the primer sequences used: CCND1 (forward, 5′-GCTGCGAAGTGGAAACCATC-3′; reverse, 5′-CCTCCTTCTGCACACATTTGAA-3′); MYC (forward, 5′-GGCTCCTGGCAAAAGGTCA-3′; reverse, 5′-CTGCGTAGTTGTGCTGATGT-3′); LEF1 (forward, 5′-TGCCAAATATGAATAACGACCCA-3′; reverse, 5′-GAGAAAAGTGCTCGTCACTGT-3′); CTNNB1 (forward, 5′-CATCTACACAGTTTGATGCTGCT-3′; reverse, 5′-GCAGTTTTGTCAGTTCAGGGA-3′); and DKK1 (forward, 5′-CCTTGAACTCGGTTCTCAATTCC-3′; reverse, 5′-CAATGGTCTGGTACTTATTCCCG-3′).
Wound healing assay
A total of 3 × 105 cells were seeded in 6-well plates. As soon as the cells are about 50% confluent, we created a wound with a pipette tip (200 μl). A gentle rinse with PBS was performed on the detached cells, and the attached cells were grown in a medium that contained no FBS. A light microscope was used to examine and photograph the wounds at 0 and 24 h after the injury.
Immunofluorescence
Cells were seeded on a confocal culture dish for 48 h. In order to fix the cells, 4% paraformaldehyde was added for 20 min. TritonX-100 in PBS buffer (0.2%) was used to increase nuclear membrane permeability and BSA in PBS buffer (1%) was used to block nonspecific binding. The dishes were then incubated with Phalloidin (1:100, YEASEN) at 4 °C overnight, followed by incubation with Alexa Fluor 594 goat anti-rabbit IgG (1:2000, CST) at room temperature for 40 min. When the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI), photographs were captured under a Leica SP8 microscope (TCS SP8, Germany).
Drug IC50 detection
We seeded 96-well plates with 3,000 cells per well. After seeding for 24 h, serially diluted NCB-0846 (0, 0.001, 0.005, 0.01, 0.025, 0.05, 0.1, 0.5, 1, and 10 μM) was added and incubated for 72 h. After adding 20 μl of CCK-8 reagent, the wells were incubated for 3 h. After measuring the absorbance at 450 nm with a spectrophotometer (CLARIO star, Germany), a spectrum analysis was conducted. IC50 values are generated by analyzing the data as follows: we normalized the readings to DMSO-treated control wells and expressed them as percentages. Percent luminescence was plotted against inhibitor concentration (logarithmic scale). Using a nonlinear regression model with log10 (inhibitor) versus response, IC50 values and dose–response curves were generated. The IC50 is defined as the concentration that achieves a half-maximal effect without completely killing the organism.
Apoptosis detection
A total of 3 × 105 cells were seeded in 6-well plates and incubated for 24 h. Serially diluted NCB-0846 (0, 0.1, 0.5, and 2.5 μM) was added to the cells and incubated for another 72 h. The cells were washed twice with PBS and centrifuged for 5 min at 2000 r/min. The cell suspension was diluted in 500-μl binding buffer. Then, Annexin V-FITC and Propidium Iodide (PI) were added to diluted cell suspension and incubated for 15 min in the dark. A total of 1 × 104 stained cells were subjected to flow cytometry. We used the BD FACSCalibur™ flow cytometer to collect data and FlowJo version 10 software was used to determine the proportion of non-apoptotic, early apoptotic, late apoptotic/necrotic, and dead cells.
Animal experiments
HuaFuKang Bioscience in Beijing provided eight-week-old BALB/c nude female mice for this study. The mice were used for the experiments after adapting to local conditions for 1 week. The mice were then randomly split into two groups with nine mice per group. A total of 6 × 105 BCPAP cells were inoculated into the subcutaneous tissues of nude mice in the medium that contained 50% Matrigel (Corning, USA). When a tumor was palpable, the tumor volume (third a week) and body weight (third a week) of each mouse were recorded and the volume of the object was calculated using the formula volume = length × width2 × 0.5. As soon as 35 days have passed since the inoculation, the mice were sacrificed and the tumors were completely isolated and photographed.
We treated subcutaneous xenografts with TNIK inhibitor by oral gavage to test whether NCB-0846 has a curative effect. Upon reaching 200 mm3 of tumor volume, the mice were randomly divided into two groups based on the tumor volume and treatment began. Each mouse was given the drug by oral gavage daily, either by 0 mg kg−1 (vehicle alone) or by 0 mg kg−1 (body weight) in DMSO/polyethylene glycol solution (10:45:45v/v) for 5 days. The tumor volume and body weight were recorded and calculated as mentioned above. On the 28th day after treatment, the mice were sacrificed and the tumors were isolated and photographed. Experimenters were blinded to cells, drugs, and solvents that were injected into the mice. Guidelines and regulations relevant to institutions and countries were followed during all experimental procedures.
Immunohistochemistry
Tumor and adjacent tissue specimens were obtained from patients at the Fourth Affiliated Hospital of Hebei Medical University. Donors were informed of the study’s purpose and the Beijing Children’s Hospital Research Ethics Board (2020-k-168) approved it. The tissues were cut at 4 μm and a solution of xylene and ethanol solutions was used to deparaffinize and rehydrate the tissues. The slides were boiled for five minutes in a 0.01-M sodium citrate buffer to recover the antigens. Afterward, the slides were sealed for 10 min with 3% hydrogen peroxide and with 5% horse serum for 1 h at room temperature (RT). The specimens were stained with the primary antibody anti-TNIK (ab224252, Abcam, USA) and incubated overnight at 4 °C. A reaction-strengthening fluid (ZSGB-Bio, China) was applied to the slides for 20 min at 37 °C, and the enzyme-labeled goat anti-rabbit IgG polymer (ZSGB-Bio, China) was then incubated for 20 min at 37 °C. Color development was carried out with DAB (ZSGB-Bio, China) for 5 min at room temperature. The slides were counterstained with Harris hematoxylin, after which they were dehydrated using a series of increasing alcohol concentrations and finally coverslipped. Representative images were captured by scanning the IHC slides with a light microscope (Nikon, 50i, Japan).
Western blot
Lentivirus-transfected cells were incubated for 48 h, harvested, and lysed for 30 min with cold RIPA buffer. The supernatant was collected by a centrifuge at 12,000 r/min for 15 min. In order to measure the protein concentration, the bicinchoninic acid protein assay (BCA) (Pierce, USA) was used. A 20-ug protein sample was separated by SDS-PAGE and electrophoresed onto a nitrocellulose filter membrane (PALL, USA). Membranes were blocked with nonfat milk (5%). The primary antibodies were incubated with the blocked membranes overnight at 4 °C. Here are the primary antibodies used in this study: anti-TNIK (Santa Cruz Biotechnology,1:1000), anti-pTNIK (Bioss,1:1000), anti-GAPDH (Abcam,1:1000), anti-caspase-3 (Beyotime,1:1000), anti-MCL1 (Beyotime,1:1000), anti-Cleaved caspase-3 (Cell signaling technology,1:1000), and anti-BCL2 (Cell signaling technology,1:1000). We incubated the membranes with secondary antibodies (IR800-IRDye®800, Abcam;1:5000), followed by a scanning measurement using the Odyssey CLx system (LI-COR Biosciences, USA).
Results
TNIK expression was increased in PTC
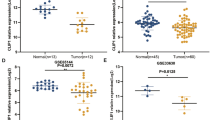
Based on the public GEO Datasets (GSE33630, GSE27155, GSE3678, GSE3467), the expression of TNIK in normal tissues was lower than that in PTC tumors. (Fig. 1A–D). Then, we performed immunohistochemical staining to detect the expression of TNIK in tumors and adjacent tissues of PTC patients. The results showed higher TNIK levels in PTC tissues (Fig. 1E). These findings firmly indicated that TNIK expression was increased in PTC.

TNIK expression positively correlated with PTC tumor. A–D Data mining of public datasets (GSE33630, GSE27155, GSE3678, and GSE3467) revealing the differential expression of TNIK in normal tissues and PTC tissues. E Immunohistochemistry (IHC) assays the expression of TNIK in adjacent tissues (normal) and PTC tumor tissues. Magnification, 100× or 400×. ***P < 0.001 vs control
TNIK promoted cell proliferation in PTC
To explore the effect of TNIK on cell proliferation, we successfully knocked down TNIK expression in TPC-1, KTC-1, and BCPAP cells by lentiviruses carrying shRNA sequences (Fig. 2A). Next, we tested the effect of down-regulated TNIK expression on the proliferation of PTC cells by CCK-8 assays, and the results showed that TNIK knockdown inhibited cell proliferation of TPC-1, KTC-1, and BCPAP cells (Fig. 2B). To verify that TNIK is responsible for tumor formation of PTC cells in vivo. We used BCPAP cells stably transfected with sh-TNIK#1 to establish a xenograft tumor model. The results showed that TNIK silencing significantly reduced the tumor volume and tumor weight (Fig. 2C, D, and E), but the two groups did not differ in body weight. (Fig. 2F). Taken together, these data indicated that TNIK facilitated cell proliferation and was indispensable for the tumorigenesis of PTC cells.

TNIK facilitated cell proliferation in vitro and in vivo. A Western blot assay indicating the expression of TNIK in PTC cells stably transfected with scramble shRNA (NC) or sh-TNIK. B CCK-8 assay depicting the change in cell viability of PTC cells stably transfected with scramble shRNA (NC) or sh-TNIK (n = 12 per time point). C–F Representative images (D), growth curves in vivo (C), tumor weight at the endpoints (E), and body weight (F) of xenografts formed by subcutaneous injection of BCPAP cells stably transfected with scramble shRNA (NC) or sh-TNIK into the dorsal flanks of nude mice (n = 9 per group). *P < 0.05 vs control
Transcriptome analysis revealed biological functions regulated by TNIK
To investigate the mechanism of TNIK silencing on PTC transcriptional responses, we performed RNA-seq in TPC-1 cells. There were 452 differentially expressed genes (177 down-regulated and 275 upregulated) when TNIK was knocked down (Fig. 3A). GO enrichment analysis was used to identify the potential biological functions associated with these differentially expressed genes. The results revealed that GO enrichment focused on maintaining location; regulating the movement of cellular components; and regulating the actin filament-based process (Fig. 3B and 3C). In addition, GSEA also revealed that genes regulated by TNIK were associated with the cytoskeleton, cell motility, and oncogenic pathways (Fig. 3D).

Transcriptome analysis revealed biological functions regulated by TNIK. A Volcano plot revealing the alteration of gene expression (fold change ≥ 1.5, P value < 0.05) in TPC cells stably transfected with scramble shRNA or sh-TNIK. B GO enrichment results of differentially expressed genes. C Differential expressed genes were functionally grouped and visualized in a network by Cytoscape. D Gene set enrichment analysis of TNIK-correlated genes in PTC derived from the publicly available dataset (GO:0060285). NES normalized enrichment score. *P < 0.001 vs control
TNIK induced cytoskeleton remodeling and promoted cell migration
Existing studies have revealed that TNIK regulated Wnt signaling in cancer [14]. Next, we examined the expression of Wnt target genes in response to TNIK. The qRT-PCR assays revealed that the knockdown of TNIK suppressed the transcriptional MYC, CCND1, DDK1, LEF1, and CTNNB1 in TPC-1 and KTC-1 cells, respectively (Fig. 4A and 4B). These results indicated that TNIK knockdown inhibited the transcription of downstream genes involved in Wnt signaling in PTC cells. According to the results of transcriptome analysis, we further examined the effect of TNIK expression on F-actin on the surface of the cell membrane during cell spreading. As judged by phalloidin staining, the representative F-actin staining of cells showed decreasing TNIK expression resulting in a reduction in the thickness of F-actin bands (Fig. 4C). We examined the effects of TNIK on cell migration using wound healing assays. “Gaps” created by micropipettes represent “wounds” and a decrease in the width of these “Gaps” indicates cell migration. The results showed that the “Gap” width in the TNIK knockdown group decreased in comparison to the control group (Fig. 4D), suggesting that TNIK enhanced cell migration in PTC.

TNIK induced cytoskeleton remodeling and promoted cell migration. A and B qRT-PCR indicating the expression of target genes of Wnt signaling pathway in TPC-1 or KTC-1 cells stably transfected with scramble shRNA (NC) or sh-TNIK. C Immunofluorescence staining of F-actin in PTC cells stably transfected with scramble shRNA (NC) or sh-TNIK. D The efficiency of migration was measured in BCPAP cells after TNIK knockdown by wound healing experiment. *P < 0.05 vs control
NCB-0846 significantly inhibited the viability of PTC cells
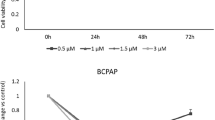
NCB-0846 is a well-characterized TNIK small-molecule catalytic inhibitor that traps TNIK in an inactive conformation and blocked TNIK autophosphorylation [9, 15]. The TNIK inhibitor of NCB-0846 was further evaluated to determine if TNIK kinase activity was critical for PTC. The results showed that NCB-0846 effectively reduced the active form of TNIK (pTNIK) within a concentration range of 0.1–2.5 μM in TPC-1 and KTC-1 cells (Fig. 5A). As shown in Fig. 5B, NCB-0846 reduced the viability of TPC-1, KTC-1, and BCPAP cells with a half-maximal inhibitory concentration (IC50) of 339 nM, 356 nM, and 767 nM, respectively. Meanwhile, NCB-0846 significantly decreased cell viability at a concentration of 500 nM. (Fig. 5C). The results supported the hypothesis that NCB-0846 targeted TNIK specifically to affect cell viability. Subsequently, we established mouse xenograft models derived from BCPAP cells to test whether TNIK preserved PTC growth in vivo. The results showed NCB-0846 obviously suppressed the growth of tumors (Fig. 5D and 5E). Treatment of NCB-0846 markedly reduced tumor volume and tumor weight in vivo (Fig. 5F and 5G), but the two groups did not differ in body weight (Fig. 5H). These results indicated that TNIK inhibitors could provide new treatment options for PTC.

NCB-0846 showed high inhibitory activity against TNIK and inhibited cancer cell growth. A Inhibition of TNIK phosphorylation by NCB-0846. TPC-1 cells and KTC-1 cells were cultured in the presence of DMSO (Control), NCB-0846 for 24 h and then analyzed by western blot. B Dose–response curves of PTC cell lines to NCB-0846. The cells were treated with increasing concentrations of NCB-0846 for 72 h. The lines represent the fitted curves, where each dot indicates the mean value ± SD of at least three independent experiments. C Cell viability of PTC cell lines treated with NCB-0846 (500 nM) for 72 h. At least three independent experiments with triplicates were conducted for each cell line. All data shown represent the mean value ± SD. D The schematic diagram depicted the drug schedule for the mice. E Tumor images from the study at the end of treatment. F–H Tumor growth curve (F), tumor weight at the endpoints (G), and Body weight (H) of BCPAP cell-derived xenografts treated with NCB-0846 (50 mg/kg, once a day (qd), five days on/two days off). Mean tumor volumes ± SEM are shown. n = 6 mice in the Control group; n = 6 mice in the NCB-0846-treated group; *P < 0.05 vs control
NCB-0846 induced cell death via apoptosis
To investigate the potential mechanism by which NCB-0846 down-regulated TNIK kinase activity to induce cell growth inhibition, we evaluated the effect of NCB-0864 on cell apoptosis. The results found that both TPC-1 and KTC-1 cells exhibit early and late apoptosis after treatment with NCB-0846 (Fig. 6A). Quantification analysis further confirmed that there was a statistically significant difference between the treatment groups and the control groups (Fig. 6B). In addition, we examined the protein expression of key genes to confirm that NCB-0846 could induce apoptosis in PTC cells. Among apoptosis-related proteins, NCB-0846 decreased the level of Bcl-2 and MCL1 and upregulated the expression of Cleaved caspase-3 compared with the untreated cells (Fig. 6C). These results indicated that NCB-0846 increased apoptosis in PTC cells.

NCB-0864 induced cell death via apoptosis. A The NCB-0864 induced apoptosis of TPC-1 cells and KTC-1 cells. Cells were treated with different doses (0.1, 0.5, and 2.5 μM) of NCB-0864 for 24 h, and the apoptotic cells were determined by Annexin V-FITC/PI analysis. B Percentage of living cells (lower left: Annexin V-FITC/PI), early apoptosis cells (lower right: Annexin V-FITC+/PI), and late apoptosis cells (upper right: Annexin V-FITC+/PI+) were shown in the flow cytometry chart. All data shown represent the means ± SD of at least three independent experiments. C Apoptosis-related genes were detected by western blot. *P < 0.05, **P < 0.01, ***P < 0.001 vs control
Discussion
In recent years, the incidence of papillary thyroid carcinoma (PTC) is on the rise [16, 17]. The etiology and molecular pathogenesis of PTC were complicated. Growing evidence supported that aberrant gene changes were the foundation upon which disease occurred. Gene rearrangements or point mutations of BRAF, RAS, or RET genes have been reported as initiating factors of PTC carcinogenesis [18, 19]. The studies in the past have reported vascular endothelial growth factor (VEGF) and its receptor (VEGFR) were over-expressed in differentiated PTC [20]. In our previous study, TNIK expression was significantly increased in PTC [11]. However, the roles of TNIK in PTC have been largely unknown, so we investigated the role of TNIK in this study. We demonstrated that TNIK significantly promoted PTC cell proliferation and tumor growth in established xenografts.
TNIK gene encodes a serine/threonine kinase and plays an important role in cancer progression [21, 22]. Yu and his colleagues found that TNIK promoted tumor growth by regulating AKT activation and cell autophagy in gastric cancer [23]. Sato et al. revealed that TNIK silencing resulted in significant inhibition of cell growth, colony, and mammosphere-forming capacity via cell cycle arrest in breast cancer [24]. Besides, TNIK could participate in cell growth, development, differentiation, and apoptosis through TGF-β, Wnt/β-catenin, and other signaling pathways [25, 26]. It remains unclear, however, how TNIK functions in PTC. We found that the expression of TNIK was increased in PTC tissues. Through CCK-8 and subcutaneous xenografts mice model assays, we verified that TNIK promoted cell proliferation in PTC. Our study indicated that TNIK might serve as a therapeutic target for PTC due to its oncogenic function.
Actin cytoskeleton usually contributes to the development of various cancers [27,28,29,30] and plays a key role in cytokinesis, cell division, and motility [31, 32]. In addition to the above effects, it also regulates cell migration and tumor morphogenesis [33]. Many molecular mechanisms are implicated in the regulatory process of the actin cytoskeleton. For instance, Rho GTPases, PAKs, ROCK, LIMKs, SSH1, and other kinases regulate actin filament dynamics and reorganization [34,35,36,37]. Ma et al. found that lncRNAs regulated the ubiquitination of actin cytoskeleton-related regulators in cancer progression [38]. In our study, we reported a novel regulatory manner of actin filaments remodeling. Through RNA-sequencing analysis, we screened that TNIK might be highly correlated with the cytoskeleton and cell motility. According to the immunofluorescence assay, we demonstrated that decreasing TNIK expression resulted in a reduction in the thickness of F-actin bands. Moreover, scratch wound healing assay further demonstrated that TNIK promoted the migration of PTC cells by regulating cytoskeleton remodeling. Consequently, TNIK regulated cell cytoskeleton remodeling, which might be the potential mechanism of PTC tumorigenesis.
In recent years, small-molecule inhibitors are the first-line therapies for advanced thyroid malignancies [39]. Regarding targeted drugs in PTC, Apatinib, targeting VEGFR2 could inhibit cell proliferation by acting on the PI3K/Akt/mTOR pathway [40]. Vemurafenib, a BRAF inhibitor, has an anti-proliferation effect in BRAF V600E-positive PTC [41]. PHA665752, c-MET inhibitor, suppressed cell growth and motility [42]. NCB-0846, TNIK kinase inhibitor, reduced cell viability and anchorage-independent growth in prostate cancer [10]. Our findings confirmed that NCB-0846 inhibited cell proliferation and promoted cell apoptosis in PTC. The anti-tumor ability of NCB-0846 suggested that targeting TNIK will be beneficial to the treatment of PTC patients.
In conclusion, TNIK was upregulated in PTC, promoted cell proliferation in TPC-1, KTC-1, and BCPAP cells, and facilitated tumor growth in established xenografts. TNIK knockdown disrupted cell cytoskeleton and motility which suppressed cell migration. NCB-0864 inhibited the proliferation of PTC cells, induced apoptosis, and restrained tumor growth in mice. This study demonstrated that TNIK was an oncogene and therapeutic target in PTC.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Yang YF, Yu B, Zhang XX, et al. Identification of TNIK as a novel potential drug target in thyroid cancer based on protein druggability prediction. Medicine. 2021;100(16):e25541.
Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973–2002. JAMA. 2006;295(18):2164–7.
Du LB, Li HZ, Wang XH, et al. Analysis of cancer incidence in Zhejiang cancer registry in China during 2000 to 2009. Asian Pac J Cancer Prev. 2014;15(14):5839–43.
Xie WC, Chan MH, Mak KC, et al. Trends in the incidence of 15 common cancers in Hong Kong, 1983–2008. Asian Pac J Cancer Prev. 2012;13(8):3911–6.
Xie SH, Chen J, Zhang B, et al. Time trends and age-period-cohort analyses on incidence rates of thyroid cancer in Shanghai and Hong Kong. BMC Cancer. 2014;14:975.
Li Z, Lin W, Zheng J, et al. Identification of immune-related lncRNAs to improve the prognosis prediction for patients with papillary thyroid cancer. Biosci Rep. 2021;41(2):BSR20204086.
Yuan R, Li Y, Fu Y, et al. TNIK influence the effects of antipsychotics on Wnt/beta-catenin signaling pathway. Psychopharmacology. 2021;238(11):3283–92.
Yu DH, Zhang X, Wang H, et al. The essential role of TNIK gene amplification in gastric cancer growth. Oncogenesis. 2014;2: e89.
Masuda M, Uno Y, Ohbayashi N, et al. TNIK inhibition abrogates colorectal cancer stemness. Nat Commun. 2016;7:12586.
Lee RS, Zhang L, Berger A, et al. Characterization of the ERG-regulated Kinome in Prostate Cancer Identifies TNIK as a Potential Therapeutic Target. Neoplasia. 2019;21(4):389–400.
Liu Y, Gao S, Jin Y, et al. Bioinformatics analysis to screen key genes in papillary thyroid carcinoma. Oncol Lett. 2020;19(1):195–204.
Ni X, Zhang J. Pediatric otolaryngology-head and neck surgery in China: present situation and future prospects. Pediatr Investig. 2019;3(3):137–40.
Cabanillas ME, Ryder M, Jimenez C. Targeted therapy for advanced thyroid cancer: kinase inhibitors and beyond. Endocr Rev. 2019;40(6):1573–604.
Masuda M, Sawa M, Yamada T. Therapeutic targets in the Wnt signaling pathway: feasibility of targeting TNIK in colorectal cancer. Pharmacol Ther. 2015;156:1–9.
Torres-Ayuso P, An E, Nyswaner KM, et al. TNIK is a therapeutic target in lung squamous cell carcinoma and regulates FAK activation through merlin. Cancer Discov. 2021;11(6):1411–23.
Raposo L, Morais S, Oliveira MJ, et al. Trends in thyroid cancer incidence and mortality in Portugal. Eur J Cancer Prev. 2017;26(2):135–43.
Al-Salamah SM, Khalid K, Bismar HA. Incidence of differentiated cancer in nodular goiter. Saudi Med J. 2002;23(8):947–52.
Nikiforova MN, Tseng GC, Steward D, et al. MicroRNA expression profiling of thyroid tumors: biological significance and diagnostic utility. J Clin Endocrinol Metab. 2008;93(5):1600–8.
Stoupa A, Kariyawasam D, Polak M, et al. Genetics of congenital hypothyroidism: modern concepts. Pediatr Investig. 2022;6(2):123–34.
Bunone G, Vigneri P, Mariani L, et al. Expression of angiogenesis stimulators and inhibitors in human thyroid tumors and correlation with clinical pathological features. Am J Pathol. 1999;155(6):1967–76.
Schouest KR, Kurasawa Y, Furuta T, et al. The germinal center kinase GCK-1 is a negative regulator of MAP kinase activation and apoptosis in the C. elegans germline. PLoS ONE. 2009;4(10):e7450.
Oehrl W, Kardinal C, Ruf S, et al. The germinal center kinase (GCK)-related protein kinases HPK1 and KHS are candidates for highly selective signal transducers of Crk family adapter proteins. Oncogene. 1998;17(15):1893–901.
Yu DH, Zhang X, Wang H, et al. The essential role of TNIK gene amplification in gastric cancer growth. Oncogenesis. 2014;2(2): e89.
Sato K, Padgaonkar AA, Baker SJ, et al. Simultaneous CK2/TNIK/DYRK1 inhibition by 108600 suppresses triple negative breast cancer stem cells and chemotherapy-resistant disease. Nat Commun. 2021;12(1):4671.
Mahmoudi T, Li VS, Ng SS, et al. The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 2009;28(21):3329–40.
Jho EH, Zhang T, Domon C, et al. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22(4):1172–83.
McGarry DJ, Armstrong G, Castino G, et al. MICAL1 regulates actin cytoskeleton organization, directional cell migration and the growth of human breast cancer cells as orthotopic xenograft tumours. Cancer Lett. 2021;519:226–36.
Jeong YJ, Hwang SK, Magae J, et al. Ascofuranone suppresses invasion and F-actin cytoskeleton organization in cancer cells by inhibiting the mTOR complex 1 signaling pathway. Cell Oncol. 2020;43(5):793–805.
Ohishi T, Yoshida H, Katori M, et al. Tankyrase-binding protein TNKS1BP1 regulates actin cytoskeleton rearrangement and cancer cell invasion. Cancer Res. 2017;77(9):2328–38.
Zhang P, Teng J, Wang L. Multiwalled carbon nanotubes inhibit cell migration and invasion by destroying actin cytoskeleton via mitochondrial dysfunction in ovarian cancer cells. Aging. 2020;12(24):25294–303.
Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326(5957):1208–12.
Akhshi TK, Wernike D, Piekny A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton. 2014;71(1):1–23.
Hall A. The cytoskeleton and cancer. Cancer Metastasis Rev. 2009;28(1–2):5–14.
Chinchole A, Lone KA, Tyagi S. MLL regulates the actin cytoskeleton and cell migration by stabilising Rho GTPases via the expression of RhoGDI1. J Cell Sci. 2022;135(20):jcs260042.
Best M, Gale ME, Wells CM. PAK-dependent regulation of actin dynamics in breast cancer cells. Int J Biochem Cell Biol. 2022;146:106207.
Grandy C, Port F, Pfeil J, et al. Influence of ROCK pathway manipulation on the actin cytoskeleton height. Cells. 2022;11(3):430.
Wang Z, Sun L, Liang S, et al. GPER stabilizes F-actin cytoskeleton and activates TAZ via PLCbeta-PKC and Rho/ROCK-LIMK-Cofilin pathway. Biochem Biophys Res Commun. 2019;516(3):976–82.
Ma X, Dang Y, Shao X, et al. Ubiquitination and long non-coding RNAs regulate actin cytoskeleton regulators in cancer progression. Int J Mol Sci. 2019;20(12):2997.
Ancker OV, Kruger M, Wehland M, et al. Multikinase inhibitor treatment in thyroid cancer. Int J Mol Sci. 2019;21(1):10.
Meng X, Wang H, Zhao J, et al. Apatinib inhibits cell proliferation and induces autophagy in human papillary thyroid carcinoma via the PI3K/Akt/mTOR signaling pathway. Front Oncol. 2020;10:217.
Jeong JH, Oh JM, Jeong SY, et al. Combination treatment with the BRAF(V600E) inhibitor vemurafenib and the BH3 mimetic navitoclax for BRAF-mutant thyroid carcinoma. Thyroid. 2019;29(4):540–8.
Chattopadhyay C, El-Naggar AK, Williams MD, et al. Small molecule c-MET inhibitor PHA665752: effect on cell growth and motility in papillary thyroid carcinoma. Head Neck. 2008;30(8):991–1000.
Funding
This work was supported by the Beijing-Tianjin-Hebei Integration Project (No. J200004, 20JCZXJC00120), Beijing Research Ward Demonstration Unit (BCRW202101), Beijing municipal science & technology commission (No. Z201100005520077), and Beijing Hospitals Authority Innovation Studio of Young Staff Funding Support (No. 202126).
Author information
Authors and Affiliations
Contributions
XN and YLG designed the experiment and revised the paper; RQZ and YBY performed most of the experiments and wrote the draft; YRY and MZ contributed to bioinformatic analysis; SCW and JLL performed tumor tissue collection and immunohistochemistry detection; XZ, YC, and LFH cultured the cells; XQZ and RLZ conducted the statistical analysis. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interest.
Ethical approval
The Ethics Committees of Beijing Children’s Hospital approved this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, R., Yu, Y., Yang, Y. et al. Therapeutic targeting of TNIK in papillary thyroid carcinoma: a novel approach for tumor growth suppression. Med Oncol 41, 160 (2024). https://doi.org/10.1007/s12032-024-02380-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-024-02380-y