
Abstract
Treatment with chimeric antigen receptor (CAR) T cells indicated remarkable clinical responses with liquid cancers such as hematological malignancies; however, their therapeutic efficacy faced with many challenges in solid tumors due to severe toxicities, antigen evasion, restricted and limited tumor tissue trafficking and infiltration, and, more importantly, immunosuppressive tumor microenvironment (TME) factors that impair the CAR T-cell function adds support survival of cancer stem cells (CSCs), responsible for tumor recurrence and resistance to current cancer therapies. Therefore, in-depth identification of TME and development of more potent CAR platform targeting CSCs may overcome the raised challenges, as presented in this review. We also discuss recent stemness-based innovations in CAR T-cell production and engineering to improve their efficacy in vivo, and finally, we propose solutions and strategies such as oncolytic virus-based therapy and combination therapy to revive the function of CAR T-cell therapy, especially in TME of solid tumors in future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Immunosuppressive tumor microenvironment (TME) due to molecular and cellular effectors, upregulation of inhibitory signaling pathways versus activating responses, and poor antigenicity of tumor cells all limited the antitumor responses delivered by both host normal immune T cells and transferred TILs (tumor infiltrated lymphocytes) during adoptive cell therapy (ACT) [1, 2]. These limitations have motivated immunologists and oncologists to introduce now approved immune checkpoint inhibitors (ICIs) and also to develop alternative ACT by engineering chimeric antigen receptors (CARs) expressed on T cells ex vivo to target tumor-associated antigens (TAAs) on the surface of tumor cells in vivo [2]. Over time and depending on the number and type of signaling domains, fourth-generation CAR T cells are developed that are enhanced to induce more enhanced cytolytic activity while producing inflammatory interleukins (ILs) (e.g., IL-12 and IL-15) and reprogrammed to have long-term survival of CAR T cells in the TME [3].
Until now, the CAR T-cell therapy was approved by FDA and limited only for hematologic tumors and targeting CD19 antigen on B cell (i.e., KYMRIAH and YESCARTA). KYMRIAH is a CD19-directed engineered autologous T-cell immunotherapy indicated for the treatment of adult patients with relapsed or refractory follicular lymphoma after two or more lines of therapy. YESCARTA is also a CD19-targeted autologous T-cell immunotherapy for adults with large B-cell lymphoma who are refractory to first-line chemo-immunotherapy or have relapsed within 12 months of first-line chemo-immunotherapy. However, preclinical studies indicated that the efficacy of CAR T-cell therapy of solid tumors is limited by several factors, including the selection of specific targetable antigens or epitopes, production of potentiated CAR, effective lymphodepletion, and infiltration, tumor homing, and survival of CAR T cells in the TME [4]. Therefore, profound solutions are needed to increase the chances of treating solid tumors with CAR T-cell immunotherapy.
Considering the survival of administered CAR T cells in vivo, selection of sources with stemness properties and reprogramming of mature cells into stem cells are examples of promising strategies under investigation, as discussed in this review. Moreover, the development of CAR T cells targeting the population of cancer stem cells (CSCs) involved in tumorigenesis, metastasis, tumor recurrence, and resistance to conventional therapies is an alternative way to eradicate established solid tumors, as investigated in recent years and summarized here. Finally, combination of CAR T-cell therapy with powerful therapies such as ICIs, oncolytic virotherapy, and chemo/radiotherapy is perspective strategy for enhancing CAR T-cell functions against resistant solid tumors that we presented this review.
CAR T cells targeting resistant CSCs and TME
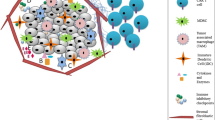
Similar to non-CSCs, CSCs express the cell surface antigens that could be targeted by CAR T cells in an HLA-independent manner, and preclinical and clinical studies have investigated to achieve an effective outcome [5, 6]. It should be noted that CSCs are heterogeneous cells for the expression or even non-expression of targeted antigens, different types of CSCs in many tumors express different pattern of TAAs, and moreover, CSCs may express the antigens shared with normal stem cell, which resulted in off-target toxicity [7] (Fig. 1a). Overcoming these challenges requires the development of new generations of CAR T cells, such as the selection of antigens with high expression levels compared to normal or CSC-specific antigens and the discovery of new CSC surface antigens [8, 9]. There are some CSCs markers that can be specifically targeted by monoclonal antibodies (mAb) for CSCs enrichment and identification and also immunotherapies by CAR T cells in clinical and preclinical studies (Table 1).

Development of CAR T cells targeting CSCs. a Convection CAR T cells developed to target common TAA on CSCs and induce apoptosis. b Modified CAR T cells developed to target specific stemness markers or antigens on CSCs and induce apoptosis. c Engineering CAR T cells to dual-target CSC-specific antigens and markers of any immunosuppressive cells in the TME such as Tregs, MDSCs, and TAMs. CAR chimeric antigen receptor, CSCs cancer stem cells, MDSCs myeloid-derived suppressor cells, TAAs tumor-associated antigens, TAMs tumor-associated macrophages, TME tumor microenvironment
CD133, or prominin-1, is a transmembrane glycoprotein found in CSCs of human tumors and drive resistance to chemotherapeutic agents that treatment with CD133-targeted CAR T cells resulted eradication of glioblastoma CSCs as well as off-tumor toxicity, as CD133 is expressed on normal neural stem cells [10]. Epithelial cell adhesion molecule (EpCAM) or CD326 is frequently overexpressed in tumor-initiating cells or CSCs and its overexpression is correlated with activation of Wnt/β-catenin signaling in CSCs [11, 12]. Therefore, EpCAM may represent a promising target for immunotherapy of EpCAM-expressing tumorigenic CSCs. For examples, CSC of liver and colorectal cancer express another transmembrane glycoprotein called EpCAM that its targeting by EpCAM-specific CAR T cells demonstrated the potential to inhibit tumor growth in EpCAM+ tumor xenografts while inducing secretion of cytotoxic cytokines such as TNF-α and IFN-γ [13]. In addition, CD123-directed CAR T cells have the potential to be highly effective in the elimination of CD123 + leukemic CSCs in a primary AML model [14].
However, targeting these markers has different safety issues and requires the identification and engineering of CAR T cells targeting only potent CSC markers such as CD271, LGR5, TIM-3, CD13, and CD105 (Fig. 1b). Overexpression of LGR5 (leucine-rich repeat-containing, G protein-coupled receptor 5) was strikingly correlated with human cancer recurrence, and application of the anti-LGR5 mAb-drug conjugate demonstrated inhibition of tumor growth in both in vitro and in vivo models [22]. In addition, the low expression of critical CSC-specific antigens limits the efficacy of therapies, and thus targeting surface markers that drive stemness or reprogramming in cancer cells enhances the function of CAR T cells to selectively target and eliminate these CSCs. For example, c-Met is a receptor tyrosine kinase that induces the expression of stem cell reprogramming factors (i.e., Nanog, SOX2, and CD133) and their targeting by kinase inhibitor [23] and CAR T cells [24] potently suppressed the growth of MET + CSCs in xenograft tumors. Considering the tumor-supportive role of tumor-associated macrophages (TAMs) in the TME of solid tumors, targeting antigens such as TIM-3 (T-cell immunoglobulin mucin-3) expressed on the surface of both TAMs and CSCs may be a promising approach for the treatment of human tumors [25] (Fig. 1c). TAMs also support CSCs in breast cancer by upregulating stemness drivers such as SOX2, OCT4, and NANOG [26]. Human myeloid-derived suppressor cells (MDSCs) are myeloid-originated progenitor cells that are classified into two subsets known as monocytic MDSCs and polymorphonuclear MDSCs. CD11b+ CD14− CD33+ MDSCs secrete cytokines and chemokines in TME providing immunosuppressive niche and thus reducing the efficacy of immunotherapy [6]. CSCs activating mTOR [27] and TGFβ1signaling pathways promoted infiltration, accumulation, and expansion of MDSCs in tumor site [28], which followed by impairment of T-cell responses. Reciprocally, MDSCs contribute to the stemness and survival of CSCs through the production of tumor-supportive PEG-E2 (prostaglandin E2) [29]. Regulatory T cells (Treg) are a T-cell-derived subset that has been shown to have multiple mechanisms of crosstalk with CSCs to promote an immunosuppressive TME [6]. Similar to MDSCs, PEG-E2 induces FOXP3+ CD4+ CD25+ Tregs to generate COX-2, suppressing the failure of effector T cells in cancer immunotherapy [30]. Tregs, mainly by secreting inhibitory cytokines such as TGF-β and IL-10, suppress CAR T cells, endogenous T cells and a variety of other immune cells [31]. Therefore, depleting (e.g., using cyclophosphamide or fludarabine) and targeting Tregs (e.g., CTLA-4 inhibition or CD25-targeting) may become an important aspect of CAR T-cell therapy for PDAC. These findings revealed that the TAM/Treg/MDSC-CSCs interaction reshapes the stemness in tumor cells resulting in tumor formation and progression and also opens the field for enhancing the antitumor potentials of CAR T cells targeting stemness markers in CSCs.
Challenges and perspectives approaches to improve CAR T cells targeting CSCs
CAR production and CAR T-cell expansion
Moreover, produced adenosine in hypoxic solid tumor mediated the reduction of IFN-supported CD8+ or CAR T-cell cytotoxicity. Therefore, the production of adenosine receptor-negative CAR T cells by applying CRISPR/Cas9 technology resulted in the generation of adenosine-resistant CAR T cells with enhanced antitumor function in vivo [32]. The antitumor responses of infused CAR T cells are also limited by their poor persistence in vivo, which could be improved by producing CAR T cells with memory or stem-like phenotypes that have long-term survival in vivo (Fig. 2a). These phenotypes could be achieved by stemness-related factor, metabolic, marker-based reprogramming of CAR T cells ex vivo. In clinical trials, CAR T cells that utilize oxidative phosphorylation (OXPHOS) have shown improved patient responses. Metabolically, strategies that redirect metabolism from glycolysis to OXPHOS/FAO or enhance mitochondrial fusion by blocking fission factors have shown promise. Restriction of glycolysis in favor of OXPHOS and FAO can be achieved by limiting glucose uptake, blocking glycolytic enzymes, or inhibiting positive regulators of glycolysis. Loss-of-function strategies, small molecule inhibitors, or upregulation of negative regulators are used in such approaches [33]. For example, glutamine promotes the differentiation of memory T cells into mature T cells and thus incubation of the source of CAR T cells with the inhibitor of glutamine uptake maintained these cells in memory-like phenotype with long persistence and better cytotoxic function in vivo [34].

Prospective methods to improve CAR T-cell persistence against CSC-specific antigens. a Short-lived T cells isolated from tumor mass, expanded ex vivo, and genetically CAR engineered plus induction of stemness factors to generate stem-like CAR T cells with long-term persistence in vivo while targeting CSC-specific markers. b T-iPSC-derived CAR T cells can be generated by reprogramming, differentiation, and genetic CAR engineering of TILs to present long and superior anti-CSC activity in vivo
Interestingly, transduction of αβ or γδ TCR into induced pluripotent stem cells (iPSCs) resulted in the production of iPSC-derived T-cell products with low cytotoxic function but long-term persistence due to their reprogrammed stemness properties [35, 36] (Fig. 2b). Indeed, the low cytotoxic function of CAR T-iPSCs is due to the rearrangement of TCR genes during iPSC manipulation, which subsequently produces the antigen specificity of effector T cells. This challenge could be overcome by using genome editing tools such as CRISPR to knock out RAG2 (recombinase gene 2), which is responsible for additional TCR rearrangement in T-iPSCs [37]. Overall, stem cell-like T cells and CAR T-iPSCs appear to be attractive products for fractionated cell therapy given their potentials to differentiate cells presenting long-term persistence in vivo such as memory and effector T cells as well as introducing a powerful resource toward improving current CAR T-cell therapy in the clinic.
Combination therapy
The immunosuppressive TME impaired the activity of transfused CAR T cells in vivo, which could be addressed by applying combination therapies. Oncolytic viruses are another live platform that is being considered as an immunotherapy approach within the ability to infect and lyse cancer cells [38]. OVs induce antitumor immune responses by releasing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) in tumor niches, followed by interaction of AMPs/DAMPs and pattern recognition receptors (PRRs) in cancer or innate immune cells and expression of proinflammatory cytokines to activate immunosuppressive TME [39, 40]. OVs can promote the intratumoral infiltration and expansion of effector immune cells [41], which could be used in cancer therapy to enhance the efficacy of CAR T-cell therapy (Fig. 3a). In addition, OVs engineered to express chemokines such as CCL5, CXCL11, and CXCR3 ligand recruited CAR T cells to the tumor site and facilitated their antitumor activity [42, 43].

Prospective pathways that Oncolytic virus (OV) and CAR T cells combination therapy can target and kill the CSCs in solid tumors. a OVs converted immunosuppressive and cold TME to the immunosuppressive and hot once by recruiting inflammatory immune cells and thus may promoted the infiltration of CAR T cells into tumor site for targeting and elimination of CSCs. b Co-treatment of cancer cells with oncolytic viruses lead to overproduction of cytokines (ILs and IFNs) that augmented the antitumor activity of infused CAR T cells. c Genetic engineering of oncolytic viruses with DNA encoding the CSC-specific marker can promote effector functions of CAR T cells to bind and kill CSCs
OVs directly lyse tumor cells, and with respect to CSCs, the Zika virus (ZIKV) and herpesvirus (HSV) have demonstrated the potential to target and kill glioblastoma CSCs [44]. OVs also induce the secretion of proinflammatory IFNs and ILs in the immunosuppressive TME or with cytokine-armed OVs, both of which contribute to the survival and expansion of CAR T cells and enhance their cytotoxic effects [38, 45, 46] (Fig. 3b). In fact, OVs activate apoptosis or lysis of tumor cells, resulting in the release of PAMPs and DAMPs, which are recognized by PRRs such as Toll-like receptors (TLRs) on immune cells, as well as the production of proinflammatory cytokines (such as type I IFNs, IL-1β/6/12, TNF-α and GM-CSF) and chemokines (such as CCL2/3/5 and CXCL10), all leading to the formation of a proinflammatory TME [47]. In addition, some CSCs are negative for markers that are selected for CAR, and OVs as carriers can provide expression of that specific marker on the surface of CSCs or tumor cells to enhance the antitumor activity of CAR T-cell therapy [48] (Fig. 3c). OVs also induce the expression of PD1/PD-L1, which sensitizes tumor cells to ICI therapy [44] and provides the combination therapies of OV, ICI, and CAR T cells. Taken together, OVs induce a switch from “cold” TME to “hot” TME by facilitating the recruitment of effector immune cells and activating systemic antitumor adaptive immunity to suppress tumor growth [49, 50]. Promising results of oncolytic virotherapy in combination with CAR T cells in preclinical studies for solid tumors lead to initiation of related ongoing clinical trials (Trials ID: NCT03740256 and NCT01953900).
Considering the plasticity of CSCs and the insufficiency of CAR T-cell monotherapy to completely and potently eliminate resistant tumor cells or CSCs, combination therapy with other standard radio/chemotherapy regimens may also be highly effective for CSC eradication. For example, treatment with local radiotherapy followed by infusion of NKG2D CAR T cells produced synergistic antitumor activity in a glioma model, indicating that radiotherapy promoted the infiltration of CAR T cells to the tumor site to exert their effector functions [51]. Similar to OVs, the combination of CSC-targeted CAR T-cell therapies with agents that induce the expression of CSC-specific antigens may overcome the obstacle posed by CSCs’ loss of the target antigen [52]. Finally, PD-1/PD-L1 overexpression resulted in CAR T-cell exhaustion in the TME [53], and the combination of CD19 CAR T-cell therapy and PD-1 blockade improved the survival and function of immunosuppressive-resistant CAR T cells in vivo [54]. In addition, M2 macrophages expressing PD-L1 limited the CAR T-cell activity that PD-L1 blockade in combination with CAR T-cell therapy resulted in a phenotypic shift toward more M1-like subsets and a loss of M2 macrophages through IFN-γ signaling, leading to enhanced antitumor activity of CAR T cells [55]. The combination of CAR T cells and ICI has proven to be safe and effective in hematological malignancies. However, the efficacy and safety of combining ICI and CAR T-cell therapies in advanced solid tumors has not been well established. With regard to the aforementioned advantages, several trials (NCT04003649, NCT03726515) are underway to evaluate the safety and efficacy of CAR T cell and ICI combinations in solid tumors, including glioblastoma. Overall, combination therapies provide durable expansion and function of CAR T cells in tumor-supportive TME, while limiting resistance mechanisms and toxicities at low desired doses of CAR T-cell infusion.
Safety issues
Besides the side effects such as cytokine release syndrome (CRS), the VH and VL domains of CAR, the scFv of a mAb or, can induce the production of anti-scFv antibodies and thus reduce their antitumor effect. Instead of the complete scFv, the construction of an extracellular domain consisting of a single variable domain on a heavy chain (VHH) resulted in nanobody as the targeting domain of CARs with the required functions for recognition and binding of cell surface-expressed target antigens while overcoming the obstacle of neutralizing antibodies [56]. CAR T cells are indicated to target and eradicate CSCs, but CSCs express antigens that are shared with normal stem cells and other normal cells, leading to off-tumor toxicity. One strategy to overcome this challenge is the direct or intratumoral administration of CAR T cells [57]. However, this method is not possible for non-surface available in tumor tissues. Other strategies involve the development of multi-CAR T cells that target more than one marker in heterogeneous CSCs [58] or modified CAR T cells expressing inhibitory CARs (iCARs) such as PD-1 and CTLA-4, which are switched off when bound to normal cells in vivo [59, 60], or the production of suicide-inducible CAR T cells to overcome off-tumor toxicity after infusion of CAR T cells [61]. Given the low density of TAA in normal cells, engineering CAR T cells with low affinity binding to TAA resulted in unbinding to normal cells and reduced cytotoxicity [62].
With regard to OV theory, patient safety is of paramount importance in cancer treatment, and treatment with oncolytic viruses appears to be the most promising in this regard. Completed trials have shown no dose-limiting toxicity. The major disadvantages of oncolytic viruses include fevers and flu-like symptoms, pre-existing antibodies, and OV replication in normal tissues, particularly the brain, especially in immunosuppressed cancer patients, which may hinder systemic delivery of the virus [63]. To overcome this problem, it is necessary to deliver the OVs to their target using a cellular vehicle, to suppress intracellular pathways associated with innate immunity by encoding one or two immunosuppressive genes of the wild-type OV in the OV vaccine, and to combine oncolytic therapy with immunosuppressive drugs [64].
Conclusion
Collectively, the suppressive TME impairs the cytotoxicity of CAR T cells, whose therapeutic potential could be enhanced by designing CAR T cells to target multiple antigens, re-expressing T engagers on the surface of memory T cells or iPSCs, reactivating the stemness or memory-induced pathway or genes in mature T cells, and combining them with other therapeutic modalities such as ICIs and OVs to enhance CAR T-cell trafficking and expansion at the tumor site, and combining them with other therapeutic modalities such as ICIs and OVs to enhance CAR T-cell trafficking and expansion at the tumor site, and also reprogramming the immunosuppressive TME at the molecular and cellular level into immunosuppressive once. Of note, the introduction of controllable keys to manage CAR T cells in case of adverse events is a critical safety recommendation for clinical establishment.
Data availability
Non applicable.
Abbreviations
- ACT:
-
Adoptive cell therapy
- CAR:
-
Chimeric antigen receptor
- CRS:
-
Cytokine-release syndrome
- CSCs:
-
Cancer stem cells
- DAMPs:
-
Damage-associated molecular patterns
- ICB:
-
Immune checkpoint blockade
- iCARs:
-
Inhibitory CARs
- ICIs:
-
Immune checkpoint inhibitors
- iPSCs:
-
Induced pluripotent stem cells
- GvHD:
-
Graft-versus-host disease
- PAMPs:
-
Pathogen-associated molecular patterns
- PBMC:
-
Peripheral blood mononuclear cell
- MDSCs:
-
Myeloid-derived suppressor cells
- OV:
-
Oncolytic virus
- TAAs:
-
Tumor-associated antigens
- TAMs:
-
Tumor-associated macrophages
- TILs:
-
Tumor-infiltrating lymphocytes
- TME:
-
Tumor microenvironment
References
Arumugam V, Bluemn T, Wesley E, Schmidt AM, Kambayashi T, Malarkannan S, et al. TCR signaling intensity controls CD8+ T cell responsiveness to TGF-β. J Leukoc Biol. 2015;98(5):703–12.
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–5.
Chmielewski M, Hombach AA, Abken H. Of CAR s and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. 2014;257(1):83–90.
D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. 2018;9(3):282.
Zhang Y, He L, Sadagopan A, Ma T, Dotti G, Wang Y, et al. Targeting radiation-resistant prostate cancer stem cells by B7–H3 CAR T cells. Mol Cancer Ther. 2021;20(3):577–88.
Mohammadzadeh H, Babaniamansour S, Majidi M, Zare A, Dehghani Firouzabadi M, Karkon-Shayan S. Merkel cell carcinoma on the right calf in association with chronic lymphocytic leukemia: a case report. Clin Case Rep. 2021;9(7):e04498.
Stingl J, Caldas C. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat Rev Cancer. 2007;7(10):791–9.
Mahmoodi S, Nezafat N, Negahdaripour M, Ghasemi Y. A new approach for cancer immunotherapy based on the cancer stem cell antigens properties. Curr Mol Med. 2019;19(1):2–11.
Afshari AR, Motamed-Sanaye A, Sabri H, Soltani A, Karkon-Shayan S, Radvar S, et al. Neurokinin-1 receptor (NK-1R) antagonists: potential targets in the treatment of glioblastoma multiforme. Curr Med Chem. 2021;28(24):4877–92.
Zhu X, Prasad S, Gaedicke S, Hettich M, Firat E, Niedermann G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget. 2015;6(1):171.
Munz M, Baeuerle PA, Gires O. The emerging role of EpCAM in cancer and stem cell signaling. Can Res. 2009;69(14):5627–9.
Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136(3):1012–24.
Zhang B-L, Li D, Gong Y-L, Huang Y, Qin D-Y, Jiang L, et al. Preclinical evaluation of chimeric antigen receptor–modified T cells specific to epithelial cell adhesion molecule for treating colorectal cancer. Hum Gene Ther. 2019;30(4):402–12.
Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GM, Cribioli E, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD 123-specific chimeric antigen receptor. Br J Haematol. 2013;161(3):389–401.
Dai H, Tong C, Shi D, Chen M, Guo Y, Chen D, et al. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: a single-arm, open-label, phase II trial. Oncoimmunology. 2020;9(1):1846926.
Kutsch N, Gödel P, Holtick U, Lohneis A, Vucinic V, Altefrohne FP, et al. A phase I dose finding trial of MB-CART20.1 in patients with relapsed or refractory B-cell non-hodgkin lymphoma. Blood. 2022;140:12980–1.
Huang H, Wu H-W, Hu Y-X. Current advances in chimeric antigen receptor T-cell therapy for refractory/relapsed multiple myeloma. J Zhejiang Univ Sci B. 2020;21(1):29.
Deng Z, Wu Y, Ma W, Zhang S, Zhang Y-Q. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 2015;16(1):1–9.
El Khawanky N, Hughes A, Yu W, Myburgh R, Matschulla T, Taromi S, et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat Commun. 2021;12(1):6436.
Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res. 2017;5(12):1152–61.
Ali S, Toews K, Schwiebert S, Klaus A, Winkler A, Grunewald L, et al. Tumor-derived extracellular vesicles impair CD171-specific CD4+ CAR T cell efficacy. Front Immunol. 2020;11:531.
Gong X, Azhdarinia A, Ghosh SC, Xiong W, An Z, Liu Q, et al. LGR5-targeted antibody-drug conjugate eradicates gastrointestinal tumors and prevents recurrence LGR5-targeted ADC eradicates gastrointestinal tumors. Mol Cancer Ther. 2016;15(7):1580–90.
Li C, Wu JJ, Hynes M, Dosch J, Sarkar B, Welling TH, et al. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology. 2011;141(6):2218–27.
Thayaparan T, Petrovic RM, Achkova DY, Zabinski T, Davies DM, Klampatsa A, et al. CAR T-cell immunotherapy of MET-expressing malignant mesothelioma. Oncoimmunology. 2017;6(12):e1363137.
Kikushige Y, Miyamoto T, Yuda J, Jabbarzadeh-Tabrizi S, Shima T, Takayanagi S-I, et al. A TIM-3/Gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell. 2015;17(3):341–52.
Li X, Bu W, Meng L, Liu X, Wang S, Jiang L, et al. CXCL12/CXCR4 pathway orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp Cell Res. 2019;378(2):131–8.
Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol. 2016;18(6):632–44.
Shidal C, Singh NP, Nagarkatti P, Nagarkatti M. MicroRNA-92 expression in CD133+ melanoma stem cells regulates immunosuppression in the tumor microenvironment via integrin-dependent activation of TGFβ. Can Res. 2019;79(14):3622–35.
Kuroda H, Mabuchi S, Yokoi E, Komura N, Kozasa K, Matsumoto Y, et al. Prostaglandin E2 produced by myeloid-derived suppressive cells induces cancer stem cells in uterine cervical cancer. Oncotarget. 2018;9(91):36317.
Mahic M, Yaqub S, Johansson CC, Taskén K, Aandahl EM. FOXP3+ CD4+ CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J Immunol. 2006;177(1):246–54.
Boroughs AC, Larson RC, Choi BD, Bouffard AA, Riley LS, Schiferle E, et al. Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. JCI Insight. 2019;4(8):6194.
Zhang H, Yu P, Tomar VS, Chen X, Atherton MJ, Lu Z, et al. Targeting PARP11 to avert immunosuppression and improve CAR T therapy in solid tumors. Nat Cancer. 2022;3:1–13.
Rad SMAH, Halpin JC, Tawinwung S, Suppipat K, Hirankarn N, McLellan AD. MicroRNA-mediated metabolic reprogramming of chimeric antigen receptor T cells. Immunol Cell Biol. 2022;100(6):424–39.
Shen L, Xiao Y, Zhang C, Li S, Teng X, Cui L, et al. Metabolic reprogramming by ex vivo glutamine inhibition endows CAR-T cells with less-differentiated phenotype and persistent antitumor activity. Cancer Lett. 2022;538:215710.
Montel-Hagen A, Seet CS, Li S, Chick B, Zhu Y, Chang P, et al. Organoid-induced differentiation of conventional T cells from human pluripotent stem cells. Cell Stem Cell. 2019;24(3):376–89.
Iriguchi S, Yasui Y, Kawai Y, Arima S, Kunitomo M, Sato T, et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat Commun. 2021;12(1):1–15.
Minagawa A, Yoshikawa T, Yasukawa M, Hotta A, Kunitomo M, Iriguchi S, et al. Enhancing T cell receptor stability in rejuvenated iPSC-derived T cells improves their use in cancer immunotherapy. Cell Stem Cell. 2018;23(6):850–8.
Keshavarz M, Ebrahimzadeh MS, Miri SM, Dianat-Moghadam H, Ghorbanhosseini SS, Mohebbi SR, et al. Oncolytic Newcastle disease virus delivered by mesenchymal stem cells-engineered system enhances the therapeutic effects altering tumor microenvironment. Virol J. 2020;17(1):1–13.
Guo ZS, Liu Z, Bartlett DL. Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol. 2014;4:74.
Shahgolzari M, Dianat-Moghadam H, Fiering S. Multifunctional plant virus nanoparticles in the next generation of cancer immunotherapies. Semin Cancer Biol. 2022;86:1076.
Liu Y, Cai J, Liu W, Lin Y, Guo L, Liu X, et al. Intravenous injection of the oncolytic virus M1 awakens antitumor T cells and overcomes resistance to checkpoint blockade. Cell Death Dis. 2020;11(12):1062.
Moon EK, Wang L-CS, Bekdache K, Lynn RC, Lo A, Thorne SH, et al. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology. 2018;7(3):e1395997.
Nishio N, Dotti G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology. 2015;4(2):e988098.
Chen L, Zhou C, Chen Q, Shang J, Liu Z, Guo Y, et al. Oncolytic Zika virus promotes intratumoral T cell infiltration and improves immunotherapy efficacy in glioblastoma. Mol Ther-Oncol. 2022;24:522–34.
Nguyen H-M, Guz-Montgomery K, Saha D. Oncolytic virus encoding a master pro-inflammatory cytokine interleukin 12 in cancer immunotherapy. Cells. 2020;9(2):400.
Chen T, Ding X, Liao Q, Gao N, Chen Y, Zhao C, et al. IL-21 arming potentiates the anti-tumor activity of an oncolytic vaccinia virus in monotherapy and combination therapy. J Immunother Cancer. 2021;9(1):e001647.
Gujar S, Pol JG, Kim Y, Lee PW, Kroemer G. Antitumor benefits of antiviral immunity: an underappreciated aspect of oncolytic virotherapies. Trends Immunol. 2018;39(3):209–21.
Rezaei R, Esmaeili Gouvarchin Ghaleh H, Farzanehpour M, Dorostkar R, Ranjbar R, Bolandian M, et al. Combination therapy with CAR T cells and oncolytic viruses: a new era in cancer immunotherapy. Cancer Gene Ther. 2021;29:1–14.
Keshavarz M, Miri SM, Behboudi E, Arjeini Y, Dianat-Moghadam H, Ghaemi A. Oncolytic virus delivery modulated immune responses toward cancer therapy: challenges and perspectives. Int Immunopharmacol. 2022;108:108882.
Azani A, Omran SP, Ghasrsaz H, Idani A, Kadkhodaei Eliaderani M, Peirovi N, et al. MicroRNAs as biomarkers for early diagnosis, targeting and prognosis of prostate cancer. Pathol Res Pract. 2023;248:154618.
Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma radiotherapy augments NKG2D CAR T cells against glioblastoma. Can Res. 2018;78(4):1031–43.
Lynn RC, Poussin M, Kalota A, Feng Y, Low PS, Dimitrov DS, et al. Targeting of folate receptor β on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood. 2015;125(22):3466–76.
Dianat-Moghadam H, Mahari A, Salahlou R, Khalili M, Azizi M, Sadeghzadeh H. Immune evader cancer stem cells direct the perspective approaches to cancer immunotherapy. Stem Cell Res Ther. 2022;13(1):1–12.
Wang J, Deng Q, Jiang YY, Zhang R, Zhu HB, Meng JX, et al. CAR-T 19 combined with reduced-dose PD-1 blockade therapy for treatment of refractory follicular lymphoma: a case report. Oncol Lett. 2019;18(5):4415–20.
Yamaguchi Y, Gibson J, Ou K, Lopez LS, Ng RH, Leggett N, et al. PD-L1 blockade restores CAR T cell activity through IFN-γ-regulation of CD163+ M2 macrophages. J Immunother Cancer. 2022;10(6):e004400.
Nix MA, Mandal K, Geng H, Paranjape N, Lin Y-HT, Rivera JM, et al. Surface proteomics reveals CD72 as a target for in vitro-evolved nanobody-based CAR-T cells in KMT2A/MLL1-rearranged B-ALLCD72 nanobody-based CAR-T in MLLr B-ALL. Cancer Discov. 2021;11(8):2032–49.
Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med. 2020;26(5):712–9.
Zhang E, Yang P, Gu J, Wu H, Chi X, Liu C, et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic malignancy. J Hematol Oncol. 2018;11(1):1–14.
Fedorov VD, Themeli M, Sadelain M. PD-1 and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5(215):215.
Liu L, Liu Y, Xia Y, Wang G, Zhang X, Zhang H, et al. Synergistic killing effects of PD-L1-CAR T cells and colorectal cancer stem cell-dendritic cell vaccine-sensitized T cells in ALDH1-positive colorectal cancer stem cells. J Cancer. 2021;12(22):6629.
Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS ONE. 2013;8(12):e82742.
Drent E, Poels R, Ruiter R, van de Donk NW, Zweegman S, Yuan H, et al. Combined CD28 and 4–1BB costimulation potentiates affinity-tuned chimeric antigen receptor-engineered T cells dual costimulation empowers very low-affinity CAR-T cells. Clin Cancer Res. 2019;25(13):4014–25.
Maroun J, Muñoz-Alía M, Ammayappan A, Schulze A, Peng K-W, Russell S. Designing and building oncolytic viruses. Future Virol. 2017;12(4):193–213.
Miest TS, Yaiw K-C, Frenzke M, Lampe J, Hudacek AW, Springfeld C, et al. Envelope-chimeric entry-targeted measles virus escapes neutralization and achieves oncolysis. Mol Ther. 2011;19(10):1813–20.
Funding
No funding is available.
Author information
Authors and Affiliations
Contributions
S.A.N, I.R, F.H, H.O.A, A.H.A, A.A, N.H.S, A.T.A, and Y.F.M contributed to investigation and writing original/revised draft. R.L and S.K.Sh contributed to conceptualization, investigation, writing original draft, writing-review & editing, visualization, supervision, and project administration. All co-authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Noraldeen, S.A.M., Rasulova, I., Lalitha, R. et al. Involving stemness factors to improve CAR T-cell-based cancer immunotherapy. Med Oncol 40, 313 (2023). https://doi.org/10.1007/s12032-023-02191-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-023-02191-7