
Abstract
Lung cancer is the leading cause of cancer-related mortality worldwide. The advent of epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) has significantly improved survival rates of patients with EGFR-mutant non-small cell lung cancer (NSCLC). However, as with other antitumor drugs, resistance to EGFR-TKIs is inevitably develops over time. Exosomes, extracellular vesicles with a 30–150 nm diameter, have emerged as vital mediators of intercellular communication. Recent studies revealed that exosomes carry non-coding RNAs (ncRNAs), including circular RNA (circRNA), microRNA (miRNA), and long noncoding RNA (lncRNA), which contribute to the development of EGFR-TKIs resistance. This review provides a comprehensive overview of the current research on exosomal ncRNAs mediating EGFR-TKIs resistance in EGFR-mutated NSCLC. In the future, detecting exosome ncRNAs can be used to monitor targeted therapy for NSCLC. Meanwhile, developing therapeutic regimens targeting these resistance mechanisms may provide additional clinical benefits to patients with EGFR-mutated NSCLC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung cancer is the second most common malignancy and the leading cause of cancer-related death worldwide, with a mortality rate of approximately 18% [1]. Non-small cell lung cancer (NSCLC) accounts for approximately 80–85% of all lung cancers, with adenocarcinoma being a common pathological subtype [2]. Epidermal growth factor receptor (EGFR) mutations, such as exon 19 deletion and exon 21 L858R insertion mutation, occur in approximately 10 to 44% of lung adenocarcinoma patients [3]. EGFR-tyrosine kinase inhibitors (EGFR-TKIs) have been approved as first-line therapy for advanced lung adenocarcinoma patients with EGFR mutation [4]. While approximately 70% of lung adenocarcinoma patients with EGFR mutation can benefit from EGFR-TKIs, about 30% do not respond due to primary resistance [4]. Furthermore, acquired resistance to EGFR-TKIs is inevitable, and most patients treated with first-line EGFR-TKIs have a progression-free survival of only 10 to 13 months [5].
The EGFR-T790M mutation is the common cause of acquired resistance to first- and second-generation EGFR-TKIs [6]. Osimertinib and other third-generation EGFR-TKIs have effectively overcome acquired resistance caused by EGFR-T790M mutation [7]. However, acquired resistance to third-generation EGFR-TKIs is also inevitable. Several previous studies [8] have revealed mechanisms of acquired resistance to the third-generation EGFR-TKIs, including EGFR-dependent mechanisms (such as EGFR C797S mutation, EGFR L718Q mutation) and EGFR-independent mechanisms (such as mesenchymal-epithelial transition factor (MET) and human epidermal growth factor receptor 2 (HER2) amplification, small cell transformation, and epithelial-mesenchymal transition (EMT)).
Exosomes, ranging in diameter from 30 to 150 nm, are a type of extracellular vesicle produced by the endoplasmic reticulum pathway. They carry various proteins, nucleic acids, metabolites, and lipids from their parent cell [9]. Exosomes play a crucial role in intercellular communication, influencing the function and physiological state of target cells, and are closely related to the occurrence and development of tumors [10]. Previous studies have shown that exosomes secreted by EGFR-TKI resistant cells can transfer resistance to EGFR-TKI sensitive cells [11].
Non-coding RNAs (ncRNAs), including circular RNA (circRNA), microRNA (miRNA), and long non-coding RNA (lncRNA), have been identified as important players in the development of lung cancer [12]. Valadi et al. [13] discovered exosomes protected miRNAs from RNase degradation. These provide conditions for exosomes to transmit ncRNA-mediated resistance between cells. In recent years, several studies have shown that ncRNAs carried by exosomes mediated resistance to EGFR-TKIs (Table 1) [14,15,16].
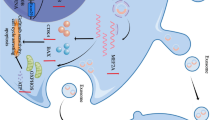
This review provides a comprehensive summary of current research on exosomal ncRNAs, including circRNA, miRNA, and lncRNA, and their role in mediated EGFR-TKIs resistance in EGFR-mutated NSCLC (Fig. 1). In the future, the detecting exosomal ncRNAs may serve as a useful tool for monitoring targeted therapy for NSCLC. Meanwhile, developing therapeutic regimens targeting these resistance mechanisms may provide additional clinical benefits to patients with EGFR-mutated NSCLC.

Mechanisms of EGFR-TKIs resistance mediated by exosomal non-coding RNAs. ROR1 RTK-like orphan receptor 1, EMT epithelial-mesenchymal transition, ATG7 autophagy-related protein 7, SOX2 OT SOX2 overlapping transcript, UCA1 urothelial carcinoma-associated 1, FOSL2 FOS-like 2
Exosomal circRNA mediate EGFR-TKIs resistance
Background
CircRNA is a covalent closed-loop non-coding RNA without 3′ or 5′-end, which is not easily degraded by endonuclease and is stably expressed in vivo [17]. Various circRNAs are involved in lung cancer progression, metastasis, and drug resistance [18]. CircRNA can act as miRNA sponges to regulate the expression of target genes [19]. For instance, Han et al. [20] demonstrated that circRNA circRAD23B regulates T-cell lymphoma invasion and metastasis 1 (TIAM1) and cyclin D2 (CCND2) by sponging miR-653-5p and miR-593-3p, respectively, and promotes the progression of NSCLC. In particular, exosomal circRNA derived from tumors plays a crucial regulatory role in developing and progressing neoplastic diseases [21].
Exosomal circRNA_102481 mediates gefitinib and erlotinib resistance
The expression of exosomal circRNA_102481 in the serum is significantly upregulated in NSCLC patients with gefitinib and erlotinib resistance [14]. Moreover, the level of circRNA_102481 and miR-30a-5p in exosomes are closely related to tumor stage, tumor differentiation status, progression-free-survival (PFS), and overall survival (OS) in NSCLC [14]. MiR-30a-5p is considered a biomarker for NSCLC and has an anti-cancer effect in various tumors by attenuating the malignant phenotype of cells [22]. The RTK-like orphan receptor 1 (ROR1) is a type-I membrane protein involved in tumor development [23]. ROR1 is overexpressed in various tumours and is associated with tumor growth and drug resistance [24]. Yang et al. [14] demonstrated that tumor-derived exosomal circRNA_102481 enhances ROR1 expression by sponging miR-30a-5p, thereby promoting gefitinib and erlotinib resistance (Table 1).
Currently, miR-30a-5p shows the potential to enhance the efficacy of EGFR-TKIs. Wang et al. [25] showed that miR-30a-5p can improve EGFR-TKIs resistance in NSCLC by inhibiting the phosphatidylinositol-3-kinase/protein kinase B (PI3K/AKT) pathway. Yan et al. [26] demonstrated that miR-30a-5p exerts inhibitory effects on the EMT and invasion of NSCLC by negatively regulating profilin-2. Therefore, miR-30a-5p may improve EGFR-TKIs resistance in NSCLC through multiple mechanisms based on current evidence. On the other hand, previous studies [27] have shown that ROR1 is also a potential target for improving EGFR-TKIs resistance. Liu et al. [28] demonstrated that ARI-1, a ROR1 inhibitor, can improve the resistance of NSCLC to EGFR-TKIs by modulating the PI3K/AKT/mammalian target of rapamycin (mTOR) signaling pathway. Notably, targeting ROR1 can also improve EGFR resistance by inhibiting EMT [29]. In conclusion, further investigation is warranted to explore the targeting of miR-30a-5p or ROR1 in NSCLC patients.
Exosomal miRNAs mediate EGFR-TKIs resistance
Background
MiRNAs are small non-coding RNAs with 18 to 24 nucleotides in lengths that regulate gene expression [30]. MiRNA can function as both oncogene and tumor suppressor gene [31]. MiRNA can regulate various biological processes, including cell proliferation, differentiation, and apoptosis [30]. MiRNAs can be used as cargo carried by exosomes and transported between cells to participate in tumor resistance [32]. Previous studies [33] have shown that exosomal miRNA could affect the gene expression profile of cells and increase the drug resistance of donor cells. Importantly, exosomal miRNA can predict EGFR-TKIs resistance [34]. Azuma et al. [35] demonstrated that exosomal miRNA from gefitinib-resistant cells can induce resistance in gefitinib-sensitive cells. Several exosomal miRNAs carried by exosomes play an important role in EGFR-TKIs resistance [15, 36,37,38].
Exosomal miR-214 mediates gefitinib resistance
MiR-214 is frequently upregulated in various tumor types, including ovarian cancer and esophageal squamous cell carcinoma, promoting tumor progression and drug resistance [39]. Zhang et al. [36] discovered that miR-214 exhibited significant upregulation in gefitinib-resistant cells and exosomes. Moreover, they demonstrated that the transfer of exosomal miR-214 from gefitinib-resistant cells to gefitinib-sensitive cells could confer the resistant phenotype, ultimately leading to the development of gefitinib resistance [36]. However, the signaling pathway through which exosomal miR-214 confers resistance to gefitinib remains unidentified [36]. Further clarification is necessary to elucidate the mechanisms by which exosomal miR-214 mediates resistance to gefitinib.
Previous studies [40] have shown that miR-214 mediates gefitinib resistance by regulating the phosphatase and tensin homolog/AKT (PTEN/AKT) pathway. Liao et al. [41] demonstrated that miR-214 expression was higher in erlotinib-resistant cell lines compared to erlotinib-sensitive cell lines in NSCLC. Down-regulation of miR-214 could reverse erlotinib resistance by enhancing homo sapiens LIM homeobox 6 (LHX6) expression [41]. Subsequently, Wang et al. [42] found that LHX6 affected EGFR-TKIs resistance by inhibiting Wnt/β-Catenin signaling pathway. These studies partially elucidate the mechanism underlying exosomal miR-214-mediated resistance to EGFR-TKIs.
It is worth mentioning that miR-214 has been reported to promote EMT and metastasis in lung adenocarcinoma [43]. Interestingly, Zhao et al. [44] proposed that miR-214 functions as a suppressor in lung cancer by targeting carboxypeptidase D. Chen et al. [45] discovered that miR-214 exerts inhibitory effects on the proliferation and progression of lung cancer by targeting Janus kinase 1 (JAK1). This paradoxical finding highlights the intricate role of miR-214 in lung adenocarcinoma.
Exosomal miR-21 mediates gefitinib resistance
MiR-21 can promote the progression of NSCLC [46] and can be used as a biomarker for tumor diagnosis and prognosis [47]. MiR-21 is overexpressed in gefitinib-resistant cells and patients with acquired EGFR-TKIs resistance [48]. The anti-tumor effect of EGFR-TKIs is mainly achieved through inhibition of EGFR downstream pathway, particularly the PI3K/AKT pathway [49]. Li et al. [48] demonstrated that miR-21 was involved in acquired resistance of EGFR-TKIs in NSCLC by downregulating PTEN and programmed cell death 4 (PDCD4) and activated the PI3K/AKT pathway. On the other hand, Liu et al. [37] showed that exosomes mediate EGFR-T790M mutation-mediated EGFR-TKIs resistance by activating the PI3K/AKT signaling pathway. Jing et al. [15] demonstrated that gefitinib-resistant cells can affect gefitinib-sensitive cells by secreting exosomes, with miR-21 being a crucial medium. Thus, exosomal MiR-21 can mediate acquired resistance to gefitinib, possibly by activating the PI3K/AKT signaling pathway.
The mechanism underlying exosomal miR-21-mediated resistance to EGFR-TKIs may be complex [50]. Firstly, exosomal miR-21 can induce resistance to EGFR-TKIs by promoting an immunosuppressive tumor microenvironment (TME). Studies [51] have shown that exosomal miR-21 derived from hypoxia pre-challenged mesenchymal stem cells can promote the progression of NSCLC and the M2-like polarization of macrophages. Furthermore, miR-21 plays a crucial role in an essential pathway associated with resistance to EGFR-TKIs. Huang et al. [52] discovered that the MAP kinase-ERK kinase/extracellular-signal-regulated kinase/miR-21 (MEK/ERK/miR-21) signaling pathway promotes resistance to osimertinib in NSCLC. Finally, miR-21 has been confirmed to be associated with EMT in lung cancer [53], which may represent one of the potential mechanisms underlying exosomal miR-21-mediated resistance to EGFR-TKIs. Targeting miR-21 may be a potential strategy for overcoming EGFR-TKIs resistance in NSCLC. Consistent with this hypothesis, Zhang et al. [54] demonstrated the improvement of EGFR-TKIs resistance in NSCLC by targeting miR-21.
Exosomal miR-522-3p mediates gefitinib resistance
The expression of miR-522-3p in H1975 cells and exosomes was significantly higher than in PC9 cells and exosomes [37]. Liu et al. [37] demonstrated that H1975 cells secreted exosomes carrying miR-522-3p to induce gefitinib resistance through the activation of the PI3K/AKT signaling pathway. Notably, EMT is also one of the mechanisms of acquired resistance to EGFR-TKIs [55]. The wound healing experiment by Liu et al. [37] showed that gefitinib resistance induced by exosomes secreted by H1975 cells was unrelated to EMT. Small clusters of drug-resistant cells may transmit drug-resistant phenotypes via exosomes in patients with NSCLC, and miR-522-3p may be one of the key mediators, which needs to be verified in further trials [56]. Importantly, Jin et al. [57] found that Bufalin could inhibit the progression of NSCLC by regulating the circ_0046264/miR-522-3p axis. Targeting miR-522-3p represents a potential strategy for improving the resistance of EGFR-TKIs in NSCLC.
Exosomal miR-210 mediates osimertinib resistance
Transforming growth factor-beta (TGF-β), as a strong inducer of EMT [58], leads to acquired resistance to EGFR-TKIs by inducing EMT [59]. Recent research has found that miR-210-3p could induce EMT in HCC827 cells independently of TGF-β [38]. Hisakane et al. [38] found that exosomal miR-210 was highly expressed in osimertinib-resistant cells and their exosomes, and the transfer of exosomal miR-210 secreted by osimertinib-resistant NSCLC cells can induce EMT and drug resistance in osimertinib-sensitive cells. This indicates that exosomal miRNA can cause EGFR-TKIs resistance by transferring resistance phenotypes and mediating EGFR-TKIs resistance by modulating the TME [60].
Exosomal miR-564 and miR-658 mediate gefitinib resistance
Azuma et al. [35] compared miRNA expression between exosomes secreted by gefitinib-sensitive cells and those secreted by gefitinib-resistant cells and found that miR-564 and miR-658 could confer a gefitinib-resistant phenotype. In addition, exosomal miR-658 may be involved in the upregulation of MET expression in gefitinib-sensitive cells, which may be one of the mechanisms by which exosomal miR-658 leads to EGFR-TKIs resistance [35]. Thus, future experiments are needed to clarify how exosomal miR-564 and miR-658 mediate gefitinib resistance phenotypes. Importantly, Yang et al. [61] found that miR-564 expression was downregulated in lung cancer and that miR-564 inhibited lung cancer progression by targeting zic family member 3 (ZIC3). This suggests that miR-564 may not be a viable target for improving EGFR-TKIs resistance in NSCLC.
Overall, the role of exosomal miRNAs in mediating EGFR-TKI resistance is complex and multifaceted. Understanding the underlying mechanisms and identifying potential therapeutic targets will be crucial for overcoming resistance and improving the efficacy of EGFR-TKIs in NSCLC.
Exosomal lncRNA mediate EGFR-TKIs resistance
Background
LncRNA is a type of non-coding RNA with a length of more than 200 nucleotides, which plays an important role in tumor development and drug resistance [62]. LncRNAs are involved in the regulation of various biological processes [63]. Exosomes can carry lncRNA to participate in information exchange between cells, including in NSCLC patients [16, 64]. Exosomal lncRNAs form tumor patients can reflect the tumor progression in real time [65], and may be involved in remodeling the TME [66].
Exosomal lncRNA MSTRG292666.16 mediate osimertinib resistance
The expression of lncRNA MSTRG292666.16 in serum exosomes is upregulated in osimertinib-resistant NSCLC patients compared with osimertinib-sensitive patients [67]. Deng et al. [67] showed that exosome-delivered lncRNA MSTRG292666.16 might be associated with osimertinib resistance. However, Deng et al. [67] did not reveal how exosomal lncRNA MSTRG292666.16 promoted osimertinib resistance. Furthermore, Wan et al. [68] showed that exosomes secreted by M2-type tumor-associated macrophages (TAMs) promoted osimertinib resistance via the MSTRG.292666.16/miR-6386-5p/MAPK8IP3 axis. This reveals the complexity of the origin of exosomal ncRNA mediating EGFR-TKIs resistance. On the other hand, targeting TAMs may be a potential way to improve EGFR-TKIs resistance in NSCLC [69]. Repolarization of TAMs may decrease the secretion of exosomes that induce resistance to EGFR-TKIs.
Exosomal lncRNA SOX2 overlapping transcript mediates osimertinib resistance
Macrophages, as a heterogeneous group of immune cells, can be classified into M1 and M2 macrophages based on their functions. M1-type macrophages secretes pro-inflammatory cytokines (such as IL-1β, IL-6, IL-12, and TNF-α), while M2-type macrophages secretes anti-inflammatory cytokines (such as IL-10 and TNF β) [70]. M2-type macrophages play a role in promoting cancer and can promote EGFR-TKIs resistance in lung cancer [71,72,73]. Blocking the crosstalk between M2-type macrophages and tumor cells can reduce the EGFR-TKIs resistance in tumor cells [66].
LncRNA SOX2 overlapping transcript (SOX2-OT) is highly expressed in serum exosomes of lung squamous cell carcinoma [74]. LncRNA SOX2-OT can regulate the expression of AKT/ERK and SOX2/ glioma-associated oncogene homolog 1(GLI-1) and promote the progression of lung cancer [75, 76]. Zhou et al. [16] showed that exosomal LncRNA SOX2-OT from H1975 cells targeted miR-627-3p, upregulated the expression of Smad2, Smad3, and Smad4, and promoted the M2-like polarization of macrophages. Finally, exosomal lncRNA SOX2-OT promotes osimertinib resistance in NSCLC cells by promoting the M2-like macrophage polarization [16]. Thus, the development of drugs targeting exosomal lncRNA SOX2-OT or inhibit M2-like macrophage polarization pathway may help partially reverse osimertinib resistance in NSCLC patients.
Exosomal lncRNA H19 mediate gefitinib and erlotinib resistance
LncRNA H19 is an imprinted gene located on human chromosome 11 [77]. It is an oncogene that promotes cell proliferation and drug resistance in NSCLC [78,79,80,81]. Lei et al. [82] found that gefitinib-resistant cells express highly levels of lncRNA H19 and can secrete RNA-carrying exosomes to promote gefitinib resistance. On the others hand, autophagy-related protein 7 (ATG7), a member of the autophagy-related proteins (ATGs) family, which is closely related to tumor progression and drug resistance [83,84,85]. ATG7 can regulate tumor progression and drug resistance by promoting autophagy [86]. ATG7 can promote chemotherapy resistance through autophagy in NSCLC [87]. Pan et al. [88] found that ATG7 may be a downstream target of miR-615-3p, and exosomal lncRNA H19 could regulate the expression of ATG7 through miR-615-3p. Furthermore, Pan et al. [88] showed that exosomal lncRNA H19 could promote erlotinib resistance via the miRNA-615-3p/ATG7 axis.
Interestingly, Chen et al. [89] discovered a significant correlation erlotinib resistance and downregulation of lncRNA H19. Furthermore, Xu et al. [90] discovered that β-Elemene can enhance the effectiveness of erlotinib by up-regulating lncRNA H19. Further investigation is required to elucidate this contradictory outcome.
Exosomal lncRNA UCA1 mediate gefitinib resistance
lncRNA UCA1 (urothelial carcinoma-associated 1) can regulate the Wnt pathway by mediating the interaction between DNA, mRNA, miRNA, and protein, thereby participate in tumor initiation and development [91]. LncRNA UCA1 can promote cisplatin and gefitinib resistance in NSCLC [92, 93]. Zhang et al. [93] showed that knockout of lncRNA UCA1 could inhibit gefitinib resistance by targeting the signal transducer and activator of transcription 3 (STAT3) signaling pathway in NSCLC. Additionally, FOS-like 2 (FOSL2), a member of the AP-1 transcription factor family [94], play a role in promotes TGF-β1-induced migration by regulating Smad3 in NSCLC [95]. Chen et al. [96] showed that exosomal lncRNA UCA1 targets FOSL2 by sponging miR-143 to promote gefitinib resistance in NSCLC. Based on the existing evidence, lncRNA UCA1 represents a potential target for improving EGFR-TKI resistance in NSCLC.
Overall, exosomal lncRNAs have emerged as important mediators of EGFR-TKI resistance in NSCLC. Their role in promoting resistance and modulating the TME highlights the complexity of the resistance mechanisms. Understanding the specific functions and mechanisms of exosomal lncRNAs in mediating resistance will be crucial for developing effective strategies to overcome EGFR-TKI resistance in NSCLC.
Conclusions
Exosomal ncRNAs have emerged as mediators of EGFR-TKIs resistance. This may be as a future exploration direction for the mechanisms of EGFR-TKIs resistance. However, several important areas require further investigation. Firstly, the origin of exosomal ncRNAs mediating EGFR-TKIs resistance is complex. Whether other sources exist beyond a small population of resistant tumor cells in TME [56] and M2-type TAMs remains to be further investigated [68]. Exploring the therapeutic potential of reducing the secretion of these exosomes is necessary [97]. For example, YAP 5-methylcytosine modification was observed to enhance exosome secretion and confer resistance to EGFR-TKIs in lung adenocarcinoma [98]. And Chen et al. [99] reversed gefitinib resistance by targeting YAP in NSCLC. Secondly, the mechanisms of EGFR-TKIs resistance mediated by exosomal ncRNAs are complex. Exosomal ncRNAs can act directly on tumor cells to induce resistance [37] and can act on M2-type macrophages in the TME, causing macrophage polarization and inducing EGFR-TKIs resistance [16]. Targeting these resistance mechanisms is a potential approach to enhance the efficacy of EGFR-TKIs. Finally, the binding mechanism of exosomes and tumor cells is not clear. Blocking the binding of specific exosomes to tumor cells may improve patient resistance to EGFR-TKIs to some extent.
Exosomal ncRNAs play an important role in EGFR-TKIs resistance. In the future, detecting exosomal ncRNAs could potentially serve as a means to monitor targeted therapy for NSCLC in the future. Meanwhile, developing therapeutic regimens targeting these resistance mechanisms may provide additional clinical benefits to patients with EGFR-mutated NSCLC.
Data availability
Not applicable.
References
Sung H, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Suster DI, et al. Molecular pathology of primary non-small cell lung cancer. Arch Med Res. 2020;51(8):784–98.
Jorge SEDC, et al. Epidermal growth factor receptor (EGFR) mutations in lung cancer: preclinical and clinical data. Braz J Med Biol Res. 2014;47(11):929–39.
Rosell R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–46.
Sullivan I, et al. Osimertinib in the treatment of patients with epidermal growth factor receptor T790M mutation-positive metastatic non-small cell lung cancer: clinical trial evidence and experience. Ther Adv Respir Dis. 2016;10(6):549–65.
Lim SM, et al. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: mechanisms and therapeutic strategies. Cancer Treat Rev. 2018;65:1–10.
Remon J, et al. Osimertinib and other third-generation EGFR TKI in EGFR-mutant NSCLC patients. Ann Oncol. 2018;29(1):20–7.
Chmielecki J, et al. Candidate mechanisms of acquired resistance to first-line osimertinib in EGFR-mutated advanced non-small cell lung cancer. Nat Commun. 2023;14(1):1070.
Chen J, et al. Tumor-derived extracellular vesicles: Regulators of tumor microenvironment and the enlightenment in tumor therapy. Pharmacol Res. 2020;159: 105041.
Steinbichler TB, et al. The role of exosomes in cancer metastasis. Semin Cancer Biol. 2017;44:170–81.
Wu S, et al. Intercellular transfer of exosomal wild type EGFR triggers osimertinib resistance in non-small cell lung cancer. Mol Cancer. 2021;20(1):17.
Ishola AA, et al. Non-coding RNA and lung cancer progression. J Chin Med Assoc. 2020;83(1):8–14.
Valadi H, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–9.
Yang B, et al. Tumor-derived exosomal circRNA_102481 contributes to EGFR-TKIs resistance via the miR-30a-5p/ROR1 axis in non-small cell lung cancer. Aging (Albany NY). 2021;13(9):13264–86.
Jing C, et al. Exosome-mediated gefitinib resistance in lung cancer HCC827 cells via delivery of miR-21. Oncol Lett. 2018;15(6):9811–7.
Zhou D, et al. Exosomal long non-coding RNA SOX2 overlapping transcript enhances the resistance to EGFR-TKIs in non-small cell lung cancer cell line H1975. Hum Cell. 2021;34(5):1478–89.
Liu J, et al. Circular RNAs: the star molecules in cancer. Mol Aspects Med. 2019;70:141–52.
Liu W, et al. Circular RNA hsa_circRNA_103809 promotes lung cancer progression via facilitating ZNF121-dependent MYC expression by sequestering miR-4302. Biochem Biophys Res Commun. 2018;500(4):846–51.
Kristensen LS, et al. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20(11):675–91.
Han W, et al. Circular RNA circ-RAD23B promotes cell growth and invasion by miR-593-3p/CCND2 and miR-653-5p/TIAM1 pathways in non-small cell lung cancer. Biochem Biophys Res Commun. 2019;510(3):462–6.
Ma S, et al. CircRNAs: biogenesis, functions, and role in drug-resistant Tumours. Mol Cancer. 2020;19(1):119.
Świtlik W, et al. miR-30a-5p together with miR-210-3p as a promising biomarker for non-small cell lung cancer: a preliminary study. Cancer Biomark. 2018;21(2):479–88.
Karvonen H, et al. Wnt5a and ROR1 activate non-canonical Wnt signaling via RhoA in TCF3-PBX1 acute lymphoblastic leukemia and highlight new treatment strategies via Bcl-2 co-targeting. Oncogene. 2019;38(17):3288–300.
Miyake N, et al. Targeting ROR1 in combination with pemetrexed in malignant mesothelioma cells. Lung Cancer. 2020;139:170–8.
Wang F, et al. Combination therapy of gefitinib and miR-30a-5p may overcome acquired drug resistance through regulating the PI3K/AKT pathway in non-small cell lung cancer. Ther Adv Respir Dis. 2020;14:1–18.
Yan J, et al. MicroRNA-30a-5p suppresses epithelial-mesenchymal transition by targeting profilin-2 in high invasive non-small cell lung cancer cell lines. Oncol Rep. 2017;37(5):3146–54.
Ghaderi A, et al. A small molecule targeting the intracellular tyrosine kinase domain of ROR1 (KAN0441571C) induced significant apoptosis of non-small cell lung cancer (NSCLC) cells. Pharmaceutics. 2023;15(4):1148–63.
Liu X, et al. Novel ROR1 inhibitor ARI-1 suppresses the development of non-small cell lung cancer. Cancer Lett. 2019;458:76–85.
Long MP, et al. Targeting ROR1 inhibits epithelial to mesenchymal transition in human lung adenocarcinoma via mTOR signaling pathway. Int J Clin Exp Pathol. 2018;11(10):4759–70.
Hill M, et al. miRNA interplay: mechanisms and consequences in cancer. Dis Model Mech. 2021;14(4):dmm047662.
Lu TX, et al. MicroRNA. J Allergy Clin Immunol. 2018;141(4):1202–7.
Li B, et al. Expression, regulation, and function of exosome-derived miRNAs in cancer progression and therapy. Faseb j. 2021;35(10):e21916–27.
Steinbichler TB, et al. Therapy resistance mediated by exosomes. Mol Cancer. 2019;18(1):58–68.
Li X, et al. Elevated exosome-derived miRNAs predict osimertinib resistance in non-small cell lung cancer. Cancer Cell Int. 2021;21(1):428–41.
Azuma Y, et al. Cancer exosomal microRNAs from gefitinib-resistant lung cancer cells cause therapeutic resistance in gefitinib-sensitive cells. Surg Today. 2020;50(9):1099–106.
Zhang Y, et al. Exosomal transfer of miR-214 mediates gefitinib resistance in non-small cell lung cancer. Biochem Biophys Res Commun. 2018;507(1–4):457–64.
Liu X, et al. Exosomes transmit T790M mutation-induced resistance in EGFR-mutant NSCLC by activating PI3K/AKT signalling pathway. J Cell Mol Med. 2020;24(2):1529–40.
Hisakane K, et al. Exosome-derived miR-210 involved in resistance to osimertinib and epithelial-mesenchymal transition in EGFR mutant non-small cell lung cancer cells. Thorac Cancer. 2021;12(11):1690–8.
Guanen Q, et al. MiR-214 promotes cell meastasis and inhibites apoptosis of esophageal squamous cell carcinoma via PI3K/AKT/mTOR signaling pathway. Biomed Pharmacother. 2018;105:350–61.
Wang YS, et al. MicroRNA-214 regulates the acquired resistance to gefitinib via the PTEN/AKT pathway in EGFR-mutant cell lines. Asian Pac J Cancer Prev. 2012;13(1):255–60.
Liao J, et al. Down-regulation of miR-214 reverses erlotinib resistance in non-small-cell lung cancer through up-regulating LHX6 expression. Sci Rep. 2017;7(1):781–90.
Wang Q, et al. LHX6 affects erlotinib resistance and migration of EGFR-mutant non-small-cell lung cancer HCC827 cells through suppressing Wnt/β-catenin signaling. Onco Targets Ther. 2020;13:10983–94.
Zhang J, et al. The potential roles of exosomal miR-214 in bone metastasis of lung adenocarcinoma. Front Oncol. 2020;10:611054–62.
Zhao X, et al. microRNA-214 governs lung cancer growth and metastasis by targeting carboxypeptidase-D. DNA Cell Biol. 2016;35(11):715–21.
Chen X, et al. MicroRNA-214 inhibits the proliferation and invasion of lung carcinoma cells by targeting JAK1. Am J Transl Res. 2018;10(4):1164–71.
Liu ZL, et al. MicroRNA-21 (miR-21) expression promotes growth, metastasis, and chemo- or radioresistance in non-small cell lung cancer cells by targeting PTEN. Mol Cell Biochem. 2013;372(1–2):35–45.
Gao W, et al. Deregulated expression of miR-21, miR-143 and miR-181a in non small cell lung cancer is related to clinicopathologic characteristics or patient prognosis. Biomed Pharmacother. 2010;64(6):399–408.
Li B, et al. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer. 2014;83(2):146–53.
Chang F, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17(3):590–603.
Bica-Pop C, et al. Overview upon miR-21 in lung cancer: focus on NSCLC. Cell Mol Life Sci. 2018;75(19):3539–51.
Ren W, et al. Extracellular vesicles secreted by hypoxia pre-challenged mesenchymal stem cells promote non-small cell lung cancer cell growth and mobility as well as macrophage M2 polarization via miR-21-5p delivery. J Exp Clin Cancer Res. 2019;38(1):62–75.
Huang WC, et al. The MEK/ERK/miR-21 signaling is critical in osimertinib resistance in EGFR-mutant non-small cell lung cancer cells. Cancers (Basel). 2021;13(23):6005–21.
Dai L, et al. miR-21 regulates growth and EMT in lung cancer cells via PTEN/Akt/GSK3β signaling. Front Biosci (Landmark Ed). 2019;24(8):1426–39.
Zhang Z, et al. Antitumor activity of anti-miR-21 delivered through lipid nanoparticles. Adv Healthc Mater. 2023;12(6): e2202412.
Zhu X, et al. EMT-mediated acquired EGFR-TKI resistance in NSCLC: mechanisms and strategies. Front Oncol. 2019;9:1044–58.
Hata AN, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22(3):262–9.
Jin J, et al. Bufalin inhibits the malignant development of non-small cell lung cancer by mediating the circ_0046264/miR-522-3p axis. Biotechnol Lett. 2021;43(6):1229–40.
Tsubakihara Y, et al. Epithelial-mesenchymal transition and metastasis under the control of transforming growth factor β. Int J Mol Sci. 2018;19(11):3672–701.
Yang X, et al. The regulatory role of APE1 in epithelial-to-mesenchymal transition and in determining EGFR-TKI responsiveness in non-small-cell lung cancer. Cancer Med. 2018;7(9):4406–19.
Choi DY, et al. Extracellular vesicles shed from gefitinib-resistant nonsmall cell lung cancer regulate the tumor microenvironment. Proteomics. 2014;14(16):1845–56.
Yang B, et al. MiR-564 functions as a tumor suppressor in human lung cancer by targeting ZIC3. Biochem Biophys Res Commun. 2015;467(4):690–6.
Zhang X, et al. Role of non-coding RNAs and RNA modifiers in cancer therapy resistance. Mol Cancer. 2020;19(1):47–72.
Sana J, et al. Novel classes of non-coding RNAs and cancer. J Transl Med. 2012;10:103–23.
Li C, et al. Tumor-derived exosomal lncRNA GAS5 as a biomarker for early-stage non-small-cell lung cancer diagnosis. J Cell Physiol. 2019;234(11):20721–7.
Zhao Y, et al. Long noncoding RNA LINC02418 regulates MELK expression by acting as a ceRNA and may serve as a diagnostic marker for colorectal cancer. Cell Death Dis. 2019;10(8):568–80.
Yin X, et al. Metformin enhances gefitinib efficacy by interfering with interactions between tumor-associated macrophages and head and neck squamous cell carcinoma cells. Cell Oncol (Dordr). 2019;42(4):459–75.
Deng Q, et al. Exosomal long non-coding RNA MSTRG.292666.16 is associated with osimertinib (AZD9291) resistance in non-small cell lung cancer. Aging (Albany NY). 2020;12(9):8001–15.
Wan X, et al. Exosomes derived from M2 type tumor-associated macrophages promote osimertinib resistance in non-small cell lung cancer through MSTRG.292666.16-miR-6836–5p-MAPK8IP3 axis. Cancer Cell Int. 2022;22(1):83–98.
Lu J, et al. Reprogramming of TAMs via the STAT3/CD47-SIRPα axis promotes acquired resistance to EGFR-TKIs in lung cancer. Cancer Lett. 2023;564:216205–20.
Isidro RA, et al. Colonic macrophage polarization in homeostasis, inflammation, and cancer. Am J Physiol Gastrointest Liver Physiol. 2016;311(1):59–73.
Xu F, et al. Astragaloside IV inhibits lung cancer progression and metastasis by modulating macrophage polarization through AMPK signaling. J Exp Clin Cancer Res. 2018;37(1):207–22.
Zhang X, et al. PD-L1 induced by IFN-γ from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol. 2017;22(6):1026–33.
Xiao F, et al. M2 macrophages reduce the effect of gefitinib by activating AKT/mTOR in gefitinib-resistant cell lines HCC827/GR. Thorac Cancer. 2020;11(11):3289–98.
Teng Y, et al. Identification of an exosomal long noncoding RNA SOX2-OT in plasma as a promising biomarker for lung squamous cell carcinoma. Genet Test Mol Biomark. 2019;23(4):235–40.
Wang Z, et al. LncRNA SOX2-OT is a novel prognostic biomarker for osteosarcoma patients and regulates osteosarcoma cells proliferation and motility through modulating SOX2. IUBMB Life. 2017;69(11):867–76.
Herrera-Solorio AM, et al. LncRNA SOX2-OT regulates AKT/ERK and SOX2/GLI-1 expression, hinders therapy, and worsens clinical prognosis in malignant lung diseases. Mol Oncol. 2021;15(4):1110–29.
Keniry A, et al. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat Cell Biol. 2012;14(7):659–65.
Ding D, et al. LncRNA H19/miR-29b-3p/PGRN axis promoted epithelial-mesenchymal transition of colorectal cancer cells by acting on Wnt signaling. Mol Cells. 2018;41(5):423–35.
Conigliaro A, et al. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol Cancer. 2015;14:155–65.
Sun H, et al. H19 lncRNA mediates 17β-estradiol-induced cell proliferation in MCF-7 breast cancer cells. Oncol Rep. 2015;33(6):3045–52.
Huang Z, et al. H19 promotes non-small-cell lung cancer (NSCLC) development through STAT3 signaling via sponging miR-17. J Cell Physiol. 2018;233(10):6768–76.
Lei Y, et al. Tumor-released lncRNA H19 promotes gefitinib resistance via packaging into exosomes in non-small cell lung cancer. Oncol Rep. 2018;40(6):3438–46.
Galluzzi L, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34(7):856–80.
Wu WK, et al. The autophagic paradox in cancer therapy. Oncogene. 2012;31(8):939–53.
Sui X, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4(10):e838–49.
O’Donovan TR, et al. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy. 2011;7(5):509–24.
Huang FX, et al. LncRNA BLACAT1 is involved in chemoresistance of non-small cell lung cancer cells by regulating autophagy. Int J Oncol. 2019;54(1):339–47.
Pan R, et al. Exosomal transfer of lncRNA H19 Promotes erlotinib resistance in non-small cell lung cancer via miR-615-3p/ATG7 axis. Cancer Manag Res. 2020;12:4283–97.
Chen C, et al. LncRNA H19 downregulation confers erlotinib resistance through upregulation of PKM2 and phosphorylation of AKT in EGFR-mutant lung cancers. Cancer Lett. 2020;486:58–70.
Xu C, et al. β-Elemene enhances erlotinib sensitivity through induction of ferroptosis by upregulating lncRNA H19 in EGFR-mutant non-small cell lung cancer. Pharmacol Res. 2023;191:106739–52.
Xue M, et al. Urothelial cancer associated 1: a long noncoding RNA with a crucial role in cancer. J Cancer Res Clin Oncol. 2016;142(7):1407–19.
Shi W, et al. LncRNA UCA1 promoted cisplatin resistance in lung adenocarcinoma with HO1 targets NRF2/HO1 pathway. J Cancer Res Clin Oncol. 2023;149(3):1295–311.
Zhang B, et al. Knockout of lncRNA UCA1 inhibits drug resistance to gefitinib via targeting STAT3 signaling in NSCLC. Minerva Med. 2019;110(3):273–5.
Tulchinsky E. Fos family members: regulation, structure and role in oncogenic transformation. Histol Histopathol. 2000;15(3):921–8.
Wang J, et al. FOSL2 positively regulates TGF-β1 signalling in non-small cell lung cancer. PLoS ONE. 2014;9(11):e112150–6.
Chen X, et al. lncRNA UCA1 promotes gefitinib resistance as a ceRNA to target FOSL2 by sponging miR-143 in non-small cell lung cancer. Mol Ther Nucleic Acids. 2020;19:643–53.
Han QF, et al. Exosome biogenesis: machinery, regulation, and therapeutic implications in cancer. Mol Cancer. 2022;21(1):207–32.
Yu W, et al. YAP 5-methylcytosine modification increases its mRNA stability and promotes the transcription of exosome secretion-related genes in lung adenocarcinoma. Cancer Gene Ther. 2023;30(1):149–62.
Chen R, et al. Exosomes-transmitted miR-7 reverses gefitinib resistance by targeting YAP in non-small-cell lung cancer. Pharmacol Res. 2021;165:105442–53.
Acknowledgements
This study was supported by the Science and Technology Project for Youth Talent of Changzhou Health Commission (QN201703), Young Talent Development Plan of Changzhou Health Commission (CZQM2020024), Major Science and Technology Project of Changzhou Health Commission (ZD202004, ZD202007), and China Postdoctoral Science Foundation (2020M670064ZX).
Funding
This study was supported by the Science and Technology Project for Youth Talent of Changzhou Health Commission (QN201703), Young Talent Development Plan of Changzhou Health Commission (CZQM2020024), Major Science and Technology Project of Changzhou Health Commission (ZD202004, ZD202007), and China Postdoctoral Science Foundation (2020M670064ZX).
Author information
Authors and Affiliations
Contributions
DC, BW, LW, RC, WZ, CF, MJ contributed to the study conception and design. The first draft of the manuscript was written by DC and all authors commented on previous versions of the manuscript. DC, BW, LW, RC, WZ, CF, MJ read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interest
The authors have no relevant financial or non-financial interests to disclose.
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cheng, D., Wang, B., Wu, L. et al. Exosomal non-coding RNAs-mediated EGFR-TKIs resistance in NSCLC with EGFR mutation. Med Oncol 40, 254 (2023). https://doi.org/10.1007/s12032-023-02125-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-023-02125-3