
Abstract
To discover new therapeutic antibodies for treatment of acute myeloid leukemia (AML) without the requirement of a known antigen, a human single-chain variable fragment (scFv) library was used to isolate novel antibody fragments recognizing HL-60 AML cells. After three rounds of biopanning, scFv-expressing phages were selected to test for binding to the target cell by flow cytometry. The clone with highest binding specificity to HL-60 cells (designated y1HL63D6) was further investigated. Fluorescent staining indicated that y1HL63D6 scFv bound to a target located on the cell surface. Whole immunoglobulin, IgG-y1HL63D6 was then generated and tested for the binding against bone marrow mononuclear cells (BMMCs) from AML patients. Significantly higher fluorescent signals were observed for some patient samples when compared to normal BMMCs or non-AML patients’ BMMCs. Next, the IgG-y1HL63D6 format was tested for antibody-dependent cell cytotoxicity (ADCC). The results demonstrated that IgG-y1HL63D6 but not the control antibody, trastuzumab, could mediate specific killing of HL-60 target cells. In conclusion, our results indicate that specific antibodies can be isolated by biopanning whole cells with a non-immunized human scFv antibody phage display library and that the isolated antibody against HL-60 cells showed therapeutic potential.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukemia (AML) is a malignant disease of the bone marrow. Overall survival in patients with AML has remained poor and remission depends on several prognostic factors, including age, genetics, and genomic stability [1]. Approximately half of older patients cannot tolerate intensive treatment and the 5-year relative survival rate for AML is low compared to the other leukemias [2]. Therefore, a more effective therapeutic method is urgently required. Targeting AML cells via cancer-restricted cell surface antigens has a potential for more effective treatment by means of immunotherapy. Several antibodies have been developed against AML surface antigens, with promising outcomes for known targets, such as CD33 [3], CLL-1 [4], CD123 [5], and CD157 [6]. However, since multiple subtypes in AML require very different therapies [7], identification of novel specific antibodies and new cell surface molecules on AML cells has potential to lead to more effective treatments. Phage display technology has been proven to be an excellent high-throughput platform for the discovery of novel lineage-specific antigens on the surface of various cancer cells [8].
Selection of antibodies against a target of interest is most commonly based on biopanning with pure target antigen coated on a solid support [9]. However, the key disadvantage of this method is the lack of natural conformation and orientation of the antigen as well as post-translational modifications. Whole-cell panning methods can be used to overcome this problem and have been successfully applied to isolate antibodies against several naturally expressed cell surface targets. These include: the follicle-stimulating hormone receptor (FSHR) expressed on L cells [10], a Müllerian inhibiting substance type II receptor on the surface of ovarian cancer antigen [11] and human T-lymphotropic virus type 1-carrying T-cell line S1T, which was derived from an adult T-cell leukemia patient [12]. Cell panning has also been applied for selection of antibodies against several membrane proteins (human CD83, canine CD117, and bat CD11b), overexpressed on Chinese Hamster Ovary (CHO) and Human Embryonic Kidney (HEK) cells [13].
The HL-60 cells used in our study are derived from a patient with acute promyelocytic leukemia (APL) but which lacks the common APL-associated t(15;17) translocation and thus has been reported as a non-APL cell line and classified as M2 according to the French–American–British classification systems [14]. Several studies have used HL-60 cells as a model to investigate cancer–drug interactions and chemical-induced differentiation [15,16,17]. For our study, we used the AML HL-60 cells to select a novel antibody against an unknown AML cell surface molecule via a whole-cell biopanning procedure. The recombinant antibody was further studied to demonstrate its potential therapeutic applications.
Materials and methods
Phage antibody library and bacterial strains
A naïve phage display scFv antibody library (Yamo I) was constructed by our laboratory [18]. M13KO7 helper phage (NEB, MA, USA) and Escherichia coli strain TG1 (Medical Research Council Laboratory, Cambridge, UK) were utilized for phage propagation and scFv production. For this library, the scFv encoding sequence was fused with C-terminal 6 × His and c-Myc tags.
Cell culture
HL-60 was purchased from American Type Culture Collection. THP-1 was purchased from Cell Lines Service. Jurkat, OCIM-1, and U937 were from University College London Cancer Institute and UCL Great Ormond Street Institute of Child Health, London, UK and Prof. Dr. Pa-thai Yenchitsomanus Siriraj Center of Research Excellence for Cancer Immunotherapy, Faculty of Medicine, Mahidol University, Thailand. HL-60 and OCIM-1 were maintained with Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 20% and 10% fetal bovine serum (FBS), respectively, plus 1X penicillin–streptomycin. Jurkat, U937, and THP-1 cells were maintained with Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FBS and 1X penicillin–streptomycin. Cells were cultured at 37 °C, 5% CO2. All cell culture media and supplements were purchased from Gibco (Eugene, OR, USA). Cells were maintained at a density between 1 × 105 and 1 × 106 cells/mL.
Affinity selection (Biopanning) of phage display scFv library on intact cells
Each round of selection consisted of a subtraction step with 5 × 106 Jurkat cells, followed by positive selection on HL-60 cells. The scheme of whole-cell affinity selection process is shown in Fig. 1. The phage library was pre-blocked with 1 mL of phosphate buffer saline (PBS) supplemented with 4% (w/v) skimmed milk (MPBS) for 30 min at room temperature with rotation. The pre-blocked phage library was then incubated with negative target Jurkat cells for 1 h at 4 °C with rotation. After incubation, cells were centrifuged at 350×g for 5 min at 4 °C. Half of the supernatant containing phage was added to 5 × 106 HL-60 cells and the other half was added to 5 × 106 of Jurkat cells as a biopanning control. The phage library was incubated for 2 h at 4 °C with rotation. Non-specific phages in the supernatant were removed after centrifugation at 350xg for 5 min at 4 °C and discarded. Cells were washed 5 times with 5 mL of 0.1% (w/v) bovine serum albumin (BSA) in PBS. Bound phages were eluted with 500 µL of 50 mM citric acid, pH 2.5 after incubation for 5 min and centrifuged at 350×g for 5 min at 4 °C. The supernatant containing phage was collected in a new tube and neutralized with 500 µL of 1 M Tris–HCl, pH 7.4. One mL of the eluted phage was used to infect 5 mL of exponentially growing E. coli TG1 (OD600 = 0.5) plus 4 mL of 2xYT media to make a total volume of 10 mL. Bacteria were incubated at 37 °C for 30 min without shaking and then plated onto a 2xYT agar plate, containing 100 µg/mL ampicillin and 1% glucose, and incubated, inverted at 37 °C overnight. Biopanning using Jurkat cells as a negative control target was performed in parallel in every step of the selection. Phage recovery was calculated as a ratio of output/input phage after each round of selection. Phage rescue from each selection round was measured and the enrichment factors were calculated as output versus input ratios as previously described [19].

Schematic representation of cell-based biopanning in this study. (1) Phage library was pre-blocked in skimmed milk. (2) Pre-blocked phages were subtracted with non-specific phage by incubation with Jurkat cells. (3–4) Unbound phages were moved into tubes containing HL-60 target cells, non-specific phages were wash out. (5) Bound phages were eluted and infected E. coli TG1. (6) Bacteria containing phagemid were plated on agar plate containing ampicillin and glucose. (7) Phages were amplified after adding helper phage for the next round of biopanning. (8) After 3 rounds of selection, single bacterial colonies were randomly picked and induced for scFv expression in a 96-deep well plate. (9–10) HL-60 binders were detected by flow cytometry
Generation of polyclonal and monoclonal scFv-pIII antibody fragments
E.coli TG1 harboring a phagemid vector was induced to express soluble scFv antibody with isopropyl β-d-1-thiogalactopyranoside (IPTG). Expression of soluble scFv-pIII antibody format was obtained by inoculating 2xYT medium containing 100 µg/mL ampicillin and 1% glucose with E. coli TG1 glycerol stock at a dilution of 1:100 from each round of biopanning or from individual colonies picked from plates from the third round of biopanning to obtain polyclonal or monoclonal scFv-pIII, respectively. Bacteria were incubated at 37 °C with 250 rpm shaking until OD600 nm reached 0.9. Cells were then pelleted by centrifugation. The supernatant was discarded, and the pellet was resuspended in fresh 2xYT containing 100 µg/mL ampicillin and 1 mM IPTG and incubated overnight at 30 °C. The following day, the culture was centrifuged at 3300×g for 30 min and the supernatant containing polyclonal or monoclonal scFv were transferred into a fresh tube and stored at 4 °C until use. Supernatant containing monoclonal scFv was purified by immobilized metal affinity chromatography (IMAC) according to the manufacturer’s protocol (Thermo Fisher Scientific, # 88222, USA.). The resins were equilibrated with equilibration Buffer (20 mM sodium phosphate, 300 mM sodium chloride, 10 mM imidazole). The crude scFv supernatants were applied to the column. The resins were washed with washing buffer (20 mM sodium phosphate, 300 mM sodium chloride, 25 mM imidazole). The scFv fragments were collected with elution buffer (20 mM sodium phosphate, 300 mM sodium chloride, 250 mM imidazole). The antibody fraction was concentrated and buffer exchanged with PBS buffer using an Amicon Ultracell, 10 kDa cut-off membrane, (Sigma, #UFC801024, USA). Protein concentration was quantified using a Nanodrop ND2000 spectrophotometer (Thermo Scientific, IL, USA).
SDS-PAGE and western blotting
Evaluation of scFv expression was determined by western blot analysis. The expressed samples were electrophoresed on 12% SDS-PAGE gels and then transferred onto PVDF membrane. The membrane was blocked in 5% (w/v) MPBS overnight at 4 °C, washed, and incubated with peroxidase-conjugated anti-c-Myc antibody (Sigma-Aldrich, USA.) at 1:2000 in 5% MPBS containing 0.05% (v/v) Tween-20 (MPBST) for 1 h at room temperature. All incubations were followed by three washes with PBST (PBS containing 0.05% Tween-20) for 5 min at room temperature. Finally, the chemiluminescent signals were developed using ECL Western blotting kit (GE Healthcare, Sweden). Amersham Hyperfilm ECL (GE Healthcare, Sweden) was used to detect the chemiluminescent signals and films were developed using an X-ray film automatic processor.
Sequencing of positive phage clones
Plasmids containing selected scFv inserts were prepared using a plasmid purification kit (QIAGEN, Germany). Automated DNA sequencing was performed by Macrogen (Korea) using pMOD forward and reverse primers (Yamo_5 (5'-CAGGAAACAGCTATGACC-3' and -96gIII (5'-CCCTCATAGTTAGCGTAACG-3'). Sequence analysis was performed using SnapGene® software (GSL Biotech). The DNA sequences were compared with human germline sequence via IMGT, the International ImMunoGeneTics information system (http://www.imgt.org) database, to identify clonal diversity. The amino acid sequences were translated by SnapGene® software.
Binding analysis of scFv fragment by flow cytometry
Binding specificity of the scFv antibody was analyzed by flow cytometry. Human cell lines were washed with 0.1% BSA in PBS. The Fc receptors on the cell surface were blocked with human immunoglobulin (IgG) for 30 min at room temperature. Cells were then washed once with 0.1% BSA in PBS. For flow cytometry analysis of polyclonal scFv, 2 × 105 cells were incubated with 200 µL of supernatant containing polyclonal scFv. After washing, the cells were incubated with 100 µL of anti-myc antibody (Sigma-Aldrich, #C3956, USA.) at 1:1000 dilution, for 1 h on ice, washed, and incubated with donkey anti-rabbit IgG-Alexa Fluor 647 (Invitrogen, #A31573, U.S.A.) at 1:1000 dilution. For flow cytometry analysis of monoclonal scFv, 2 × 105 cells were incubated with 10 µg/mL of monoclonal scFv for 1 h on ice. After washing, the cells were incubated with 100 µL anti-6X His tag antibody Dylight 488 (Abcam, #ab117512, UK) at 1:1000 dilution in 0.1% BSA in PBS, for 1 h on ice. All incubations were followed by washing one time with 0.1% BSA in PBS and centrifugation at 330×g for 5 min at 4 °C. Finally, the cells were resuspended in 300 µL of 0.1% BSA in PBS containing 1 µg/mL of propidium iodide (PI) (Invitrogen, #P3566, USA) and analyzed by a flow cytometer (Attune NxT Acoustic Focusing Cytometer (Thermo Fisher Scientific# P3566, USA.)). PI staining was used to exclude dead cells. Ten thousand PI-negative cells were acquired.
Cloning and expression of IgG-y1HL63D6 antibody
The variable heavy (VH) sequence of clone y1HL63D6 was amplified by polymerase chain reaction (PCR) using Pfu DNA polymerase. PCR amplifications were performed in a total volume of 50 µL, consisting of 1X Pfu DNA polymerase buffer, 0.2 mM dNTP (NEB, #N0447L, USA), 1.8 U of Pfu DNA polymerase (Promega, #M7741, USA), 100 ng of DNA template, and 1 µM of VH gene-specific forward primer with NheI (152VHNheIFW-5’GCCGCTAGCCAGGTGCAGCTGGTGCAGTC3’) and reverse primers with NheI (152VHNheIRv5’GGTGCTAGCTGAGGAGACAGTGACCGTGGT3’). The amplification steps involved initial denaturation at 95 °C for 2 min, followed by 30 cycles of denaturation at 95 °C for 1 min, annealing at 63 °C for 30 s, polymerization at 72 °C for 1 min in each cycle, and a final polymerization at 72 °C for 5 min. The PCR product was purified and digested with NheI (NEB, #R3131S, USA) and then ligated into NheI digested pKR-CH vector [20]. The y1HL63D6 variable light (VL) sequence and the constant gene of human IgG1 lambda light chain, containing flanking 5’ NheI and 3’ AgeI restriction sites, were synthesized (GeneArt® gene synthesis services, Thermo Fisher Scientific, USA.) and subcloned into pTT28 vector (NRC-BRI, Canada) at the corresponding restriction sites. The integrity of the constructs was confirmed by Automated DNA sequencing (Macrogen, Korea).
The Expi293 Expression System (Thermo Fisher Scientific, # A14635, USA.) was used to express whole IgG according to the manufacturer’s instructions. Twenty-five µg of heavy (HC) and light (LC) chain expression vectors were used at ratio of 1:3 for transfections. At seven-day post-transfection, supernatant was collected after centrifugation to remove cells and antibodies were purified using HiTrap MabSelect PrismA affinity chromatography (Cytiva, #17549851, Sweden), according to the manufacturer’s instructions. The final elution peaks were analyzed by SDS-PAGE. The eluted antibodies were concentrated and dialyzed against PBS for further experiments.
Confocal fluorescence microscopy
HL-60 cells were washed once with 0.1% BSA in PBS. Cells were blocked with human IgG for 30 min at room temperature followed by washing with 0.1% BSA in PBS. 1 × 106 cells were co-incubated with 2,000 µg/mL of purified IgG-y1HL63D6 antibody and 2.5 µg/mL of anti-CD33 (BioLegend, # 303402, U.S.A.) in a final volume of 100 µL for 1 h on ice. Cells were then washed and incubated with 1:250 of goat F(ab')2 anti-human IgG-Fc-DyLight 650 (Abcam, #ab98593, UK) and 1:200 goat anti-mouse IgG-Alexa Fluor 488 (Thermo Fisher Scientific, #A11029, USA.) for 1 h on ice in the dark. After that, nuclei were stained with 1:500 Hoechst 33342 (Thermo Fisher Scientific, #H3570, USA) for 30 min, washed and cells were dropped on a glass slide, and then covered with coverslip. Stained cells were visualized under confocal fluorescence microscope (Nikon A1R, Japan).
Binding analysis of IgG-y1HL63D6 against cell lines and patient-derived samples by flow cytometry
HL-60, U937, THP-1, OCIM-1, Jurkat, and peripheral blood mononuclear cells (PBMCs) were incubated with 200 µg/mL of IgG-y1HL63D6 or isotype control antibody (Trastuzumab) for 1 h on ice, followed by washing with 0.1% BSA in PBS. Cells were then stained with anti-human IgG-Fc-DyLight 488 (Abcam, #ab98590, UK) on ice for 1 h. Cells were then washed and resuspended with 0.1% BSA in PBS containing 1 µg/mL PI. Samples were analyzed by flow cytometer. The gating strategy used for analysis is as illustrated in Fig. S1.
Bone marrow samples from AML and non-AML patients were collected at primary diagnosis under the approval of the Ethical Review Board of Sanphasitthiprasong Hospital, Ubon Ratchathani, Thailand. Bone marrow mononuclear cells (BMMCs) were isolated using Ficoll® Paque Plus according to manufacturer’s protocol (Cytiva, #17144002, Sweden). BMMCs were blocked with human IgG for 30 min at room temperature and washed with 0.1% BSA in PBS. The cells were stained with 200 µg/mL of IgG-y1HL63D6 and 2.5 µg/mL of anti-CD33 antibody (BioLegend, # 303402, USA.) on ice for 1 h. Cells were then washed and stained with 5 µg/mL of anti-human CD45-APC antibody (BioLegend, #368511, USA) and goat anti-mouse IgG (H + L)-Alexa Fluor 555 (Thermo Fisher Scientific # A32727, USA.) at 1:500 dilution for 1 h on ice in the dark. Ten thousand PI-negative cells were acquired using Attune™ NxT focusing flow cytometer. MFI values were determined using Attune™ NxT software. The gating strategy used is described in Fig. S2. The MFI ratio was calculated by MFI of IgG-y1HL63D6 divided by MFI of Trastuzumab, which was used an isotype control. The average value of MFI in each group were reported.
Antibody-dependent cell-mediated cytotoxicity (ADCC) Assay
HL-60, OCIM-1, and Jurkat were used as target cells and freshly isolated PBMCs from healthy donors were used as effector cells. Peripheral blood samples of healthy donors were obtained under the approval of the research ethics board of the Suranaree University of Technology, Nakhon Ratchasima, Thailand. PBMCs were separated by Ficoll density gradient centrifugation. The target cells were labeled with 5 µM carboxyfluorescein succinimidyl ester (CFSE) dye (Invitrogen, # 65085084, U.S.A.) for 15 min. The labeling reaction was stopped by adding 5 mL of cold PBS + 10% FBS. Cells were centrifuged, supernatant discarded, and then resuspended in IMDM medium containing 10% FBS. The PBMCs and target cell numbers were adjusted to 3 × 107 and 6 × 105 cells/mL, respectively. 100 µL of target cells was preincubated with 100 µL of 300 µg/mL IgG at room temperature for 15 min and then media containing effector cells were added to a final volume of 300 µL to reach an effector-to-target cell ratio (E:T) of 50∶1. Pooled PBMCs from 5 donors or from individual donors were used as effector cells. After 4 h of incubation at 37 °C in a humidified CO2 incubator, cells were washed and stained with Fixable Viability Dye (FVD) eFluor™ 780 (Invitrogen, # 65286040, USA) for 30 min at 4 °C. For maximum lysis, CFSE-labeled target cells were heated at 75 °C for 15 min. After washing, the cells were analyzed by flow cytometry. Target cells were gated using a SSC vs CFSE plot and CFSE + cells (at least 10,000 events) were assessed for binding to FVD. The gating strategy used is shown in Fig. S3. The proportion of cell death was determined based on the percentage of CFSE + /FVD + cells. The percentage of specific cell death of labeled target cells is calculated using the following formula: Percentage of specific cell death = (Percent experimental lysis – Percent spontaneous lysis) / (Percent maximum lysis – Percent spontaneous lysis) × 100.
Statistical analyses
The data were recorded as mean ± standard error of mean (SEM) and were analyzed by two-way analysis of variance (ANOVA). Dunnett’s multiple comparisons test was applied for analyses of non-parametric data of Figs. 2c and 6a. Mann–Whitney test was applied for analyses of non-parametric data of Fig. 5b. For Fig. 6b, parametric data were analyzed with Unpaired t with Welch’s correction test, after normal distribution was indicated. Statistical testing was performed using GraphPad Prism 8 software (GraphPad Software Inc., USA.). Tables of data with mean ± SEM, sample sizes, and the exact P-values of each experiment can be found in Table S1.

Enrichment of phage after repeated rounds of biopanning against whole cells. a Flow cytometry was carried to determine the binding of polyclonal scFv-pIII antibodies to HL-60 cells from each round of biopanning. Bound scFv-pIII was detected with rabbit anti-myc antibody. Donkey anti-Rabbit IgG, Alexa Fluor 647 was used to detect binding of the rabbit anti-myc antibody. b Representative scFv clones in culture supernatant were detected by western blot analysis using anti-myc conjugated with peroxidase. Lanes 1–6 represented different individual clones of monoclonal scFv-pIII antibody. c The selected scFv clones were expressed from E.coli TG1, purified, and incubated with HL-60, Jurkat, and PBMCs. The scFv were labeled with anti-6X His tag antibody-Dylight 488. Y-axis indicate the percentage of gated cells that showed fluorescent signal (Mean ± SEM, n = 3). Significant differences between sample groups were assessed by Two-way ANOVA Dunnett’s multiple comparisons test (**P < 0.01, ****P < 0.0001, and ns; not significant). Clone 3D6, designated as y1HL63D6, was selected for further investigation
Results
Affinity selection of specific scFv antibody against HL-60 cells
To isolate scFv antibody fragments against human AML cells, a human pre-immune phage display scFv antibody library (Yamo I) [18] was used for biopanning against viable HL-60 AML cells (Fig. 1). The library was subtracted by pre-incubation with the T acute lymphoblastic leukemia (ALL) cell line, Jurkat, followed by selection on intact HL-60 cells. Three rounds of selection were performed, and the recovery ratio is calculated based on the titer of the input and recovered phage as shown in Table S2. Results showed that the ratio between HL-60 target cell and Jurkat increased from 0.53 to 3.01 and 237.5-fold after each round of selection, demonstrating the enrichment of phage specifically binding to the target AML cells.
Binding properties of polyclonal and monoclonal scFvs
To confirm the enrichment of phage from each round of biopanning against HL-60 cells, polyclonal scFvs from each round were expressed as soluble scFv-pIII from TG1 E. coli supernatant and binding to HL-60 cells was assessed by flow cytometry analysis. The polyclonal scFv pool from the third round of selection showed the highest median fluorescent intensity (MFI) = 711) when compared to those from the first (MFI = 333) and second rounds (MFI = 367) (Fig. 2a). These results indicated that scFv antibodies from the third round contained clones that bind to HL-60. Western blot analysis of 6 representative scFv clones obtained from the third round of biopanning demonstrated different expression levels and size of various scFv-pIII clones (Fig. 2b). Binding properties of individual clones isolated by cell-based biopanning on HL-60 cells were determined by flow cytometry. Of 276 clones analyzed, 4 clones showed a high signal against HL-60 (Fig.S4). The amino acid sequences of these clones, designated y1HL63C10, y1HL63D6, y1HL63D12, and y1HL63G3, were analyzed by automated DNA sequencing. Clone y1HL63C10 was not productive, containing a stop codon and was out of frame. Clone y1HL63D6, y1HL63D12, and y1HL63G3 showed complete full-length scFv sequences. Therefore, a total of 3 different scFv clones were obtained. Amino acid sequence analysis indicated that all light chains are lambda type. The complementarity-determining region (CDR) of these clones are indicated in Table S3. Bioinformatic analysis revealed that these scFv antibodies belong to different immunoglobulin families of heavy chain or light chain and represented unique clones as summarized in Table S3.
Cross-reactivity analysis of the selected scFv clones
Binding of the three identified clones against HL-60, Jurkat, and PBMCs was investigated by flow cytometry (Fig. 2c). The results indicated that clone y1HL63D6 (also designated 3D6) and y1HL63G3 (also designated 3G3) bound specifically to HL-60 with a similar binding pattern, while clone y1HL63D12 (also designated 3D12) bound to HL-60 and Jurkat cells but not to PBMCs. Based on these data, clone y1HL63D6, which showed higher levels of binding against HL-60 than y1HL63G3, was selected for further study.
Specific binding of IgG-y1HL63D6 to AML cell lines
The y1HL63D6 scFv was reformatted to IgG1 for subsequent functional studies. The VH and VL DNA fragments were cloned into pKR-CH and pTT28 vector which contains constant regions of heavy and light chain, respectively (Fig.S5a-b). The vectors were transfected into Expi293F cells and the expressed antibody was purified from the supernatant using protein A affinity column chromatography. The SDS-PAGE from different steps of the purification showed two bands at 50 kDa and 25 kDa, under reducing conditions, corresponding to the HC and LC of IgG, respectively (Fig.S5c). The yield after purification was approximately 120 mg/L.
Specific binding of IgG-y1HL63D6 was assessed against 4 AML cell lines: HL-60, U937, THP-1, and OCIM-1 as well as against Jurkat (T-ALL) and PBMCs by flow cytometry. Fluorescent intensity analysis indicated that 3 out of 4 AML cell lines (HL-60, U937, and THP-1) expressed a common cell surface target antigen at different levels (Fig. 3a). We used an MFI ratio greater than 1.5 as a cut-off value for a positive cell staining [6]. While the biopanning target, HL-60, showed the highest (MFI ratio = 22.4) value, U937 and THP-1 showed MFI ratios of 2.81 and 1.88, respectively. OCIM-1 showed an MFI ratio lower than 1.5 (MFI ratio = 1.01), similar to the control cell line (Jurkat) that was used for negative selection (Fig. 3b), indicating that these two cell lines do not possess the HL-60 cell surface antigen targeted by the IgG-y1HL63D6 antibody.

Cross-reactivity of IgG-y1HL63D6 to different AML cell lines. Flow cytometry was used to assess the binding of IgG-y1HL63D6 to different AML cell lines as indicated. Trastuzumab, a therapeutic antibody that binds to HER2, was used as a human IgG1 isotype control. a Overlay histogram of IgG-y1HL63D6 (red) and Trastuzumab (black) was plotted. b Median fluorescent intensity (MFI) ratio of each cell lines was plotted in Mean ± SEM. n = 3. The dot line indicates an MFI ratio of 1.5, cut-off value for bona fide binding
Next, confocal fluorescence microscopy was performed to determine the localization of IgG-y1HL63D6 on HL-60 cells. The cells were co-stained with anti-CD33, a cell surface marker for HL-60 cells, for comparison. Localization of IgG-y1HL63D6 on the cell surface could be clearly observed on HL-60 cells (Fig. 4a), but the binding pattern of IgG- y1HL63D6 was distinct from that of anti-CD33 (Fig. 4c–e). The negative control, trastuzumab, did not bind to HL-60 cells (Fig. 4f, h–j). Quantification of the mean fluorescence intensity of individual cells, stained with IgG- y1HL63D6 or trastuzumab are shown in supplementary Fig.S6a–b, which indicated that the MFI of HL-60 stained with IgG-y1HL63D6 (MFI = 317) was significantly higher than that of Trastuzumab (MFI = 21).

Surface staining of HL-60 using IgG-y1HL63D6. HL-60 cells were stained with mouse anti-human CD33 and IgG-y1HL63D6 antibody (upper panels) or isotype control, trastuzumab (lower panels). Anti-Human IgG-Fc-DyLight 650 (pink) was used to detect binding of IgG-y1HL63D6 and trastuzumab. Anti-Mouse IgG-Alexa Fluor 488 (green) was used to detect binding of the mouse anti-human CD33. Nuclei were stained with Hoechst 33,342 (blue). Cell images were obtained using an Apo TIRF 60 × Oil DIC N2 objective of Nikon A1R confocal laser microscope. Scale bars = 10 μm at × 600 magnification (a, b, c, f, g, and h). Zoomed images of a representative cell (dashed yellow boxes) are shown (d, e, i, and j)
Binding of IgG-y1HL63D6 against patient samples
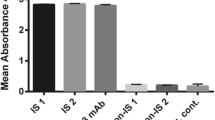
To further investigate a potential therapeutic application of the isolated antibody, binding ability of IgG-y1HL63D6 to patient samples was investigated. The binding to BMMCs from patients diagnosed with AML (n = 4), ALL (n = 2), and healthy donors (n = 2) were assessed by flow cytometry (Fig. 5a). Since leukemic blast cells express low to intermediate CD45 levels, the CD45-positive populations were gated and further gated for CD33 positivity to select only myeloid blasts for the analysis of specific interactions. The MFI ratio was defined by the MFI of the IgG-y1HL63D6 divided by MFI of Trastuzumab, which was used as isotype control. The results indicated that the average MFI ratio of BMMCs from AML patients was 1.35 ± 0.18 (n = 4) and were significantly higher than those of the non-AML BMMCs (MFI ratio of 0.97 ± 0.27, n = 4) (Fig. 5b). The AML sample that showed an MFI ratio of 1.63 was diagnosed as M2 subtype, the same as HL-60 cells. The sample that showed an MFI ratio of 1.16 was diagnosed as M3 subtype; while, the two samples where the FAB subtype was not determined (N/A) showed MFI ratios of 1.27 and 1.35.

Binding of IgG-y1HL63D6 to samples from AML patients. Flow cytometry was used to assess the binding of IgG-y1HL63D6 to samples obtained from patients and healthy volunteers. Trastuzumab, therapeutic antibody that binds to HER2, was used as an isotype control. a Overlay histogram of IgG-y1HL63D6 (red) and Trastuzumab (black) was plotted. b Median fluorescent intensity (MFI) ratio of AML BMMC samples and non-AML BMMC samples were plotted as Mean ± SEM. Significant differences between sample groups were assessed by the Mann–Whitney test (*P < 0.05). The AML subtype of the sample was identified as M2 and M3 or were not identified (N/A). The dotted line indicates an MFI ratio of 1.5, cut-off value for bona fide binding
Antibody-dependent cell-mediated cytotoxicity (ADCC) assay
Lastly, the ADCC activity of the recombinant IgG-y1HL63D6 antibody was investigated. HL-60, OCIM-1, and Jurkat were used as target cells with freshly isolated and pooled PBMCs from 5 donors (Fig. 6a) or PBMCs from 5 individual heathy donors (Fig. 6b), as a source of effector cells. The ADCC activity was analyzed at an antibody concentration of 100 µg/mL to ensure saturated antigen binding. The cells were co-incubated with PBMCs at a 50:1 effector-to-target ratio. As AML cells have been shown to be negative for HER2 expression [21], the therapeutic IgG1 antibody, trastuzumab which targets HER2, was used as a negative control. Flow cytometry analysis demonstrated that IgG-y1HL63D6 could significantly increase the death of the CFSE-labeled target cells when compared to trastuzumab. The cytotoxic effect of the antibody was specific to HL-60 (12.84%) only, as no cytotoxicity was detected with OCIM-1 (-1.35%) or Jurkat (-1.7%) cells when pooled PBMCs were used as effector cells (Fig. 6a), indicating that IgG-y1HL63D6 could mediate ADCC activity via targeting of a specific epitope on the surface of HL-60 cells. Notably, a variation in ADCC activity of samples from different individual donors was observed (Fig. 6b) and the percentage of HL-60 cell death induced by IgG-y1HL63D6 using PBMC from different individual donors varied between 13.3% and 38.0%. However, in all cases, IgG-y1HL63D6-mediated cytotoxicity was significantly higher than those of the negative control. These data indicate that IgG-y1HL63D6 antibody has a potential to be further developed for therapeutic purposes.

ADCC assay for IgG-y1HL63D6 induced killing of HL-60 via normal human PBMC Freshly isolated PBMCs from healthy human donors were co-cultured with CFSE-labeled HL-60 cells at E/T ratio of 50:1 in the presence of 100 µg/mL IgG-y1HL63D6 or Trastuzumab (isotype control) at 37 °C for 4 h. Specific cell killing was indicated as the percentage of dead target cells relative to maximum cell death (heating at 75 °C for 15 min). a PBMC from pool of 5 donors against CFSE-labeled HL-60, OCIM-1, and Jurkat (data points represent the Mean ± SEM of three independent triplicate experiments). b PBMCs from 5 individual donors (represented by 5 different colors) against CFSE-labeled HL-60 (data are Mean ± SEM of triplicate experiments). Statistical differences in multiple comparison were determined using Two-way ANOVA Dunnett’s multiple comparisons test (***P < 0.001 and ****P < 0.0001). Statistical differences between groups were determined using Unpaired t test (*P < 0.05)
Discussion
The residual leukemic stem cells after chemotherapy may cause relapse, leading to poor prognosis in AML patients [22]. More effective and less toxic therapeutic options are required to eliminate minimal residual cells [23] and we proposed that novel antibodies that can target a molecule on the surface of leukemic blasts would be beneficial for more effective treatment. We applied phage display technology to obtain such an antibody, using whole-cell biopanning to select for targets in the natural conformation and HL-60 as a representative of AML cells [24]. To deplete the number of irrelevant phage antibody and enhance specific binders, we extended enrichment over three rounds of selection. Each round consisted of negative selection with a non-AML cell line, i.e., Jurkat, which is an immortalized line of human T lymphocyte from lymphoid linage, followed by positive selection with HL-60. As cells express large number of potential epitopes, selections are extensively driven by the density of the target antigens, subtraction of non-specific phage in each biopanning would increase enhancement ratio of specific to non-specific phage [25]. Increase in enrichment ratio after each round of biopanning in this study (Table S2) has previously been observed in the isolation of CHO-cells expressing the human membrane protein CD36 [26]. However, since depletion relies on affinity and density of target antigen on both target and negative cells, depletion does not always ensure elimination of non-specific clones. This could explain why clone y1HL63D12 cross-reacted with Jurkat, the subtractive cell line, even after the third round of biopanning (Fig. 2c). Temperature is also an important parameter when designing an affinity selection experiment. In this study, we performed the biopanning at 4 °C to preserve surface membrane antigens and prevent internalization of phage particles.
Two scFvs clones, y1HL63D6 and y1HL63G3, had similar binding patterns, and they showed specific binding to HL-60 and notably did not cross-react with Jurkat, a representative of lymphoid leukemia. The scFvs also did not bind to normal PBMCs. We propose that these scFvs have the potential to be developed as a targeted therapy for AML. Amino acid sequence analysis demonstrated that the VH CDR3 residues had a similar pattern, with varying number of amino acids between the AR and DAFDI motifs: ARX7DAFDI for clone y1HL63D6 and ARX5DAFDI for clone y1HL63G3 (Table S3). It has been demonstrated that diversity of the VH CDR3 effects the antibody specificity [27]. Therefore, this amino acid sequence may be responsible for specific binding to the target on the surface of HL-60 cells. Binding assays using flow cytometry indicated that the full-length IgG, IgG-y1HL63D6 binds not only to HL-60 but also to other AML cell lines, U937 and THP-1, but not to OCIM-1, highlighting the heterogenicity of AML [28]. These results are in accordance with the finding that the trend of MFI ratio of IgG-y1HL63D6 against AML patients was significantly higher than those of non-AML leukemia and normal BMMCs (Fig. 5). The MFI ratio of AML samples varied between samples. These results indicated substantial inter-patient heterogeneity expression [29], emphasizing the importance of personalized therapy [30].
According to the French–American–British (FAB) classification systems, HL-60 are classified as AML M2 (acute myeloblastic leukemia with maturation) [24]. U937 and THP-1 are classified as AML M5 (monocytic cell lines) [31], while OCIM-1 are classified as (AML M6, (erythroleukemia blasts) [32]. Interestingly, the sample that showed the highest MFI ratio was isolated from an AML M2 patient, the same subtype as HL-60, the target cell for biopanning. Therefore, IgG-y1HL63D6 might specifically target to epitope that express on myeloblastic and monocytic cells. The binding of IgG-y1HL63D6 and Trastuzumab to different cell populations, based on the side scatter and CD45 expression, showed that the IgG-y1HL63D6 bound to monocytes derived from patient BMMCs (Fig.S7a), but it did not bind to monocytes derived from normal BMMCs (Fig.S7b); whereas, the binding was higher than that of monocytes from normal PBMCs (Fig.S7c). Identities of the cell surface epitopes of the isolated antibodies remain to be explored by immunoprecipitation followed by LC–MS/MS analysis or CRISPR-based method [33]. In addition, more work needs to be done to determine cross-reactivity of the antibodies to normal cells and tissues. Once the nature of the target antigen is known, the binding affinity of isolated antibody could be further improved by various techniques of affinity maturation as well [34].
In this study, Trastuzumab, a well-known therapeutic antibody against cancer, was used as an isotype control because it has been reported that HL-60, U937 cell lines, and AML patient-derived leukemic cells showed negative HER2 expression [21]. Although it has been reported that THP-1 weakly expressed HER2 at the mRNA level [35], our flow cytometry analyses showed the Trastuzumab staining against both HL-60 and THP-1 cell lines was equivalent to three other irrelevant antibodies which included anti-Aflatoxin [36], anti-Zearalenone [37], and anti-Bradyrhizobium [38] as shown in Fig.S8a and b, respectively. These results demonstrated that Trastuzumab was an appropriate control, even if we cannot rule out the possibility that certain populations of patient’s samples might express HER2, which could bind to Trastuzumab, resulting in underestimated MFI ratio.
ADCC is known as an influential factor in the efficacy of cancer therapy [39]. Effector cells especially NK cells are superior in engaging monoclonal antibodies that bind on the surface membrane of target cells for ADCC activity [40]. In the present study, IgG-y1HL63D6 was found to induce ADCC activity only in HL-60, but not OCIM-1 and Jurkat, when PBMCs were used as effector cells. These results suggest that IgG-y1HL63D6 could specifically bind to molecules commonly expressed on HL-60 and mediate cell death, most likely by NK cell-mediated ADCC [41]. The cytotoxicity observed in this study was moderate because PBMCs were used as effector cells instead of isolated NK cells. In addition, we found that when pooled PBMCs was used as effector cells, the cytotoxicity level was lower (~ 12%) than when using individual PBMC (~ 22%). This was likely because of allogeneic response of pooled PBMCs, which could stimulate T cells to kill non-HLA matched cells [42]. Therefore, the remaining effector cells from pooled PBMC was lower than those from individual PBMC, resulting in lower percentage of specific cell death. Nevertheless, the cytotoxic effect of IgG-y1HL63D6-mediated cytotoxicity on HL-60 cells was significantly higher than those of isotype antibody control. Variability in cytotoxic capacity among donors could be explained by the disparity in expression of NK cell receptors within an individual [43]. In addition, the IgG1 antibody in this study has not been Fc engineered. ADCC activity of the isolated antibody could be further improved by Fc engineering [44, 45]. Furthermore, the IgG could have therapeutic potential as an antibody drug conjugate and the scFv obtained from this study could be applicable for use as a chimeric antigen receptor in CAR T-cell therapy or as a bispecific T-cell engager when linker to anti-CD3.
Conclusion
In summary, a human antibody, designated as y1HL63D6, that can specifically target certain population of patient AML blasts has been isolated from biopanning of a naïve human phage display scFv library against the HL-60 cell line. Potential therapeutic application was demonstrated by ADCC assay of the converted full-length IgG format. This biopanning strategy and the isolated antibody represented a promising approach for the expansion of novel AML treatment.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AML:
-
Acute myeloid leukemia
- scFv:
-
Single-chain variable fragment
- IgG:
-
Immunoglobulin G
- VH:
-
Variable heavy
- VL:
-
Variable light
- HC:
-
Heavy chain
- LC:
-
Light chain
- BMMCs:
-
Bone marrow mononuclear cells
- PBMCs:
-
Peripheral blood mononuclear cells
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- MFI:
-
Median fluorescent intensity
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30.
American cancer society. cancer facts & figures 2021. American Cancer Society. 2021. https://www.cancer.org/research/cancer-facts-statistics. Accessed 8 Mar 2022.
Caron PC, et al. Biological and immunological features of humanized M195 (Anti-CD33) monoclonal antibodies. Cancer Res. 1992;52(24):6761.
Bakker ABH, et al. C-Type Lectin-Like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004;64(22):8443–50. https://doi.org/10.1158/0008-5472.CAN-04-1659.
Jin L, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor α chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5(1):31–42. https://doi.org/10.1016/j.stem.2009.04.018.
Krupka, C., et al. Targeting CD157 in AML using a novel, Fc-engineered antibody construct. Oncotarget. 2017;8(22):35707–35717. https://doi.org/10.18632/oncotarget.16060
Kantarjian H, et al. Acute myeloid leukemia: current progress and future directions. Blood Cancer J. 2021;11(2):41. https://doi.org/10.1038/s41408-021-00425-3.
Sidhu SS. Phage display in pharmaceutical biotechnology. Curr Opin Biotechnol. 2000;11(6):610–6. https://doi.org/10.1016/S0958-1669(00)00152-X.
Zhang, Y., et al. Prokaryotic expression of MLAA-34 and generation of a novel human ScFv against MLAA-34 by phage display technology. Oncotarget. 2017;8(24):39077–39086. https://doi.org/10.18632/oncotarget.16590
Crepin R, et al. Whole-cell biopanning with a synthetic phage display library of nanobodies enabled the recovery of follicle-stimulating hormone receptor inhibitors. Biochem Biophys Res Commun. 2017;493(4):1567–72. https://doi.org/10.1016/j.bbrc.2017.10.036.
Yuan QA, et al. Isolation of anti-MISIIR scFv molecules from a phage display library by cell sorter biopanning. Cancer Immunol Immunother. 2008;57(3):367–78. https://doi.org/10.1007/s00262-007-0376-2.
Muraoka S, et al. Effective induction of cell death on adult T-cell leukaemia cells by HLA-DRbeta-specific small antibody fragment isolated from human antibody phage library. J Biochem. 2009;145(6):799–810. https://doi.org/10.1093/jb/mvp039.
Jones ML, et al. Targeting membrane proteins for antibody discovery using phage display. Sci Rep. 2016;6:26240. https://doi.org/10.1038/srep26240.
Drexler HG, et al. Leukemia cell lines: in vitro models for the study of acute promyelocytic leukemia. Leuk Res. 1995;19(10):681–91. https://doi.org/10.1016/0145-2126(95)00036-N.
Corcoran A, et al. Biological evaluation of double point modified analogues of 1,25-Dihydroxyvitamin D(2) as potential anti-leukemic agents. Int J Mol Sci. 2016;17(2):91. https://doi.org/10.3390/ijms17020091.
Li Q, et al. Subcellular localization of DJ-1 in human HL-60 leukemia cells in response to diallyl disulfide treatment. Mol Med Rep. 2016;14(5):4666–72. https://doi.org/10.3892/mmr.2016.5831.
Takahashi, N., et al. Retinoylation (covalent modification by retinoic acid) of Rho-GDIβ in the human myeloid leukemia cell line HL60 and its functional significance. Biochim Biophys Acta. 2016;1861(12, Part A):2011–2019. https://doi.org/10.1016/j.bbalip.2016.10.001
Pansri P, Jaruseranee N, Rangnoi K, Kristensen P, Yamabhai M. A compact phage display human scFv library for selection of antibodies to a wide variety of antigens. BMC Biotechnol. 2009;9(1):6. https://doi.org/10.1186/1472-6750-9-6.
Keller T, et al. Selection of scFv antibody fragments binding to human blood versus lymphatic endothelial surface antigens by direct cell phage display. PLoS ONE. 2015;10(5): e0127169. https://doi.org/10.1371/journal.pone.0127169.
Rangnoi K, et al. Binding characteristic of various antibody formats against aflatoxins. ACS Omega. 2021;6(39):25258–68. https://doi.org/10.1021/acsomega.1c03044.
Bühring HJ, et al. The receptor tyrosine kinase p185HER2 is expressed on a subset of B-lymphoid blasts from patients with acute lymphoblastic leukemia and chronic myelogenous leukemia. Blood. 1995;86(5):1916–23. https://doi.org/10.1182/blood.V86.5.1916.bloodjournal8651916.
van Rhenen A, et al. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res. 2005;11(18):6520–7. https://doi.org/10.1158/1078-0432.CCR-05-0468.
Brian B, Eytan MS. Which are the most promising targets for minimal residual disease-directed therapy in acute myeloid leukemia prior to allogeneic stem cell transplant? Haematologica. 2019;104(8):1521–31. https://doi.org/10.3324/haematol.2018.208587.
Dalton WT Jr, et al. HL-60 cell line was derived from a patient with FAB-M2 and not FAB-M3. Blood. 1988;71(1):242–7. https://doi.org/10.1182/blood.V71.1.242.242.
Mehdipour T, et al. Tailoring subtractive cell biopanning to identify diffuse gastric adenocarcinoma-associated antigens via human scFv antibodies. Immunology. 2020;159(1):96–108. https://doi.org/10.1111/imm.13129.
Hoogenboom HR, et al. Selection-dominant and nonaccessible epitopes on cell-surface receptors revealed by cell-panning with a large phage antibody library. Eur J Biochem. 1999;260(3):774–84. https://doi.org/10.1046/j.1432-1327.1999.00214.x.
Xu JL, Davis MM. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity. 2000;13(1):37–45. https://doi.org/10.1016/S1074-7613(00)00006-6.
Lagunas-Rangel FA, et al. Acute myeloid leukemia-genetic alterations and their clinical prognosis. Int J Hematol Oncol Stem Cell Res. 2017;11(4):328–39.
Horibata S, et al. Heterogeneity in refractory acute myeloid leukemia. Proc Natl Acad Sci U S A. 2019;116(21):10494. https://doi.org/10.1073/pnas.1902375116.
Collignon A, et al. A chemogenomic approach to identify personalized therapy for patients with relapse or refractory acute myeloid leukemia: results of a prospective feasibility study. Blood Cancer J. 2020;10(6):64. https://doi.org/10.1038/s41408-020-0330-5.
Chanput W, P.V., Wichers H. THP-1 and U937 Cells. In: The Impact of Food Bioactives on Health: in vitro and ex vivo models [Internet], C.P. Verhoeckx K, López-Expósito I, et al. Editor. Cham (CH): Springer; 2015. Chapter 14.
Drexler HG, Matsuo Y, MacLeod RAF. Malignant hematopoietic cell lines: in vitro models for the study of erythroleukemia. Leuk Res. 2004;28(12):1243–51. https://doi.org/10.1016/j.leukres.2004.03.022.
Zotova A, Zotov I, Filatov A, Mazurov D. Determining antigen specificity of a monoclonal antibody using genome-scale CRISPR-Cas9 knockout library. J Immunol Methods. 2016;439:8–14. https://doi.org/10.1016/j.jim.2016.09.006.
Tabasinezhad M, Talebkhan Y, Wenzel W, Rahimi H, Omidinia E, Mahboudi F. Trends in therapeutic antibody affinity maturation: from in-vitro towards next-generation sequencing approaches. Immunol Lett. 2019;212:106–13. https://doi.org/10.1016/j.imlet.2019.06.009.
Park S, et al. Quantitative RT-PCR assay of HER2 mRNA expression in formalin-fixed and paraffin-embedded breast cancer tissues. Int J Clin Exp Pathol. 2014;7(10):6752–9.
Rangnoi K, et al. Enhancement and analysis of human antiaflatoxin B1 (AFB1) scFv antibody-ligand interaction using chain shuffling. J Agric Food Chem. 2018;66(22):5713–22. https://doi.org/10.1021/acs.jafc.8b01141.
Sompunga P, et al. Generation of human and rabbit recombinant antibodies for the detection of Zearalenone by phage display antibody technology. Talanta. 2019;201:397–405. https://doi.org/10.1016/j.talanta.2019.04.034.
Khaing, K.K., et al. Application of Recombinant Human scFv Antibody as a Powerful Tool to Monitor Nitrogen Fixing Biofertilizer in Rice and Legume. Microbiol Spectr. 2021;9(3):e0209421. https://doi.org/10.1128/Spectrum.02094-21
Zahavi D, et al. Enhancing antibody-dependent cell-mediated cytotoxicity: a strategy for improving antibody-based immunotherapy. Antib Ther. 2018;1(1):7–12. https://doi.org/10.1093/abt/tby002.
Hassenrück F, et al. Sensitive detection of the natural killer cell-mediated cytotoxicity of anti-CD20 antibodies and its impairment by B-Cell receptor pathway inhibitors. Biomed Res Int. 2018;2018:1023490. https://doi.org/10.1155/2018/1023490.
Wang W, et al. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. 2015;6:368–368. https://doi.org/10.3389/fimmu.2015.00368.
Fujiwara H, et al. Tissue-restricted T cell alloresponses across HLA barriers: selection and identification of leukemia-restricted CTL in HLA-mismatched stimulator–responder pairs. Bone Marrow Transpl. 2003;32(4):371–8. https://doi.org/10.1038/sj.bmt.1704142.
Horowitz, A., et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med. 2013;5(208):208ra145. https://doi.org/10.1126/scitranslmed.3006702
Romain G, et al. Antibody Fc engineering improves frequency and promotes kinetic boosting of serial killing mediated by NK cells. Blood. 2014;124(22):3241–9. https://doi.org/10.1182/blood-2014-04-569061.
Vasu S, et al. Decitabine enhances anti-CD33 monoclonal antibody BI 836858-mediated natural killer ADCC against AML blasts. Blood. 2016;127(23):2879–89. https://doi.org/10.1182/blood-2015-11-680546.
Acknowledgements
The authors would like to thank Miss Yuwadee Kitprasong, medical technologist at Sanphasitthiprasong hospital, for patient sample collection. We would also like to thank to all participants who volunteered for this study. We are in dept. of Prof. Dr. Pa-thai Yenchitsomanus from Siriraj Center of Research Excellence for Cancer Immunotherapy, Mahidol University, Thailand for several cell lines and grateful to MY lab members and KAC lab members for excellent technical assistances and advice.
Funding
This research was supported by Thailand Science Research and Innovation (TSRI) (TRF Senior Research grant number RTA6180012) and BIOTEC, the National Science and Technology Development Agency (NSTDA) (grant number P-18–50127), and Ministry of Higher Education, Science, Research and Innovation (MHESI) (grant number 256101A3040017). TS was co-supported by the Royal Golden Jubilee PhD program of Thailand (rgj.trf.or.th) and Suranaree University of Technology (www.sut.ac.th) [grant number PHD/0098/2553]. She also was supported by Newton Fund from British Council, as well as grants from MY Lab. MY was also supported by the Distinguished Research Professor Grant (NRCT 808/2563) of the National Research Council of Thailand.
Author information
Authors and Affiliations
Contributions
Thitima Sumphanapai performed the experiments, data collection, and writing of the initial draft. Surasak Sawatnatee provided patient samples and had a consultant/advisory role. Jenny Yeung conceived the experiments, supervised assay techniques, and revised the manuscript. Kerry Chester conceived the experiments and provided the critical review, commentary, and revision. Montarop Yamabhai conceived the experiments, conceptualization, supervision, edited the manuscript, and acquisition of the financial support for the project leading to this publication. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval
The patient samples were collected under the approval of the Ethical Review Board of Sanphasitthiprasong hospital, Ubon Ratchathani, Thailand. The peripheral blood samples of healthy donors were obtained under the approval of the research ethics board of the Suranaree University of Technology, Nakhon Ratchasima, Thailand, in accordance with the Declaration of Helsinki.
Consent to participate
All samples analyzed in this study were obtained after informed consent of the donors.
Consent to publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sumphanapai, T., Chester, K., Sawatnatee, S. et al. Targeting acute myeloid cell surface using a recombinant antibody isolated from whole-cell biopanning of a phage display human scFv antibody library. Med Oncol 39, 205 (2022). https://doi.org/10.1007/s12032-022-01806-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-022-01806-9