
Abstract
Lapatinib, a novel oral dual tyrosine kinase inhibitor blocking HER1 and HER2 pathways, has presented beneficial effects on breast cancer with positive HER2. However, its efficacy is largely limited by the occurrence of acquired drug resistance. In this study, we aimed to explore the underlying molecular mechanisms of Lapatinib resistance using bioinformatics strategies. The gene expression profile of SKBR3-R (acquired Lapatinib-resistant) and SKBR3 (Lapatinib-sensitive) cell line was downloaded from gene expression omnibus database. Then, the differentially expressed genes (DEGs) were selected using dChip software. Furthermore, gene ontology (GO) and pathway enrichment analyses were carried out by using DAVID database. Finally, the protein–protein interaction network was constructed, and the hub genes in the network were analyzed by using STRING database. A total of 300 DEGs, such as HSPA5, MAP1LC3A and RASSF2, were screened out. GO functional enrichment analysis showed that the genes were associated with cell membrane component-related, stimulus-related and binding-related items. KEGG pathway analysis indicated that three dysfunctional pathways, including PPAR signaling pathway, cytokine–cytokine receptor interaction and pathways in cancer, were enriched. Protein–protein interaction network construction revealed that some hub genes, such as PPARG, TGFBI, TGFBR2, TIMP1, CTGF, UBA52 and JUN, might have an association with Lapatinib resistance. The present study offered new insights into the molecular mechanisms of Lapatinib resistance and identified a series of important hub genes that have the potential to be the targets for treatment of Lapatinib-resistant breast cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most common cancer for women in the world [1]. Approximately 20% of breast cancers over-express human epidermal growth factor receptor 2 (HER2), which is related to not only aggressive tumor behavior but also poor patient outcome [2]. Lapatinib, an orally dual tyrosine kinase inhibitor targeting HER1 and HER2, presented dramatically therapeutic effects in patients with HER2-positive breast cancer, including reduction in recurrence risk and prolongation of overall survival [3, 4]. However, the vast majority of patients with initial excellent responses to Lapatinib eventually develop drug resistance, which has presented a severe clinical challenge [5].
It is still totally unclear about the molecular mechanisms of acquired Lapatinib resistance. Previous studies have described that HER2 alteration and multiple bypass signaling might have a correlation with this progress. For example, the abnormal expression and/or activation of signaling molecules includes PI3K [6], RON [7], SRC [8], PP2A [9], CXCR4 [10], NF-κB [11], FOXM1 [12], PI3K/AKT and MAPK/ERK [13], which participated in Lapatinib resistance through various mechanisms. Generally speaking, these studies demonstrated that multiple gene alterations occurred in Lapatinib-resistant cells. Therefore, it is difficult to treat breast cancer with Lapatinib resistance by interrupting single molecular alteration or pathway.
In the past several years, a variety of therapeutic measures attempting to reverse Lapatinib resistance have been explored [14]. However, owing to the unclear pathogenesis, investigations on the treatments are still at an early stage and the treatment effects are unsatisfactory. In fact, the majority of the studies concentrated on any certain molecular target, ignoring that Lapatinib resistance involves aberrant expressions of multiple genes. It reflected that traditional therapeutic approaches targeting any certain single gene are insufficient to illuminate the nature of resistance, and thus, it is urgent to investigate the resistance-related multiple gene variations with more powerful genome-wide technologies, which may shed new light on clarification of the nature of Lapatinib resistance and then help look for novel therapeutic strategies.
Microarray is a high-throughput tool for efficiently performing global gene expression profiles, which has been widely utilized for exploring the mechanisms underlying a series of disorders. Using microarray, previous studies have described the gene expression profiles of acquired Lapatinib-resistant breast cancer cells [15, 16]. Accordingly, to better understand the intrinsic mechanisms, it is necessary to carry out further research about gene expression profiles of acquired Lapatinib resistance.
In the present study, using bioinformatics methods, public microarray data were downloaded for identifying differentially expressed genes (DEGs) between acquired Lapatinib-resistant and Lapatinib-sensitive breast cancer cells. The functions of DEGs were further assessed by gene ontology (GO) annotation, pathway enrichment and protein–protein interaction network construction. We aimed to obtain better insights into the mechanisms and clarify potential therapeutic targets for overcoming Lapatinib resistance.
Methods
Identification of differentially expressed genes (DEGs) from public microarray data
To obtain DEGs in acquired Lapatinib-resistant breast cancer cells compared to Lapatinib-sensitive breast cancer cells, we downloaded the public gene expression profile (GSE38376) from the gene expression omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). This dataset was deposited by Ram et al. [16], containing information from HER2-positive breast cancer cell line SKBR3 (Lapatinib-sensitive) and SKBR3-R (acquired Lapatinib-resistant cell line). Then, the dataset was analyzed using dChip software (v.2011.01) (http://www.hsph.harvard.edu/). T test was used to screen out Lapatinib resistance-related DEGs between SKBR3-R and SKBR3 cells, with a threshold of P value <0.05 and fold change ≥1.5.
Functional enrichment analysis of DEGs
Using database such as annotation, visualization and integrated discovery (DAVID) database, the functional enrichment analyses of the DEGs, including gene ontology (GO) function analysis and Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis, were carried out. In the GO analysis, the categories include cellular component (CC), biological process (BP) and molecular function (MF) terms, and P value <0.01 was regarded as statistically significant differences. In the KEGG pathways analysis, enriched pathways were identified according to the hypergeometric distribution with a P value <0.1.
Protein–protein interaction network construction by STRING
Through inputting ID number of DEGs, the products of these 300 DEGs were analyzed by the online tool, STRING, to predict the interaction among them. A combined score of not <0.7 (high confidence score) was considered significant. The hub protein was selected based on its association with other proteins. The DEGs with more association with other DEGs indicate important roles in the PPI interaction network.
Results
Identification of DEGs between Lapatinib-sensitive and acquired Lapatinib-resistant breast cancer cells
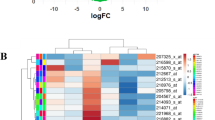
The analysis has been conducted as shown in Fig. 1. Based on the public microarray dataset GSE38376, the t test in dChip software was used to analyze the gene expression profiles and identify the DEGs between Lapatinib-sensitive SKBR3 cells and acquired Lapatinib-resistant SKBR3-R cells with the described criteria. As a result, a total of 300 DEGs were screened out, including 146 up-regulated and 154 down-regulated DEGs. The top ten down-regulated DEGs and up- regulated DEGs are listed in Table 1.

The flowchart of the bioinformatics analysis
Functional annotation and pathway enrichment of DEGs
To investigate the altered biological function of the DEGs, data were clustered through gene ontology (GO) analysis in DAVID. A P value of <0.01 was considered significant. The enriched GO terms that were divided into CC, BP and MF ontologies are illustrated as Figs. 2, 3 and 4, respectively.

GO enrichment of DEGs in cellular component ontology

GO enrichment of DEGs in biological process ontology

GO enrichment of DEGs in molecular function ontology
In the CC ontology, we found that the majority of enriched categories were associated with cell membrane components, such as extracellular region (45 genes), vesicle (20 genes) and plasma membrane part (45 genes). In addition, the other enriched CC GO terms contained extracellular matrix (13 genes) and actin cytoskeleton (11 genes). In the BP ontology, the most significant GO categories were stimulus-related items such as response to extracellular stimulus (13 genes) and response to steroid hormone stimulus (11 genes). Besides, the other enriched categories comprised items related to cell proliferation, cell adhesion, cell motion and anti-apoptosis: for instance negative regulation of cell proliferation (15 genes), biological adhesion (21 genes), cell motion (8 genes) and anti-apoptosis (9 genes). In the MF ontology, the binding-related items constitute the majority of enriched GO categories, including oxygen binding (5 genes), extracellular matrix binding (4 genes) and protein binding/bridging (6 genes).
Furthermore, the enrichment of 3 dysfunctional pathways was assessed by the KEGG pathway analysis, which included PPAR signaling pathway (4 genes), cytokine–cytokine receptor interaction (8 genes) and pathways in cancer (9 genes) (Table 2).
Protein–protein interaction network construction
STRING (a database of known and predicted protein interactions) was used to predict protein interactions among the DEGs. Firstly, the 300 DEGs were submitted to the STRING Web site to get PPI data. Then, the PPIs, with combined scores greater than 0.7, were selected for constructing PPI networks. In the PPI networks, 7 node proteins, including PPARG, TGFBI, TGFBR2, TIMP1, CTGF, UBA52 and JUN, showed a strong association with other node proteins (more than 5), indicating that they have higher hub degrees (Table 3; Fig. 5). These hub genes (proteins) might play crucial roles in the Lapatinib resistance of breast carcinoma.

Protein–protein interaction network diagram of the 7 node proteins including PPARG, TGFBI(5), TGFBR2, TIMP1, CTGF, UBA52 and JUN, showing more association with other node proteins (more than 5), indicating that they have higher hub degrees
Discussion
Lapatinib resistance is a huge challenge for treating patients with breast cancer. Therefore, it is very necessary to explore the mechanisms of Lapatinib resistance and develop feasible treatment strategy for it. Through gene expression profiling by microarray technology, the key genes associated with drug resistance could be discovered, which could be further utilized to explore novel diagnostic and therapeutic strategies. In this study, we identified DEGs between Lapatinib-resistant and Lapatinib-sensitive breast cancer cells. Then, we analyzed their functions by GO annotation and pathway enrichment. Finally, the interrelationship of them was investigated using protein–protein interaction network construction. As a result, a series of hub genes were identified, which might play crucial roles in Lapatinib resistance and might be potential targets for treatment of Lapatinib-resistant breast cancer.
DChip is a powerful software for probe-level and high-level analysis of gene expression microarrays. Using this tool, we obtained 300 DEGs between Lapatinib-resistant and Lapatinib-sensitive breast cancer cells, including 146 up-regulated and 154 down-regulated DEGs. Among the DEGs, a proportion of them showed remarkable fold change (more than 5) in Lapatinib-resistant cells compared with sensitive cells, such as HBE1, PI3, ZNF704, CYP1B1, CFD and KLHL13, with 7.45, −5.96, −5.62, −9.63, −6.32 and −8.25 fold change, respectively. Therefore, we hypothesized that the DEGs have the potential to be biomarkers for distinguishing or predicting Lapatinib resistance. However, the mechanism remains unclear and further verification experiments are needed. In addition, evidence indicates that some DEGs may contribute to drug resistance in various carcinomas. For examples, HSPA5 (heat shock 70 kDa protein 5), listed in the top ten up-regulated DEGs, has been proven to participate in chemotherapy resistance of B-lineage acute lymphoblastic leukemia [17] and breast cancer [18] cells. MAP1LC3A, another gene listed as one of the top ten up-regulated DEGs, could activate LC3A through demethylation, thereby mediating autophagy that contributed to the acquired resistance to EGFR tyrosine kinase inhibitors in lung cancer [19]. Besides, MAP1LC3A has been also shown to be correlated with herceptin resistance [20]. RASSF2, listed as one of the top ten down-regulated DEGs, is a novel tumor suppressive protein. Loss of RASSF2 could enhance tumorigenicity of lung cancer cells and confer their resistance to chemotherapy [21]. Taken together, the DEGs might contribute to Lapatinib resistance through complex mechanisms.
Through GO annotation in DAVID database, we analyzed the biological function of the DEGs. First, in CC oncology, we found that the majority of the DEGs were enriched in cell membrane-related items, and other DEGs were enriched in non-membrane-related items such as extracellular matrix and actin cytoskeleton. The results reflected that Lapatinib resistance might occur through complex cellular molecular mechanisms involved in both membrane and non-membrane structure. Previous evidence suggested that actin cytoskeleton [22] and ion pumps [23] conferred chemoresistance of various tumor cells. Then, in GO category of BP, our data showed that the most significant items were stimulus-related ones such as extracellular response. Our data also revealed that other enriched categories included items associated with cell proliferation, cell adhesion, cell motion and anti-apoptosis. It is easy to understand that drug resistance could be interpreted as a cellular adaptive response to drugs as alien chemicals. In response to the stimulus of Lapatinib, cancer cells can develop resistance by gene mutation and natural selection mechanism. In the evolution process, a series of genetic changes occurred in cancer cells, which participate in drug resistance through different mechanisms such as affecting cell proliferation, adhesion, motion and apoptosis. As a result, the resistance phenotype emerged, including suppressing cell death signals and maintaining proliferation ability under the condition of drug exposure. In addition, cancer cells with drug resistance phenotype frequently have the enhanced cell migration and invasion capability. The DEGs might have influences on migration and invasion by participating in cell motility and cytoskeleton organization. Taken together, these results showed that the DEGs might participate in various biological processes about Lapatinib resistance of breast cancer. In the MF portion, the most enriched oncologies are three binding-related items, including oxygen binding, extracellular matrix binding and protein binding/bridging. These data reflected that the DEGs may affect the binding among protein, oxygen and extracellular matrix and then influence energy metabolism and signal translation in cancer cells. It has been reported that energy metabolism mechanism and aberrant signal translation participated in drug resistance especially Lapatinib resistance in breast cancer [24]. Accordingly, the DEGs might play important roles in Lapatinib resistance through binding-related mechanisms, which is worth testing in further studies.
Pathway analysis may reveal more precise biological functions of genes than GO analysis. In the present study, three pathways were enriched, including cytokine–cytokine receptor interaction, pathways in cancer and PPAR signaling pathway, respectively. It is easily comprehensible that cytokine–cytokine receptor interaction and pathways in cancer might take part in cancer development including drug-resistant phenotype. In addition, recent studies have demonstrated that PPARγ is up-regulated in many tumor types including breast cancer and involved in cell proliferation, invasion, metastasis and chemoresistance [25]. Currently, it is unknown whether PPAR signaling pathway is involved in Lapatinib resistance. Further studies are needed to explore the roles of the DEGs enriched in the PPAR signaling pathway.
Through PPI network construction, a series of hub proteins have been observed to form a local network, including PPARγ, TGFBI, TGFBR2, TIMP1, CTGF, UBA52 and JUN, of which several DEGs have been reported to associate with cancer development or drug resistance. PPARγ, peroxisome proliferator-activated receptor gamma, was an important component of PPAR signaling pathway, and its agonists could reverse multidrug resistance (MDR) of breast cancer cells by interrupting the aberrant PPARγ/PTEN/PI3K/Akt pathway [26]. This finding was in accordance with the results of the pathway analysis in which PPAR signaling pathway was enriched. TGFBI and TGFBR2 are crucial molecules of TGF-beta pathway. Evidence reflected that the cross talk between TGFB and HER2 may result in increased cancer cell proliferation, survival and invasion, accelerated metastasis in animal models and occurrence of resistance to chemotherapy and HER2-targeted therapy [27]. TIMP-1, tissue inhibitor of metalloproteinase 1, increases expression of proteins such as CK2a, CDK1 and PLK1 and hence associates with chemoresistance of breast cancer cells [28]. Meanwhile, CTGF, connective tissue growth factor, was found to be over-expressed in breast cancer and associated with resistance to paclitaxel and doxorubicin [29]. UBA52 is the abbreviation of ubiquitin A-52 residue ribosomal protein fusion product 1. As we know, ubiquitin could conjugate to target proteins and perform different functions depending on the Lys residue of the ubiquitin that is linked, including cell cycle regulation, lysosomal degradation and kinase modification. Reports showed that E3 ubiquitin ligase HOIP attenuates apoptotic cell death induced by cisplatin in ovarian cancer [30]. Low expression of the E3 ubiquitin ligase CBL confers chemoresistance in human pancreatic cancer cells [31], which demonstrated that ubiquitin or ubiquitin-like signaling might play crucial roles in drug resistance of cancers. Moreover, Jun, an oncogene, has been found to participate in drug resistance in many types of cancers [32]. Taken together, these results supported the notion that the hub genes might play important roles in Lapatinib resistance. However, it is unclear whether the above hub genes can be used to be targets for reversing Lapatinib resistance of breast cancer. The results of this study are preliminary findings, and further evaluations for the potential roles of the hub genes are needed.
In summary, the study provides preliminary research for the mechanisms of Lapatinib resistance of breast cancer. DEGs of Lapatinib-resistant breast cancer cells were screened out by computational bioinformatics methods. Then, the aberrant pathways in Lapatinib-resistant cancer cells were identified. Furthermore, several key hub DEGs were selected as potential targets for reversing Lapatinib resistance. The results of the present study may give a valuable indication for both the basic research and clinical treatment of Lapatinib-resistant breast cancer. However, further investigations are still needed to confirm our hypothesis.
References
Schroeder RL, Stevens CL, Sridhar J. Small molecule tyrosine kinase inhibitors of ErbB2/HER2/Neu in the treatment of aggressive breast cancer. Molecules. 2014;19(9):15196–212. doi:10.3390/molecules190915196.
Creedon H, Byron A, Main J, Hayward L, Klinowska T, Brunton VG. Exploring mechanisms of acquired resistance to HER2 (human epidermal growth factor receptor 2)-targeted therapies in breast cancer. Biochem Soc Trans. 2014;42(4):822–30. doi:10.1042/BST20140109.
Araki K, Fukada I, Horii R, Takahashi S, Akiyama F, Iwase T, et al. Lapatinib-associated mucocutaneous toxicities are clinical predictors of improved progression-free survival in patients with human epidermal growth factor receptor (HER2)-positive advanced breast cancer. Breast Cancer Res Treat. 2014;148(1):197–209. doi:10.1007/s10549-014-3148-7.
Figueroa-Magalhaes MC, Jelovac D, Connolly RM, Wolff AC. Treatment of HER2-positive breast cancer. Breast. 2014;23(2):128–36. doi:10.1016/j.breast.2013.11.011.
Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012;17(1):1–16.
Brady SW, Zhang J, Seok D, Wang H, Yu D. Enhanced PI3K p110alpha signaling confers acquired lapatinib resistance that can be effectively reversed by a p110alpha-selective PI3K inhibitor. Mol Cancer Ther. 2014;13(1):60–70. doi:10.1158/1535-7163.MCT-13-0518.
Wang Q, Quan H, Zhao J, Xie C, Wang L, Lou L. RON confers lapatinib resistance in HER2-positive breast cancer cells. Cancer Lett. 2013;340(1):43–50. doi:10.1016/j.canlet.2013.06.022.
Formisano L, Nappi L, Rosa R, Marciano R, D’Amato C, D’Amato V, et al. Epidermal growth factor-receptor activation modulates Src-dependent resistance to lapatinib in breast cancer models. Breast Cancer Res. 2014;16(3):R45. doi:10.1186/bcr3650.
McDermott MS, Browne BC, Conlon NT, O’Brien NA, Slamon DJ, Henry M, et al. PP2A inhibition overcomes acquired resistance to HER2 targeted therapy. Mol Cancer. 2014;13:157. doi:10.1186/1476-4598-13-157.
De Luca A, D’Alessio A, Gallo M, Maiello MR, Bode AM, Normanno N. Src and CXCR4 are involved in the invasiveness of breast cancer cells with acquired resistance to lapatinib. Cell Cycle. 2014;13(1):148–56. doi:10.4161/cc.26899.
Bailey ST, Miron PL, Choi YJ, Kochupurakkal B, Maulik G, Rodig SJ, et al. NF-kappaB activation-induced anti-apoptosis renders HER2-positive cells drug resistant and accelerates tumor growth. Mol Cancer Res. 2014;12(3):408–20. doi:10.1158/1541-7786.MCR-13-0206-T.
Gayle SS, Castellino RC, Buss MC, Nahta R. MEK inhibition increases lapatinib sensitivity via modulation of FOXM1. Curr Med Chem. 2013;20(19):2486–99.
Lee YY, Kim HP, Kang MJ, Cho BK, Han SW, Kim TY, et al. Phosphoproteomic analysis identifies activated MET-axis PI3K/AKT and MAPK/ERK in lapatinib-resistant cancer cell line. Exp Mol Med. 2013;45:e64. doi:10.1038/emm.2013.115.
Thery JC, Spano JP, Azria D, Raymond E, Penault Llorca F. Resistance to human epidermal growth factor receptor type 2-targeted therapies. Eur J Cancer. 2014;50(5):892–901. doi:10.1016/j.ejca.2014.01.003.
Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Can Res. 2009;69(17):6871–8. doi:10.1158/0008-5472.CAN-08-4490.
Komurov K, Tseng JT, Muller M, Seviour EG, Moss TJ, Yang L, et al. The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol Syst Biol. 2012;8:596. doi:10.1038/msb.2012.25.
Uckun FM, Qazi S, Ozer Z, Garner AL, Pitt J, Ma H, et al. Inducing apoptosis in chemotherapy-resistant B-lineage acute lymphoblastic leukaemia cells by targeting HSPA5, a master regulator of the anti-apoptotic unfolded protein response signalling network. Br J Haematol. 2011;153(6):741–52. doi:10.1111/j.1365-2141.2011.08671.x.
Chang YW, Chen HA, Tseng CF, Hong CC, Ma JT, Hung MC, et al. De-acetylation and degradation of HSPA5 is critical for E1A metastasis suppression in breast cancer cells. Oncotarget. 2014;5:10558.
Nihira K, Miki Y, Iida S, Narumi S, Ono K, Iwabuchi E, et al. An activation of LC3A-mediated autophagy contributes to de novo and acquired resistance to EGFR tyrosine kinase inhibitors in lung adenocarcinoma. J Pathol. 2014;234(2):277–88. doi:10.1002/path.4354.
Koukourakis MI, Giatromanolaki A, Bottini A, Cappelletti MR, Zanotti L, Allevi G, et al. Prospective neoadjuvant analysis of PET imaging and mechanisms of resistance to Trastuzumab shows role of HIF1 and autophagy. Br J Cancer. 2014;110(9):2209–16. doi:10.1038/bjc.2014.196.
Clark J, Freeman J, Donninger H. Loss of RASSF2 enhances tumorigencity of lung cancer cells and confers resistance to chemotherapy. Mol Biol Int. 2012;2012:705948. doi:10.1155/2012/705948.
Mithraprabhu S, Khong T, Spencer A. Overcoming inherent resistance to histone deacetylase inhibitors in multiple myeloma cells by targeting pathways integral to the actin cytoskeleton. Cell Death Dis. 2014;5:e1134. doi:10.1038/cddis.2014.98.
Eljack ND, Ma HY, Drucker J, Shen C, Hambley TW, New EJ, et al. Mechanisms of cell uptake and toxicity of the anticancer drug cisplatin. Metallomics. 2014;6(11):2126–33. doi:10.1039/c4mt00238e.
Lozy F, Karantza V. Autophagy and cancer cell metabolism. Semin Cell Dev Biol. 2012;23(4):395–401. doi:10.1016/j.semcdb.2012.01.005.
Cizkova K, Konieczna A, Erdosova B, Lichnovska R, Ehrmann J. Peroxisome proliferator-activated receptors in regulation of cytochromes P450: new way to overcome multidrug resistance? J Biomed Biotechnol. 2012;2012:656428. doi:10.1155/2012/656428.
To KK, Tomlinson B. Targeting the ABCG2-overexpressing multidrug resistant (MDR) cancer cells by PPARgamma agonists. Br J Pharmacol. 2013;170(5):1137–51. doi:10.1111/bph.12367.
Chow A, Arteaga CL, Wang SE. When tumor suppressor TGFbeta meets the HER2 (ERBB2) oncogene. J Mammary Gland Biol Neoplasia. 2011;16(2):81–8. doi:10.1007/s10911-011-9206-4.
Hekmat O, Munk S, Fogh L, Yadav R, Francavilla C, Horn H, et al. TIMP-1 increases expression and phosphorylation of proteins associated with drug resistance in breast cancer cells. J Proteome Res. 2013;12(9):4136–51. doi:10.1021/pr400457u.
Wang MY, Chen PS, Prakash E, Hsu HC, Huang HY, Lin MT, et al. Connective tissue growth factor confers drug resistance in breast cancer through concomitant up-regulation of Bcl-xL and cIAP1. Can Res. 2009;69(8):3482–91. doi:10.1158/0008-5472.CAN-08-2524.
Mackay C, Carroll E, Ibrahim AF, Garg A, Inman GJ, Hay RT, et al. E3 ubiquitin ligase HOIP attenuates apoptotic cell death induced by cisplatin. Can Res. 2014;74(8):2246–57. doi:10.1158/0008-5472.CAN-13-2131.
Kadera BE, Toste PA, Wu N, Li L, Nguyen AH, Dawson DW, et al. Low expression of the E3 ubiquitin ligase CBL confers chemoresistance in human pancreatic cancer and is targeted by epidermal growth factor receptor inhibition. Clin Cancer Res. 2014. doi:10.1158/1078-0432.CCR-14-0610.
Wu Q, Yang Z, Nie Y, Shi Y, Fan D. Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches. Cancer Lett. 2014;347(2):159–66. doi:10.1016/j.canlet.2014.03.013.
Acknowledgement
This study was funded by the special foundation for the 1130 Project of Xinqiao Hospital of Third Military Medical University (2012).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Zhang, L., Huang, Y., Zhuo, W. et al. Identification and characterization of biomarkers and their functions for Lapatinib-resistant breast cancer. Med Oncol 34, 89 (2017). https://doi.org/10.1007/s12032-017-0953-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-017-0953-y