
Abstract
In this study, we intend to identify key immune-related genes (IRGs) in gliomas using the TCGA and GTEx databases. Following collection of the RNA-seq data of lower-grade glioma (LGG) and glioblastoma (GBM) patients from the TCGA and GTEx databases, the differentially expressed IRGs (DE-IRGs) were screened. The ESTIMATE algorithm was utilized to evaluate StromalScore and ImmuneScore of LGG and GBM samples and a multifactorial Cox risk model was constructed to identify the related risk genes. The core IRGs of LGG and GBM were screened through a PPI network, followed by exploration of their correlation with glioma prognosis. The relationship between IRGs and immune cells in LGG and GBM was detected. In vitro assays were performed to detect the effect of CXCL9 on glioma cell development. We screened 403 and 492 DE-IRGs in LGG and GBM. StromalScore and ImmuneScore were related to overall survival in LGG, but not in GBM. CXCL9 was identified as a core gene in LGG and GBM and shared association with the prognosis of gliomas. Furthermore, a correlation was found between CXCL9 and immune infiltration of LGG and GBM. Glioma cell proliferation and invasion could be inhibited by silencing of CXCL9. Overall, CXCL9 is correlated to the prognosis of glioma patients and may accelerate glioma development via immune regulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gliomas are frequently occurring primary intracranial tumors that contribute to significant mortality and morbidity (Ostrom et al. 2014). Gliomas can be categorized into lower-grade glioma (LGG) and glioblastoma (GBM); arising de novo or from lower-grade lesions, GBM is quite malignant and presents with poor prognosis despite surgery and treatment (Bao et al. 2021; Wang and Bettegowda 2015). The development of glioma is influenced by the complicated interactions between tumor and immune cells, and response of the immune system to cancer can exert effects on the survival, proliferation, and invasion in glioma (Alghamri et al. 2021). Tumor-infiltrating immune cells are components of microenvironment that regulate tumor progression, and significant advances have been made in terms of immune cell infiltration in central nervous system tumors including gliomas (Domingues et al. 2016). Intriguingly, gene therapy, which can regulate the tumor microenvironment (TME), has been highlighted for treatment of glioma (Banerjee et al. 2021). In this context, it is of significance to seek novel gene targets for treatment of glioma based on regulation of immune infiltration.
The ESTIMATE algorithm–based calculation of immune and stromal scores can expedite the quantification of the immune and stromal parts in tumors, and differentially expressed genes (DEGs) of the two scores were found to be implicated in TME and might affect the prognosis of glioma (Jia et al. 2018). The glioma stromal/immune scores share close correlation with the grade and histology as well as survival time of glioma (Li et al. 2020a). The C-X-C motif (CXC) chemokines are chemotactic molecules and confirmed as promising prognostic markers as well as therapeutic targets for different cancers (Mollica Poeta et al. 2019; Yang et al. 2021). It should be noted that the bioinformatics prediction in our study through the joint analysis of TCGA and GTEX databases identified the CXC chemokine CXCL9 as a key differentially expressed immune-related gene (DE-IRG) in both LGG and GBM. CXCL9 shares association with immune cell infiltration as well as immune-associated biomarkers (Liang et al. 2021). Notably, the overexpression of CXCL9 in solid tumors is correlated with CD8+ T cell infiltration, and downregulation of CXCL9 expression may result in attenuation of tumor growth (Dangaj et al. 2019). Interestingly, CXCL9 is previously identified as a differentially upregulated gene in GBM (Sreekanthreddy et al. 2010). Another study has reported that CXCL9 is expressed in intracranial GL261 tumors in vivo and that CXCL9 is capable of facilitating glioma cell growth in vitro (Liu et al. 2011). Furthermore, CXCL9 expression can be increased by astrocytoma cells upon co-culture with activated peripheral blood mononuclear cells, which regulates the proliferation of T cells (Jehs et al. 2011). In view of the aforementioned reports, we thus propose a hypothesis that CXCL9 functioning as a IRG might affect immune infiltration in LGG and GBM and impact the prognosis of the patients, and intended to validate it in the present study, in hope of finding a novel target for improvement of the prognosis of glioma through immunotherapy.
Materials and Methods
Data Collection and Processing
Through the TCGA database, the FPKM data and corresponding clinical information for patients with LGG and GBM were downloaded, where the LGG dataset contained only 529 tumor samples, and the GBM dataset contained 5 normal tissue samples and 169 tumor samples. Samples of 1152 normal brain tissue were obtained from the GTEx database. The list of human IRGs was obtained from the ImmPort database, containing a total of 2498 IRGs.
Through the GENCODE database, the Ensembl number of genes was converted to Official Symbol. Then, data from TCGA and GTEx were merged, and the expression profiles were normalized, followed by batch effect removal using the “sva” package in the R software. LGG and GEM datasets of TCGA data were collected for analyzing clinical characteristics of samples (WHO.grade, IDH.status, 1p/19q.codeletion, Gender, Primary.therapy.outcome, Race, Age, and Histological.type). High and low expression was grouped according to the median of CXCL9. The statistical method was chi-square test, and the basic R package was used to draw the baseline table (Supplemental Table 1).
Evaluation of the StromalScore and the ImmuneScore Using the ESTIMATE Algorithm
The “Estimate” package (Version 1.0.13) in R language was utilized to calculate the StromalScore and ImmuneScore of LGG and GBM samples. StromalScore represented infiltration of stromal cells in tumor tissues and ImmuneScore represented infiltration of immune cells in tumor tissues. The LGG and GBM samples were divided into high/low StromalScore and high/low ImmuneScore groups according to the median values of StromalScore or ImmuneScore, respectively.
Differential Gene Expression Analysis
The “limma” package (Version 3.42.2) in R language and Wilcoxon test were used to calculate normal samples and DEGs between high/low StromalScore and ImmuneScore groups. |logFC|> 1 and FDR < 0.05 were considered the screening criteria for DEGs. The “pheatmap” package (Version 1.0.12) was used to delineate the heatmaps of DEGs and correlation.
Kaplan–Meier Survival Analysis
With the use of the “survival” package (Version 3.1–11) in R language, Kaplan–Meier survival analysis was performed to compare differences in overall survival between high/low StromalScore and ImmuneScore groups in LGG and GBM and the relationship between key IRGs with overall survival of LGG and GBM patients.
Multifactorial Cox Risk Model
A multivariate Cox risk scale model was constructed using the R language “survival” package. DE-IRGs in LGG and GBM and those in StromalScore and ImmuneScore were merged, followed by construction of a multivariate Cox risk ratio model to evaluate gene expression and risks of StromalScore and ImmuneScore. Genes associated with StromalScore and ImmuneScore were selected with significance threshold FDR < 0.05 as the screening condition.
Protein–Protein Interaction (PPI) Network Construction
Using the online tool String, the PPI network (minimum required interaction score: 0.4) was constructed. Next, 11 genes associated with StromalScore and ImmuneScore risks in LGG were obtained. The PPI network of these 11 genes was constructed, and then images were delineated and the core degree was calculated using the software Cytoscape.
Calculation of the Immune Cell Content of the Samples Using the CIBERSORT Algorithm
The immune cell content of each LGG and GBM sample was calculated using the CIBERSORT algorithm, with the help of the “preprocessCore” package (Version 1.48.0), the “parallel” package (Version 3.6.3), and the “e1071” package (Version 1.7–3). Pearson correlation analysis was used to analyze the correlation between genes and immune cell types. Samples with p < 0.05 were also included in the correlation analysis of genes and immune cell types.
Cell Treatment
Normal human glial cell line HEB (CC-Y1197) and human glioma cell lines U251MG (CC-Y1526) and A172 (CC-Y1025) were purchased from Shanghai Biological Technology Co., Ltd. Enzyme Research (Shanghai, China). Cells were cultured in DMEM supplemented with 10% FBS (Gibco) at 37 °C with 5% CO2. Small interfering RNA (siRNA) CXCL9 was used to inhibit CXCL9 expression. The si-Crtl was used as a control. The siRNA was designed and synthesized by RIBIOBIO (Shanghai, China), si-Ctrl (product ID: siN0000001-1–5), and si-CXCL9 (product ID: siG000004283A-1–5). X-tremeGENE HP DNA Transfection Reagent (06366236001, Yayu Biotechnology, Shanghai, China). siRNA was transfected into U251MG and A172 cells according to the manufacturer’s instructions. Cells were harvested at 48 h after transfection.
RT-qPCR
TRIzol reagent (15596018, Thermo Fisher Scientific, Rockford, IL) was used to extract the total RNA, followed by reverse transcription using the RevertAid RT reverse transcription kit (K1691, Thermo Fisher Scientific). RT-qPCR was performed using fluorescence qPCR (7500, ABI Company, Oyster Bay, NY). Primer sequences are displayed in Supplemental Table 2. GAPDH was used as an internal reference. The 2−ΔΔCt method was used to assess the relative expression levels of the genes (Ouyang et al. 2015).
CCK-8 Assay
CCK-8 assay kits (C0037, Beyotime, Shanghai, China) were used for cell proliferation detection. In brief, cells in the logarithmic growth phase were seeded into a 96-well plate (1 × 104 cells/well), and 10 μL of CCK-8 solution was added to each well. After 2 h of incubation at 37 °C, the absorbance at 450 nM was measured with a microtiter plate reader. Measurements were performed at 0, 24, 48, 72, and 96 h after siRNA transfection.
Transwell Assay
Cell invasion and migration assays were performed using transwell chambers (pre-coated with/without Matrigel) (Chen et al. 2020). Invading and migrating cells were counted and photographed.
Scratch Test
Straight lines were drawn evenly on the back of the 6-well plate, with an interval of 0.5–1 cm. At least 5 lines were threaded through each well. Glioma cells in logarithmic phase were digested with 0.25% trypsin and dispersed in DMEM/F-12 medium containing 10% fetal bovine serum to obtain single cell suspensions which were inoculated into the 6-well plate at 1.0 × 105 cells/well. The distance was determined from images captured under a microscope at 0 h and 24 h after wounding.
Statistical Analysis
All data were processed using the GraphPad Prism 9.0 statistical software. Measurement data from three independent experiments were expressed in the form of mean ± standard deviation. Independent sample t-test was performed for comparison between two groups. Data among multiple groups at different time points were compared using ANOVA or two-way ANOVA and Bonferroni post hoc test. p < 0. 05 and p < 0. 01 indicated that the difference was significant.
R language Version 3.6.3 and Cytoscape Version 3.6.1 were used in the study. The integrated development environment RStudio (Version 1.2.5033) was utilized for R language compilation and Perl language (Version 5.28.1) for file processing.
Results
Joint Analysis of the TCGA and GTEx Databases Identified DE-IRGs in Both LGG and GBM
To identify the functions of IRGs in glioma, we first performed differential analysis on TCGA and GTEx data and identified 8136 DEGs (including 3870 upregulated genes and 4266 downregulated genes) in LGG (Fig. 1A) and 8860 DEGs (including 3518 upregulated genes and 5342 downregulated genes) in GBM (Fig. 1B). Furthermore, based on the list of immune genes in ImmPort, 403 DE-IRGs (including 224 upregulated genes and 179 downregulated genes) were extracted from the DEGs in LGG (Fig. 1C) and 492 DE-IRGs (including 309 upregulated genes and 183 downregulated genes) from those in GBM (Fig. 1D). The above results suggested that IRGs may play a key role in regulating occurrence and development of LGG and GBM.

Expression heatmaps of DEGs and DE-IRGs in LGG and GBM. A Heatmap of DEGs in LGG (N = 1152, T = 529). B Heatmap of DEGs in GBM (N = 1157, T = 169). C Heatmap of DE-IRGs in LGG (N = 1152, T = 529). D Heatmap of DE-IRGs in GBM (N = 1157, T = 169). Upregulated genes are shown in red, downregulated genes in green, and genes with the same expression levels in black. N, normal; T, tumor
StromalScore and ImmuneScore Were Significantly Associated with LGG Prognosis
To understand the infiltration of stroma and immune cells in glioma, we evaluated StromalScore and ImmuneScore in LGG and GBM patients by the ESTIMATE algorithm and found that the StromalScore for LGG patients ranged from − 1749.60 to 1705.96 (Fig. 2A) and the ImmuneScore ranged from − 1686.78 to 2434.25 (Fig. 2B). The StromalScore for GBM patients ranged from − 1389.42 to 1433.87 (Fig. 2C) and the ImmuneScore ranged from − 1000.30 to 2697.66 (Fig. 2D).

Relationship between StromalScore/ImmuneScore and glioma prognosis. A The StromalScore distribution of LGG. The abscissa displays samples from large to small by score. B The ImmuneScore distribution of LGG. C The StromalScore distribution of GBM. D The ImmuneScore distribution of GBM. E Overall survival curve between high/low StromalScore of LGG. Red lines indicate the survival curves of the high StromalScore group, and blue lines indicate the survival curves of the low StromalScore group. F Overall survival curve between high/low ImmuneScore of LGG. G Overall survival curve between high/low StromalScore of GBM. H, Overall survival curve between high/low ImmuneScore of GBM
To assess the relationship of StromalScore and ImmuneScore with survival, we screened samples with survival time greater than 100 in LGG and GBM and classified LGG and GBM patients into high and low groups according to the median StromalScore and ImmuneScore. The Kaplan–Meier survival analysis found that LGG patients with high StromalScore and ImmuneScore had poor prognosis (Fig. 2E, F). However, StromalScore and ImmuneScore were not associated with prognosis of patients with GBM (Fig. 2G, H). Collectively, StromalScore and ImmuneScore were significantly correlated with LGG prognosis.
StromalScore and ImmuneScore Risk Genes Were Identified Through Multivariate Cox Regression Analysis
To assess the relationship of DE-IRGs with StromalScore or ImmuneScore, we first performed differential analysis using the “limma” package in R language to identify DEGs between high/low StromalScore and ImmuneScore groups. For LGG patients, there were 91 DEGs in the high and low StromalScore groups, including 89 upregulated genes and 2 downregulated genes in the high StromalScore group (Fig. 3A); there were 96 DEGs in the high and low ImmuneScore groups, including 89 upregulated genes and 7 downregulated genes in the high ImmuneScore group (Fig. 3B). For GBM patients, there were 83 DEGs in the high and low StromalScore groups, with 81 genes upregulated and 2 genes downregulated in the high StromalScore group (Fig. 3C); however, there were no DEGs in high and low ImmuneScore group (image not shown).

Multivariate Cox regression analysis for prediction of StromalScore and ImmuneScore risk genes in gliomas. A Volcano map of DEGs between high/low StromalScore of LGG. Red dots indicate genes that are highly expressed in the high StromalScore group, green dots indicate genes that are highly expressed in the low StromalScore group, and black dots indicate genes with no significant differences. B Volcano map of DEG between high/low ImmuneScore of LGG. C DEGs between high/low StromalScore of GBM. D Venn diagram of the intersection of DE-IRGs in LGG and DEGs of StromalScore and ImmuneScore. E Multivariate Cox regression model to screen risk genes of StromalScore in LGG. A hazard ratio > 1 is generally considered a high risk; otherwise, it indicates a low risk. Only the genes at significant risk are shown in the figure. F Multivariate Cox regression model to screen risk genes of ImmuneScore in LGG. G Venn diagram of the intersection of DE-IRG of GBM and DEG of StromalScore. H Multivariate Cox regression model to screen risk genes of StromalScore in GBM
The DE-IRGs in LGG and the DEGs of StromalScore and ImmuneScore were further merged to obtain 68 intersection genes (Fig. 3D). Then, the multivariate Cox regression analysis of these genes showed that 25 IRGs in LGG clearly affected StromalScore, including 9 high-risk genes and 16 low-risk genes (Fig. 3E). Meanwhile, 28 IRGs apparently affected ImmuneScore, including 4 high-risk genes and 24 low-risk genes (Fig. 3F).
Combination of DE-IRGs in GBM and DEGs in StromalScore yielded 65 intersection genes (Fig. 3G). Multivariate Cox regression analysis found that 18 IRGs were associated with StromalScore, including 7 high-risk genes and 11 low-risk genes (Fig. 3H). From this, we further identified the StromalScore and ImmuneScore risk genes in the LGG and GBM.
Five Core IRGs Were Screened in Gliomas Based on the PPI Network
In order to better understand the interaction between risk genes, we took the intersection of StromalScore and ImmuneScore risk genes in LGG (Fig. 4A) and used the online tool String to build the PPI network of these 11 genes (Fig. 4B), which found FCGR2B, CXCL9, and CSF1R as the core genes. Next, we further constructed a PPI network for the StromalScore risk genes of GBM and IL10 and CCL2 were identified as the core genes (Fig. 4C). Thus, we proposed that FCGR2B, CXCL9, and CSF1R might be key IRGs in LGG, while IL10 and CCL2 might be key IRGs in GBM.

PPI network of the IRGs. A Venn diagram of the intersection of StromalScore and ImmuneScore risk genes in LGG. B PPI network of 11 risk genes in LGG. The brighter color indicates the higher core degree of the gene. C PPI network of 18 risk genes related to StromalScore in GBM
CXCL9 Was Associated with Poor Prognosis and Immune Infiltration in LGG and GBM
Differential analysis revealed that CXCL9 and CSF1R were upregulated in LGG and that FCGR2B was downregulated in LGG (Fig. 5A). However, both IL10 and CCL2 were upregulated in GBM (Fig. 5B). Based on the median value of gene expression, LGG samples were divided into samples with high and low gene expression. The plotted overall survival curve found that CXCL9 significantly reduced the survival rate of LGG patients (Fig. 5C), and CCL2 obviously diminished that of GBM patients (Fig. 5D), while other genes had no significant correlation with the overall survival of LGG and GBM. Furthermore, we found that CXCL9 was also significantly upregulated in GBM (Fig. 5E); since CCL2 was not a significant DEG in LGG, CXCL9 might be more important than CCL2 in gliomas. In addition, the survival curves based on combination of the survival data of LGG and GBM and CXCL9 expression data found that the upregulated CXCL9 expression markedly reduced the survival rate of glioma patients (Fig. 5F).

Analysis of the effect of key IRGs on the prognosis of LGG and GBM. A Expression of CXCL9, CSF1R, and FCGR2B in LGG (N = 1152, T = 529). B IL10 and CCL2 expression in GBM (N = 1157, T = 169). C Overall survival curves for the high and low CXCL9 expression groups in LGG. The red line indicates the high CXCL9 expression, and the blue line indicates the low expression of CXCL9. D Overall survival curves of the high and low CCL2 expression groups in GBM. The red line indicates the high level of CCL2 expression, and the blue line indicates the low expression of CCL2. E The expression of CXCL9 in GBM (N = 1157, T = 169). F Overall survival curves of the high and low CXCL9 expression groups in the combined data of LGG and GBM. The red line indicates the high CXCL9 expression, and the blue line indicates the low expression of CXCL9. N, normal; T, tumor
To further determine the correlation of CXCL9 with immune cells in LGG and GBM, we calculated the immune cell content of the LGG and GBM samples using the CIBERSORT algorithm. Correlation analysis results showed that CXCL9 was correlated with 13 types of immune cells in GBM, including plasma cells, T cells CD8, T cells CD4 naïve, T cells CD4 memory resting, T cells CD4 memory activated, T cells gamma delta, monocytes, macrophages M0, macrophages M1, dendritic cells resting, dendritic cells activated, mast cells resting, and mast cells activated (Fig. 6A). Besides, CXCL9 was correlated with T cells CD4 memory resting, T cells gamma delta, NK cells resting, macrophages M0, and macrophages M1 in LGG (Fig. 6B).

Correlation analysis between CXCL9 and immune cell infiltration in LGG and GBM. A Correlation between the expression of CXCL9 and immune cell content in LGG. B Correlation between CXCL9 and immune cell content in GBM
The above results indicated that CXCL9, as an immune-related prognostic biomarker, was associated with the immune infiltration of LGG and GBM.
Silencing of CXCL9 Inhibited the Malignant Features of Glioma Cells
We further validated the biological function of CXCL9 in glioma, and the RT-qPCR results showed that CXCL9 expression was elevated in glioma cell lines U251MG and A172 as compared with that in normal glial cell lines HEB (Fig. 7A). Glioma cell lines were transfected with si-CXCL9, and the silencing efficiency was verified by RT-qPCR (Fig. 7B). CCK-8 results showed that CXCL9 silencing inhibited U251MG and A172 cell proliferation (Fig. 7C). In addition, the transwell assay and scratch test also revealed that CXCL9 silencing inhibited the invasion and migration of U251MG and A172 cells (Fig. 7D–F). These results demonstrated that the high expression of CXCL9 was an unfavorable factor for glioma.

Silencing of CXCL9 inhibits glioma cell proliferation, invasion, and migration. A The expression level of CXCL9 in HEB normal glial cells and glioma cells. B Silencing efficiency of CXCL9. C The growth of glioma cells treated with si-CXCL9 as detected by CCK-8 assay. D The effect of si-CXCL9 on glioma cell invasion as detected by transwell assay. E The effect of si-CXCL9 on glioma cell migration as detected by transwell assay. F The effect of si-CXCL9 on glioma cell migration as detected by the scratch test. p < 0. 05 and p < 0. 01. The experiments were repeated in triplicate
Discussion
Malignant glioma has a high mortality partially due to the morphology of infiltrative growth (Siminska et al. 2018). In this study, we aimed to identify key IRGs in gliomas by analyzing the transcriptional profiles in the TCGA and GTEx databases, and found the correlation of CXCL9 with the prognosis of glioma patients and the promoting potential of it in the progression of glioma through immune regulation.
In the first place, we performed joint analysis of the TCGA and GTEx databases to identify DE-IRGs in both LGG and GBM, which suggested the important involvement of IRGs in the regulation of LGG and GBM progression. Intriguingly, we further revealed that StromalScore and ImmuneScore were significantly associated with the prognosis of LGG. IRGs are pivotal regulators of immune infiltration (Lu et al. 2021). Immune cell infiltration and IRGs can participate in the TME of GBM (Huang et al. 2020a, b). As previously reported, IRGs can be applied to diagnose and assess the prognosis of in LGG (Tan et al. 2020; Zhang et al. 2020), which is consistent with our results. Immune cell infiltration scores serve as reliable prognostic predictors for LGG, and the prognostic IRGs might be applied as targets for immunotherapy study (Wang et al. 2021). Immune scores and stromal scores were revealed to be negatively correlated with the overall survival of glioma patients in grade II/III, and the higher risk score indicated the greater ESTIMATE score, suggesting accelerated immune cell infiltration and stromal composition (Li et al. 2020b). It has been also documented that immune/stromal scores have association with the prognosis of LGG, evidenced by the higher median survival of the patients with lower stromal scores and immune scores (Liang and Huang 2020). These previous reports can provide support for our results on the correlation of IRGs with the development of gliomas.
Furthermore, we performed multivariate Cox regression analysis to identify StromalScore and ImmuneScore risk genes, and then five core IRGs were screened in gliomas based on the PPI network. Importantly, a significant association was found in the current study between CXCL9 and the poor prognosis and immune infiltration in GBM and LGG (in GBM: plasma cells, T cells CD8, T cells CD4 naïve, T cells CD4 memory resting, T cells CD4 memory activated, T cells gamma delta, monocytes, macrophages M0, macrophages M1, dendritic cells resting, dendritic cells activated, mast cells resting, and mast cells activated; in LGG: T cells CD4 memory resting, T cells gamma delta, NK cells resting, macrophages M0, and macrophages M1). Accumulating evidence has highlighted that CXCL9 might participate in immune regulation in gliomas. For instance, chemo-attractant CXCL9 is observed to be poorly expressed in LGG relative to HGG, with the involvement of T cell regulation (Weenink et al. 2019). Additionally, the expression of CXCL9 can be induced by astrocytoma cells after co-culture with activated peripheral blood mononuclear cells, which might aid in regulating immune responses mediated by T cells in the brain (Jehs et al. 2011). However, unlike our study, this report did not involve the systematic analysis on the CXCL9-related immune cell types in gliomas. Moreover, the expression of CXCL9 also presents positive correlation with infiltration of immune cells (CD8+ T cells and CD4+ T cells, macrophages, and dendritic cells) in cutaneous melanoma patients (Huang et al. 2020a). Besides, CXCL9 is related to the alteration of immune cell pattern in TME of pancreatic adenocarcinoma and can facilitate tumor progression (Gao et al. 2020). Clearly, the function of CXCL9 in immune regulation has been also evidenced in other malignancies.
Another key result demonstrated in our study was that silencing of CXCL9 could suppress the proliferation, invasion, and migration of glioma cells. CXC chemokines are able to modulate tumor cell proliferation and invasion in multiple cancer types by acting to the TME (Li et al. 2020c). Notably, CXCL9 is capable of accelerating in vitro glioma cell growth (Liu et al. 2011). As previously reported, upregulation of CXCL9 transcripts is found in GBM samples in comparison to that in normal brain samples (Sreekanthreddy et al. 2010). The chemokine network including CXCL9 is suggested to be an important part of the TME in GBM (Urbantat et al. 2021). In addition, expression of CXCL9 is detected in intracranial GL261 tumors in vivo and CXCL9 can result in enhancement of glioma cell growth in vitro (Liu et al. 2011). It was thus concluded that CXCL9 could retard the progression of glioma cells.
Conclusion
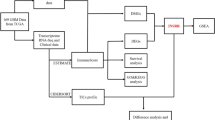
Taken together, the current study demonstrated that CXCL9, serving as an immune gene in glioma, may promote the occurrence and development of glioma through immune regulation (Fig. 8). This study provides a potential target for immunotherapy of glioma patients. However, the specific mechanism of CXCL9 regulating immune infiltration in glioma remains unclear, and the clinical feasibility of the application of CXCL9-based therapy warrants further validation. Due to lack of time and funds, and the difficulty in collecting clinical samples, we only verified the regulatory effect of CXCL9 on glioma cell activity in vitro. In the future study, we will explore the clinical correlation, single cell immune characteristics, immune infiltration, and immune regulation analysis of CXCL9 with glioma.

Schematic diagram of the molecule mechanism of CXCL9 affecting glioma progression through immunomodulation. CXCL9, as the immune gene in glioma patients, may promote the occurrence and development of glioma through immune regulation
Data Availability
The data underlying this article will be shared on reasonable request to the corresponding author.
References
Alghamri MS, McClellan BL, Hartlage MS, Haase S, Faisal SM, Thalla R, Dabaja A, Banerjee K, Carney SV, Mujeeb AA, Olin MR, Moon JJ, Schwendeman A, Lowenstein PR, Castro MG (2021) Targeting neuroinflammation in brain cancer: uncovering mechanisms, pharmacological targets, and neuropharmaceutical developments. Front Pharmacol 12:680021. https://doi.org/10.3389/fphar.2021.680021
Banerjee K, Nunez FJ, Haase S, McClellan BL, Faisal SM, Carney SV, Yu J, Alghamri MS, Asad AS, Candia AJN, Varela ML, Candolfi M, Lowenstein PR, Castro MG (2021) Current approaches for glioma gene therapy and virotherapy. Front Mol Neurosci 14:621831. https://doi.org/10.3389/fnmol.2021.621831
Bao Z, Wang Y, Wang Q, Fang S, Shan X, Wang J, Jiang T (2021) Intratumor heterogeneity, microenvironment, and mechanisms of drug resistance in glioma recurrence and evolution. Front Med 15(4):551–561. https://doi.org/10.1007/s11684-020-0760-2
Chen M, Liu X, Xie P, Wang P, Liu M, Zhan Y, Wang H, Feng Y, Li Y (2020) Circular RNA circ_0074026 indicates unfavorable prognosis for patients with glioma and facilitates oncogenesis of tumor cells by targeting miR-1304 to modulate ERBB4 expression. J Cell Physiol 235(5):4688–4697. https://doi.org/10.1002/jcp.29347
Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, Lanitis E, Duraiswamy J, Tanyi JL, Benencia F, Conejo-Garcia J, Ramay HR, Montone KT, Powell DJ Jr, Gimotty PA, Facciabene A, Jackson DG, Weber JS, Rodig SJ, Hodi SF, Kandalaft LE, Irving M, Zhang L, Foukas P, Rusakiewicz S, Delorenzi M, Coukos G (2019) Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell 35(6):885-900.e810. https://doi.org/10.1016/j.ccell.2019.05.004
Domingues P, Gonzalez-Tablas M, Otero A, Pascual D, Miranda D, Ruiz L, Sousa P, Ciudad J, Goncalves JM, Lopes MC, Orfao A, Tabernero MD (2016) Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav Immun 53:1–15. https://doi.org/10.1016/j.bbi.2015.07.019
Gao HF, Cheng CS, Tang J, Li Y, Chen H, Meng ZQ, Chen Z, Chen LY (2020) CXCL9 chemokine promotes the progression of human pancreatic adenocarcinoma through STAT3-dependent cytotoxic T lymphocyte suppression. Aging (Albany NY) 12(1):502–517. https://doi.org/10.18632/aging.102638
Huang B, Han W, Sheng ZF, Shen GL (2020a) Identification of immune-related biomarkers associated with tumorigenesis and prognosis in cutaneous melanoma patients. Cancer Cell Int 20:195. https://doi.org/10.1186/s12935-020-01271-2
Huang S, Song Z, Zhang T, He X, Huang K, Zhang Q, Shen J, Pan J (2020b) Identification of immune cell infiltration and immune-related genes in the tumor microenvironment of glioblastomas. Front Immunol 11:585034. https://doi.org/10.3389/fimmu.2020.585034
Jehs T, Faber C, Juel HB, Nissen MH (2011) Astrocytoma cells upregulate expression of pro-inflammatory cytokines after co-culture with activated peripheral blood mononuclear cells. APMIS 119(8):551–561. https://doi.org/10.1111/j.1600-0463.2011.02773.x
Jia D, Li S, Li D, Xue H, Yang D, Liu Y (2018) Mining TCGA database for genes of prognostic value in glioblastoma microenvironment. Aging (Albany NY) 10(4):592–605. https://doi.org/10.18632/aging.101415
Li Y, Deng G, Qi Y, Zhang H, Gao L, Jiang H, Ye Z, Liu B, Chen Q (2020a) Bioinformatic profiling of prognosis-related genes in malignant glioma microenvironment. Med Sci Monit 26:e924054. https://doi.org/10.12659/MSM.924054
Li Y, Deng G, Zhang H, Qi Y, Gao L, Tan Y, Hu P, Wang Y, Liu B, Chen Q (2020b) Weighted gene correlation network analysis identifies microenvironment-related genes signature as prognostic candidate for Grade II/III glioma. Aging (Albany NY) 12(21):22122–22138. https://doi.org/10.18632/aging.104075
Li Y, Wu T, Gong S, Zhou H, Yu L, Liang M, Shi R, Wu Z, Zhang J, Li S (2020c) Analysis of the prognosis and therapeutic value of the CXC chemokine family in head and neck squamous cell carcinoma. Front Oncol 10:570736. https://doi.org/10.3389/fonc.2020.570736
Liang H, Huang C (2020) Identification of tumor microenvironment-related genes in lower-grade gliomas by mining TCGA database. Transl Cancer Res 9(8):4583–4595. https://doi.org/10.21037/tcr-20-1079
Liang YK, Deng ZK, Chen MT, Qiu SQ, Xiao YS, Qi YZ, Xie Q, Wang ZH, Jia SC, Zeng LHY (2021) CXCL9 is a potential biomarker of immune infiltration associated with favorable prognosis in ER-negative breast cancer. Front Oncol 11:710286. https://doi.org/10.3389/fonc.2021.710286
Liu C, Luo D, Reynolds BA, Meher G, Katritzky AR, Lu B, Gerard CJ, Bhadha CP, Harrison JK (2011) Chemokine receptor CXCR3 promotes growth of glioma. Carcinogenesis 32(2):129–137. https://doi.org/10.1093/carcin/bgq224
Lu Y, Li K, Hu Y, Wang X (2021) Expression of immune related genes and possible regulatory mechanisms in Alzheimer’s disease. Front Immunol 12:768966. https://doi.org/10.3389/fimmu.2021.768966
Mollica Poeta V, Massara M, Capucetti A, Bonecchi R (2019) Chemokines and chemokine receptors: new targets for cancer immunotherapy. Front Immunol 10:379. https://doi.org/10.3389/fimmu.2019.00379
Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, Langer CE, Pekmezci M, Schwartzbaum JA, Turner MC, Walsh KM, Wrensch MR, Barnholtz-Sloan JS (2014) The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol 16(7):896–913. https://doi.org/10.1093/neuonc/nou087
Ouyang S, Song Y, Tian Y, Chen Y, Yu X, Wang D (2015) RNF8 deficiency results in neurodegeneration in mice. Neurobiol Aging 36(10):2850–2860. https://doi.org/10.1016/j.neurobiolaging.2015.07.010
Siminska D, Kojder K, Jezewski D, Kojder I, Skorka M, Gutowska I, Chlubek D, Baranowska-Bosiacka I (2018) The pathophysiology of post-traumatic glioma. Int J Mol Sci 19(8):2445. https://doi.org/10.3390/ijms19082445
Sreekanthreddy P, Srinivasan H, Kumar DM, Nijaguna MB, Sridevi S, Vrinda M, Arivazhagan A, Balasubramaniam A, Hegde AS, Chandramouli BA, Santosh V, Rao MR, Kondaiah P, Somasundaram K (2010) Identification of potential serum biomarkers of glioblastoma: serum osteopontin levels correlate with poor prognosis. Cancer Epidemiol Biomarkers Prev 19(6):1409–1422. https://doi.org/10.1158/1055-9965.EPI-09-1077
Tan YQ, Li YT, Yan TF, Xu Y, Liu BH, Yang JA, Yang X, Chen QX, Zhang HB (2020) Six immune associated genes construct prognostic model evaluate low-grade glioma. Front Immunol 11:606164. https://doi.org/10.3389/fimmu.2020.606164
Urbantat RM, Vajkoczy P, Brandenburg S (2021) Advances in chemokine signaling pathways as therapeutic targets in glioblastoma. Cancers (basel) 13(12):2983. https://doi.org/10.3390/cancers13122983
Wang J, Bettegowda C (2015) Genomic discoveries in adult astrocytoma. Curr Opin Genet Dev 30:17–24. https://doi.org/10.1016/j.gde.2014.12.002
Wang Z, Xie F, Wu Y, Wang L, Bai Y, Long J, Wang X (2021) Differential genes and scoring criteria among immunogenomic clusters of lower-grade gliomas. Int Immunopharmacol 101(Pt B):108376. https://doi.org/10.1016/j.intimp.2021.108376
Weenink B, Draaisma K, Ooi HZ, Kros JM, Sillevis Smitt PAE, Debets R, French PJ (2019) Low-grade glioma harbors few CD8 T cells, which is accompanied by decreased expression of chemo-attractants, not immunogenic antigens. Sci Rep 9(1):14643. https://doi.org/10.1038/s41598-019-51063-6
Yang X, Wei Y, Sheng F, Xu Y, Liu J, Gao L, Yang J, Sun X, Huang J, Guo Q (2021) Comprehensive analysis of the prognosis and immune infiltration for CXC chemokines in colorectal cancer. Aging (Albany NY) 13(13):17548–17567. https://doi.org/10.18632/aging.203245
Zhang M, Wang X, Chen X, Zhang Q, Hong J (2020) Novel immune-related gene signature for risk stratification and prognosis of survival in lower-grade glioma. Front Genet 11:363. https://doi.org/10.3389/fgene.2020.00363
Author information
Authors and Affiliations
Contributions
YXL and BL designed the study. CJ and XBL collated the data, carried out data analyses, and produced the initial draft of the manuscript. ZPZ contributed to drafting the manuscript. All authors have read and approved the final submitted manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable
Consent for Publication
Not applicable
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liao, Y., Liu, B., Jin, C. et al. Exploring the Correlation of Abnormal CXCL9 Expression with Immune Infiltration in Glioma and Patient Prognosis Based on TCGA and GTEx Databases. J Mol Neurosci 72, 2413–2424 (2022). https://doi.org/10.1007/s12031-022-02083-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-022-02083-x