
Abstract
Brain-derived neurotrophic factor (BDNF), or abrineurin, is a member of the neurotrophin family of growth factors that acts on both the central and peripheral nervous systems. BDNF is also well known for its cardinal role in normal neural maturation. It binds to at least two receptors at the cell surface known as tyrosine kinase B (TrkB) and p75NTR. Additional neurotrophins that are anatomically linked with BDNF include neurotrophin-3 (NT-3), neurotrophin-4 (NT-4), and nerve growth factor (NGF). It is evident that BDNF levels in patients with Alzheimer’s disease (AD) are altered. AD is a progressive disorder and a form of dementia, where the mental function of an elderly person is disrupted. It is associated with a progressive decline in cognitive function, which mainly targets the thinking, memory, and behavior of the person. The degeneration of neurons occurs in the cerebral cortex region of brain. The two major sources responsible for neuronal degeneration are protein fragment amyloid-beta (Aβ), which builds up in the spaces between the nerve cells, known as plaques, disrupting the neuron signaling pathway and leading to dementia, and neurofibrillary tangles (NFTs), which are the twisted fibers of proteins that build up inside the cells. AD is highly prevalent, with recent data indicating nearly 5.8 million Americans aged 65 and older with AD in 2020, and with 80% of patients 75 and older. AD is recognized as the sixth leading cause of death in the USA, and its prevalence is predicted to increase exponentially in the coming years. As AD worsens over time, it becomes increasingly important to understand the exact pathophysiology, biomarkers, and treatment. In this article, we focus primarily on the controversial aspect of BDNF in AD, including its influence on various other proteins and enzymes and the current treatments associated with BDNF, along with future perspectives.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dementia is a neurological condition characterized by cognitive deficit and impairment. It can be classified into four major types: (i) Alzheimer’s disease, (ii) frontotemporal dementia, (iii) vascular dementia, and (iv) Lewy body dementia. Alzheimer’s disease (AD) is a neurodegenerative syndrome which is seen primarily in the elderly. According to recent studies, about 1% of people aged 65 or older suffer from AD. Notably, AD is a major cause of senility in about 60–70 % of patients (see Fig. 1). It is also estimated that the total population suffering from dementia worldwide will double in the coming years. The onset of AD cannot be determined merely by the progress of its pathophysiological alterations but also by the alterations in the cerebral cortex region—the ability of the brain to undergo the therapeutic changes observed in AD without manifesting clinical signs and symptoms [1]. Over the past decade, researchers have successfully identified excess production of amyloid beta (Aβ) as a contributory factor in AD pathogenesis; however, the mechanism involved in Aβ production and CNS irregularities in AD is still unclear [2]. Neuronal mechanisms including tau hyperphosphorylation, neuroinflammation, and various other processes are reported to have a substantial impact on the pathophysiology of multifactorial AD, which at advanced stages becomes fatal. Despite achieving positive results in phase II trials for more than 50 new drugs for AD in the past decade, phase III trials have been less successful. At present, treatments for AD are limited to treating symptoms and slowing the progression of the disease [3].

Graphical representation of types of dementia by percentage. Reference: Adopted from http://seniorsfirstbc.ca/for-professionals/dementia/
Neurotrophins are a family of growth factors responsible for the regulation of neuronal progress, differentiation, and durability in the peripheral and central nervous systems. These proteins trigger and regulate neurogenesis, which is the ability to generate new nerve cells from existing neural stem cells. Neurotrophins include NGF, BDNF, NT-3, and NT-4 [4]. BDNF and NT-4 bind to the TrkB receptor, while NGF and NT-3 bind to TrkA and TrkC receptors [5]. Among the four, BDNF is the most active neurotrophin, which is encoded by the BDNF gene and is located on chromosome 11, regions p13–14, in humans [6]. Since BDNF plays a fundamental role in the growth and plasticity of the cerebrum, it is reported to be extensively involved in psychiatric disorders including AD [7]. Studies by various researchers to determine cerebral BDNF concentrations in patients suffering from AD illustrate conflicting results among the various tests; therefore, AD is seen as an important area for research and development. Molecular dysregulation has been associated with the development of AD; thus, it may provide significant evidence for the interpretation of the pathogenetic processes along with the detection of novel biomarkers for early, precise and selective determination of AD [8]. Certain exercise and niacin appear to improve BDNF and TrkB levels [9,10,11]. Although the research and development of effective therapeutic agents for neurodegenerative disorders constitutes one of the most ethically and economically costly challenges of modern medicinal chemistry, the socioeconomic importance of developing such drugs justifies any associated costs, and motivates the continued search for new therapies [12].
METHODS
Previously published papers were searched from major databases including ScienceDirect, Elsevier, and Frontiers, using the following key words or combinations: AD, BDNF, neurodegeneration, amyloid beta. A total of 112 English-language articles were included for analysis in the present work.
Alzheimer’s Disease
AD is a chronic neurodegenerative disorder involving the degeneration of neurons in the cortex of the brain, causing dementia in 70–80% of patients [13]. It leads to loss of consciousness, impaired thinking, disorientation, behavioral changes and eventually death. AD is widely seen in the elderly population (aged 60–65 years). Characterized by a dominant inheritance pattern, EOAD is associated with classical Mendelian patterns of inheritance with an age-dependent penetrance, while late-onset AD (LOAD) also has a strong genetic component [14]. The primary cause of this disorder is not yet clear; however, plaques and neurofibrillary tangles (NFTs) are two major factors often associated with its progression [13]. Common symptoms include confusion, language difficulties, and personality changes.
The neuronal cell membrane consists of a protein molecule known as amyloid precursor protein (APP). APP consists of two fragments, one existing inside the cell membrane and the other external to the cell membrane. The APP molecule is responsible for the vital function of growth and repair of the neuron after an injury. The protein disintegrates into the two halves via its two enzymes, alpha-secretase and gamma-secretase. These enzymes are peptide fragments which are soluble in the membrane, thus leading to the normal functioning of the brain. However, in some cases the gamma-secretase enzyme combines with beta-secretase and together cleaves the APP molecule, giving rise to an insoluble peptide fragment. This fragment is responsible for the generation of a monomer, Aβ. Aβ monomers are chemically stickier and collectively bind to the peripheral neurons, forming Aβ plaques between the neurons and disrupting neuron–neuron signaling. Amyloid plaques are also capable of initiating an immune response and inducing inflammation causing degeneration of the adjacent neurons. The condition in which amyloid plaques accumulate on the walls of blood capillaries of the cerebrum is called cerebral amyloid angiopathy (CAA). CAA is also responsible for increasing the risk of brain hemorrhage or rupture and severe blood loss. Moreover, it is evident that Aβ accumulates in the mitochondria of brain cells in AD and also inhibits enzyme activity and utilization of glucose by neurons [15].
Neurofibrillary tangles (NFTs) are a primary marker for AD; the formation of these tangles occurs inside the neurons. Neurons are held together by cytoskeletons which are partly constructed of microtubules. The primary function of the cytoskeleton includes the transport of nutrients and other molecules. A special protein called tau protein ensures the stability of the microtubules. Although it is not yet clear, it is believed that Aβ plaque formed outside the neuron initiates the pathway inside the neuron, which further activates the kinase, which is an enzyme responsible for transferring of the phosphate group to the tau proteins. The tau proteins then begin to alter their initial structure, and become detached from microtubules, aggregate with other tau proteins, and ultimately become tangled. This is recognized as a characteristic feature of AD NFTs. The signaling pathway is disrupted due to NFTs, which can lead to apoptosis. As the neurons die, large-scale changes begin to occur in the brain, including atrophying of the brain tissue, causing enlargement of the ventricles and the fluid-filled cavities. The gyri, which are characteristic ridges of the brain, are diminished, leading to widening of the sulci (grooves between the gyri). The comparison of a normal aged brain with that of an AD patient’s brain is shown in Fig. 2.

Comparison of an Alzheimer’s patient’s brain (left) with a normal aged brain (right)
Regardless of the specific AD philosophy of the amyloid hypothesis or tau theory, additional theories which are accepted worldwide include the following: (i) metal dysmetabolism and fluctuating signal transduction led by chemical factors, (ii) the resulting vascular disturbances triggered by deprived blood distribution in the cerebrum, (iii) pre-existing syndromes including diabetes mellitus, hypertension, and hypercholesteremia, (iv) genetic vulnerability caused by APP mutations, presenilin (PSEN1 and PSEN2) genes and the allelic deviations in apolipoprotein-E (APOE), (v) mitochondrial and immune deficiency, (vi) interruption in the synthesis of NGFs, (vii) environmental factors and immune mediators [12], and (viii) sleep deficiency, which enhances Aβ plaque accumulation [16], while efficient sleep stimulates soluble Aβ clearance [17]. APOE acts as a regulator of lipoprotein metabolism and is the strongest risk factor for LOAD. It is located on chromosome 19 and is responsible for encoding three common alleles (ε2, ε3, ε4), among which APOEε4 is associated with increased AD risk. This pathway is implicated in AD pathogenesis due to its dominant role in cholesterol metabolism. It is considered that one APOEε4 allele increases the risk of AD risk threefold, and two APOEε4 alleles increase AD risk 12-fold. APOEε4 is also known to be linked to a dose-dependent decrease in age at onset. Conversely, APOEε2 is found to be associated with a lower risk for AD and later age at onset [14]. Besides APOE and PSEN, genome-wide association studies (GWAS) have identified several other genes that contribute to risk for AD, including CASS4, CELF1, FERMT2, HLA-DRB5, INPP5D, MEF2C, NME8, PTK2B, SORL1, ZCWPW1, SLC24A4, CLU, PICALM, CR1, BIN1, MS4A, ABCA7, EPHA1, and CD2AP [18]. All of these genes are associated with relevant mechanisms and pathways, but await replication in large studies to further determine their efficacy as significant markers for AD [19]. Obesity and systemic inflammation may also interfere with immunological progression which results in the development of AD [20].
According to the latest genetic studies, immune-related genes increase the risk for AD [21], and involve neuroinflammation. The coding variants in the triggering receptor expressed on myeloid cells 2 (TREM2) gene confer the highest AD risk, thus indicating the high risk associated with microglial neuroinflammation in AD development. TREM2 is a type I transmembrane receptor expressed in a subset of myeloid lineage cells including microglia, dendritic cells, osteoclasts, monocytes, and tissue macrophages [22, 23]. Between homozygous mutations and heterozygous variants, the latter is associated with neurodegenerative syndromes such as late-onset AD [23, 24]. As the anti-inflammatory role of TREM2 is reported, it is suspected of interfering with the brain’s ability to prevent the buildup of plaque and thus promotes the development of AD [25].
Currently, the definitive diagnosis of AD is only possible postmortem, while enhanced techniques and criteria in the future may aid in early diagnosis. The only possible clinical diagnosis currently is a probable diagnosis which requires a detailed history of type and indications from the patient and neuropsychological assessment along with other tests [25]. An operationalized medical analysis with criteria including the NINCDS-ADRDA (National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association) has good sensitivity and specificity (>80%) for distinguishing patients with AD and individuals without dementia; however, the ability to distinguish between AD and other types of dementias is less precise (23–88%) [25].
Brain-Derived Neurotrophic Factor
The brain-derived neurotrophic factor, which is also referred to as abrineurin, is a member of the neurotrophin family of growth factors which also includes NGF, NT-3, and NT-4. Among these, BDNF is the only neurotrophin which is abundantly present in the mammalian brain [26]. Yves-Alain Barde and Hans Thoenen were the first neurobiologists to separate and isolate it from the pig brain in 1989, and subsequently its biochemical structure was elucidated [26]. Apart from the hippocampus, cortex, and basal forebrain, BDNF is also expressed in the retina, kidneys, prostate, motor neurons, and skeletal muscles [27]. It is synthesized in the endoplasmic reticulum (ER) [28] and is encoded by the BDNF gene located on chromosome sequence 11 in humans. There are eight different promoters that control BDNF transcription, and each one corresponds to different transcripts comprising one of eight untranslated 5′ exons (I–VIII) spliced to the 3′ encoding exon. Promoter IV action, regulating the translation of exon IV-comprising mRNA sequence, is strongly triggered by calcium ion and is chiefly controlled by a Cre regulatory factor, signifying recognized functions for the transcription factor CREB and the basis of BDNF's action-reliant events [29] . It affects the development and differentiation of the nerve cells along with modulation of both short- and long-lasting synaptic interactions. BDNF exists in two major forms, proBDNF and mature BDNF; the terminal domain of pro-BDNF is cleaved by a different protein convertase enzyme to develop into a biologically active mature BDNF [28]. BDNF operates on the nerve cells of the central and peripheral nervous system and is responsible for vital functions such as learning, memory, and higher thinking [30]. BDNF has several single-nucleotide polymorphisms (SNPs) including rs6265, C270T, rs7103411, rs2030324, rs2203877, rs2049045, and rs7124442, among which Val66Met (rs6265) is the most commonly studied SNP, since the Val66Met mutation results in the reduction of hippocampal tissue, which further causes learning and memory disorders [31] including AD and Parkinson’s, leading to neurodegeneration [32]. The outcomes of GWAS indicate an association of Val66Met and the promoter region of the BDNF gene C270T with increased risk of AD [33,34,35]. The regions affected by the underlying mechanisms causing decreased concentrations of BDNF mRNA in an AD brain are the basal forebrain, hippocampal region, and temporal and parietal cortices [36].
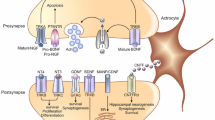
BDNF triggers the mechanism of action by binding to the cell surface of its two receptors, tyrosine kinase β (TrkB) and p75NTR, which are present in the cells and gray matter of the spinal cord [28] and are highly responsive to this growth factor Fig. 3. TrkB exists in two forms, full-length TrkB (TrkB-FL) and truncated TrkB. The promotion of survival and differentiation of basal forebrain cholinergic neurons is demonstrated by BDNF [37]. BDNF induces the release of acetylcholine (ACh) in the cholinergic neurons; ACh is a neurotransmitter which is impaired in AD patients. Several studies have found an association between BDNF and the pathogenesis of AD. Both BDNF and proBDNF are dramatically decreased during the final stage of AD [38]. BDNF is an exemplar marker of synaptic repair therapy, as it regulates all three aspects of synaptic physiology—protection, repair of existing synapses, and stimulation of new synapse formation—even during the presence of numerous toxins [32].

The binding of the TrkB receptor with BDNF induces cell survival, while the binding of p75NTR to proBDNF induces apoptosis
BDNF is widely known for its contribution to resilience and antidepressant action; it is a common genetic locus of risk for psychological diseases and is one of the most prominent molecules studied within psychiatry [35]. The role of BDNF supporting the survival and differentiation of nerve cells by fusing and initiating TrkB has also been well recognized. Alterations in BDNF have been widely identified by researchers around the globe in clinical conditions such as major depression and bipolar and anxiety disorders, and altered expression and impaired functioning of BDNF have been confirmed by many studies [6, 7, 39, 40]. BDNF is also a shared risk factor for susceptibility to those pathologies, by acting on biological mechanisms such as inflammation, neuroplasticity or hypothalamic–pituitary–adrenal axis functions which are all altered in these psychiatric disorders [6].
Effect of BDNF on Alzheimer’s Disease, Tyrosine Kinase B and Tau
The study of the cause and pathological processes of AD is still under investigation, while numerous postmortem studies have already suggested that BDNF levels in an AD brain are lower than those present in a normal human brain. Clearly, during AD, three of the seven transcripts of the BDNF gene are underexpressed [41]. However, studies which are described on the basis of the degree of pathology show contrary results, where early-stage AD is not necessarily associated with enduring and extremely severe conditions. During postmortem examination of subjects with early-stage AD, elevated TrkB expression in the hippocampus was observed, while BDNF expression remained unaltered [42]; however, in patients suffering from severe AD, the concentrations of both BDNF and TrkB were decreased in the cerebral cortex and hippocampal regions. Evidence of protection against the risk of AD later in life by higher levels of peripheral BDNF was cited by Weinstein et al. In addition, the risk for AD was lowered by 33% due to higher BDNF levels by one standard deviation [43].
The potential of the brain to learn and think is often altered in AD patients due to the disruption of BDNF in the blood and CNS. Peng et al. applied western blotting to determine the decrease in BDNF. The comparative levels of BDNF proteins in the parietal cortex of patients were analyzed during the experiment and were clinically categorized into three categories: (i) no cognitive impairment (NCI), (ii) mild cognitive impairment (MCI), and (iii) mild to moderate AD. The experimental results demonstrated a decrease in proBDNF by 21% and 30% in the AD and MCI groups, respectively, in comparison with the NCI group, in agreement with previous results indicating a 40% decrease during final-stage AD. Mature BDNF was observed to be decreased by 62% and 34 % in the AD and MCI groups, respectively. The notable reduction in both mature and proBDNF precede the decrease in choline acetyltransferase activity observed later in AD. The results indicated that a decrease in both types of BDNF occurs during the initial stage of AD progression and corresponds to impaired cognitive function, suggesting that both proBDNF and BDNF have a specific role in synaptic loss and cellular dysfunction underlying cognitive impairment in AD [38]. Apart from BDNF, two other signaling molecules implicated in neurodegeneration are glycogen synthase kinase-3β (GSK3β) and cAMP response element-binding protein (CREB) [44, 45]. The association of platelet GSK3β activity, lymphocyte CREB activity, and plasma BDNF with AD has been established. AD patients with depression were found to have increased GSK3 β and CREB activity. The increase in CREB was confirmed to correlate with the occurrence of depressive symptoms in patients or with relatively low BDNF levels in patients suffering from depression [46].
Tyrosine Kinase-B (TrkB) is a neurotrophin receptor vital for neuronal maturation and synaptic plasticity in the brain [47]. According to studies, during AD progression, distinct cholinergic neurons in the nucleus basalis (NB) exhibit substantial downregulation of TrkA, TrkB, and TrkC expression. The tangle-bearing neurons observe a large decline in TrkB-FL immunoreactivity; however, no change in the level is observed for p75NTR [48]. The binding and activation of BDNF with TrkB receptors that are present on both the presynaptic and the postsynaptic membrane initiates autophosphorylation and dimerization of tyrosine deposits in the intracellular region of the receptor and activates pathways that signal the cytoplasmic region including mitogen-activated protein kinase, phosphatidylinositol-3 kinase, and phospholipase C-γ consecutively [6, 47].
The neurotrophic deficiency in AD is the consequence of several mechanisms initiated by the Aβ peptide acting at different levels of the BDNF/TrkB signaling pathway. Aβ activity was found to induce the overproduction of calpain in the postmortem AD brain. Calpain is a calcium-activated protease that exists as an inactive proenzyme in the cytosol. (When the intracellular calcium level is overloaded, it initiates the conversion of the proenzyme to its active form. Activated calpain then cleaves cytoplasmic and nuclear substrates, leading to apoptosis). Activation of this calcium-activated protease through Aβ in neuronal cultures induced a decrease in TrkB-FL by cleavage with the adjacent receptor Shc docking site. This activity generates truncated TrkB-FL, which may act as a neurotrophin sink or prevailing negative receptor, and the intracellular fragment with the entire tyrosine kinase domain. Earlier it was proposed that the proteolytic fragments produced by Trk receptors may regulate cell functions such as transcription or survival/apoptosis equilibrium. Furthermore, Aβ may correspondingly induce the upregulation of truncated TrkB isoforms in AD via transcriptional processes. Selective TrkB pre-mRNA linking for generation of TrkB-Shc transcripts is increased via serine/arginine-rich splicing factor 3 (SRSF3), whose mRNA levels are amplified in AD and SH-SY5Y cells treated with Αβ filaments. The decline in BDNF concentration is ultimately the result of abnormal transcription, primarily because of CREB impairment in the hippocampal and frontal cortex of AD sufferers by overlying processes. Initially, this transcription factor is proteolyzed by calpain, resulting in a truncated protein with moderate action. Moreover, protein kinase A (PKA), which is a chief CREB controller, is interrupted in the cerebrum of AD patients via Aβ activity. The inhibition of PKA signaling and, consequently, CREB is attributed to calpain-dependent proteolysis of PKA RII subunits and downregulation of PKA O-GlcNAcylation. Similarly, Aβ reduces CREB activity by GSK3β overstimulation, which is supported by dual processes, reduced GSK3β suppressing phosphorylation of Ser9 through PKA and calpain proteolysis to yield a truncated GSK3β with augmented kinase activity. Ultimately, Aβ also lowers CREB activity by decreasing N-methyl-D-aspartate receptor (NMDAR) concentration and calpain-facilitated cleavage of DARPP-32, a chief inhibitor of PP1, the phosphatase which controls CREB dephosphorylation and deactivation [36].
Additional mechanisms significantly disrupting BDNF/TrkB signaling in AD are the elimination of MAPK/ERK and PI3K/Akt pathways by sublethal levels of Aβ, without hindering TrkB-FL and PLC-γ initiation, and the interruption of BDNF-induced TrkB endocytosis. The exposure to Aβ oligomer can damage receptor endocytosis and downstream Akt commencement by means of GSK3β-mediated dynamin 1 phosphorylation. In addition, the oligomers cause a deficit in BDNF-facilitated TrkB retreated handling by interrupting ubiquitin and calcium homeostasis. Finally, dysfunction of mitochondria which is caused by Aβ is a primary feature in AD related to discrepancies in BDNF axonal transport [36]. Figure 4 illustrates the dysfunction of BDNF/TrkB signaling and the various modifications in an AD brain.

Dysfunction of BDNF/TrkB signaling in AD. AD patients display decreased levels of BDNF in numerous areas of the brain due to reduced gene expression (a). The activation of GSK3β, which leads to tau hyperphosphorylation, emerges as a consequence of subsequent decline in neurotrophic signaling (b). By the process of transcription factor SRSF3, the expression of truncated TrkB isoforms is favored in the AD cerebrum (c). The activity of GSK3β and calpain, which cleaves the full-length TrkB (TrkB-FL) receptor near the receptor Shc docking site, is stimulated by the Aβ peptide. (d). CREB activity is inhibited by various mechanisms including a reduction in N-methyl-D-aspartate receptor (NMDAR) levels and (e) increase in PP1 action. f32, TrkB-FL calpain fragment of 32 kDa; P, pau-phosphorylation continues; tTrkB, calpain truncated TrkB-FL
Tau proteins are fragments abundantly present in the neurons of the CNS, and have primary roles such as stabilization of the microtubules in the axons. They may undergo various modifications under the pathological and physiological changes and stimulate neurodegenerative disorders such as AD [49]. It has been established that the NFTs in the brain of an AD patient are due to modified tau proteins, and the enhanced proliferation of tau is linked with disease progression. The accumulation of tau begins at the entorhinal cortex (EC) and the hippocampal region of the brain and prolongs disease progression. Studies have suggested that tau proteins can accumulate in the brain by oligomer “seeds” that travel across the synapse. The severity of the disease can be directly linked with the abnormal tau protein present in the brain. Rosa et al. demonstrated the effect of tau proteins on the levels of BDNF in animal and cellular models. Two types of transgenic mouse models of human tau expression and human tau (hTau40)-transfected human neuroblastoma (SH-SY5Y) cells were used to determine the effects of excess or pathologically modified wild-form human tau on BDNF expression. Notable downregulation of BDNF mRNA in comparison to controls was similarly observed in transgenic mouse models both with and without NFTs, along with hTau40-SH-SY5Y cells. Correspondingly, the overexpression of Aβ significantly downregulated BDNF expression in transgenic mice. However, the BDNF levels exhibited were similar to those in wild-type mice when crossed with tau knockout mice. Such findings reveal that excess or pathologically modified wild-type human tau is responsible for downregulation of BDNF. In addition, it was found that neither a mutation in tau nor the presence of NFTs is required for toxicity. These results suggest that amyloid-β-induced BDNF downregulation is at least partially mediated by tau. . Therefore, AD treatments targeting Aβ alone may not be effective without taking into account the influence of tau pathology on neurotrophic pathways [50].
BDNF as a Biomarker for Cognitive Reserve Against Alzheimer’s Disease
Disease biomarkers are biological modifications of particular characteristics that provide analytical or prognostic value about a disease, and also aid in predicting patient response to a particular treatment [51, 52]. As AD tends to progress slowly in the early years, the early detection of a biomarker for treatment or prevention of AD has become an urgent goal. An analysis in 2010 described the association of increased BDNF levels only with poorer visual and verbal memory among AD patients and not elderly controls [53]. It was also reported that the severity of disease was independent of increased concentrations of BDNF in AD patients [54]. Increased BDNF levels are found in the early stage of AD [55, 56]. In 2013, the Turkish researchers Gezen-Ak et. al. investigated the BDNF levels in patients using enzyme-linked immunosorbent assay (ELISA), which showed a decrease in the level of BDNF in the EOAD and LOAD groups in comparison with age-matched control individuals (EOAD: age of onset < 65; n = 22 and LOAD: age of onset > 65; n = 54) [57]. These findings were in contrast to those observed by O’Bryant et al., who found no such difference when patients with mild to moderate AD were compared with controls [58]. In 2016, Buchman and collaborators investigated the association between BDNF levels and cognitive decline in the dorsolateral prefrontal cortex (DLPFC) in a group of 535 elderly patients followed for cognitive decline and dementia over a 6-year period and brain autopsy at death. The strongest association of BDNF with cognitive decline was found among those with high levels of AD neuropathology, with 50% slower decline in the 90th versus the 10th percentile of BDNF expression. The correlation was greatest in people with dementia. This decrease in BDNF levels in individuals with pathologically confirmed AD was not associated with macroscopic infarcts, Lewy body disease, or hippocampal sclerosis. The association between BDNF expression and cognitive decline remained in a model adjusting for age, gender, education and neuropathology. The effect of AD pathology on cognitive impairment was also associated with BDNF expression level, with the strongest effect related to high levels of AD pathology. In individuals with the highest AD pathology (90th percentile), cognitive decline was around 40% slower with 90th vs. 10th percentile BDNF expression. Consequently, this study highlights the role of BDNF as a potential substantial contributor to slowing cognitive decline in the elderly, especially in the setting of progressive AD neuropathology [59].
While BDNF levels have shown differing effects among various AD patients in different studies, with no changes in levels observed in some, it is believed that this may be at least partly due to heterogeneity in patient enrollment standards and methodological biases [60]. It is also assumed that the increased BDNF expression during the initial stage is a compensatory mechanism [61]. Another pathological marker of the AD cerebrum that is strongly related to AD pathogenesis is synaptic loss [62, 63]. Because BDNF is involved in the regulation in neurite outgrowth and synaptic plasticity, a decline in BDNF concentration is also involved in its pathophysiology [39]. However, in contrast to neuronal loss, synaptic dysfunction and synapse loss are mutable, highly dynamic, and plastic [39, 64, 65]. BDNF may serve as a marker specifically associated with the presence and/or progression of the mnemonic symptoms which are common to other pathological conditions sharing alterations in this cognitive domain.
In addition to BDNF, cerebrospinal fluid (CSF) biomarkers are also used to enhance the clinical analysis of AD [66, 67]. CSF is a clear body fluid located within the tissues surrounding the brain and spinal cord of all vertebrates. It behaves as a “liquid cushion” that provides simple mechanical and immunological protection to the brain within the skull, and it can be obtained via lumbar puncture [68, 69]. Various studies of CSF include the isoforms of Aβ peptides and phosphorylation epitopes of the tau protein. A systematic review found that AD could be differentiated from other dementias by the detection of lower concentrations of Aβ1–42 and higher concentrations of total tau or hyperphosphorylated tau at threonine 231 and 181 in comparison with age-matched controls. These results were established in a detailed critical analysis showing sensitivity of 83% and specificity of 72%. Furthermore, the results of a longitudinal study showed that amalgamation of CSF (total tau, hyperphosphorylated tau, and Aβ1–42) at baseline exhibited sensitivity of 95% and specificity of 83% for the detection of early AD in patients with MCI [25, 70]. Nevertheless, there still exists a high degree of inconsistency among CSF studies, and standardized analytical practices are needed [71]. Early diagnosis of AD, where correct diagnosis is most challenging, could be performed by developing a group of CSF biomarkers that might be used to generate AD-specific biomarker data. The combined use of CSF biomarkers for Aβ1–42, tau, and phosphorylated tau with innovative markers such as Aβ1–38 has improved diagnostic accuracy [25, 72]. Additional potential candidate biomarkers include markers of inflammation and oxidative stress.
BDNF is also indicated as a common factor in the majority of the effective treatments for these mnemonic symptoms [73]. Initially, the role of BDNF as a disease biomarker for neurodegenerative/neuropsychiatric disorders was rejected for various reasons: first, fluctuations in BDNF levels are found in other pathological conditions, thus raising questions regarding its discriminatory capability and effectiveness; secondly, contradictory results have been obtained from numerous studies; and finally, considering the mutual dysregulation in various pathological conditions, it could be assumed that BDNF may be a biomarker specifically associated with the development and progression of mnemonic symptoms common in many disorder and is related to pathologically shared deficits in this cognitive domain. Taking the above into consideration, it can be concluded that in today’s world, BDNF alone cannot be utilized as a specific and reliable biomarker for AD.
BDNF as a Therapeutic Treatment in Alzheimer’s Disease
Several potential treatments for other neurodegenerative disorders have been used for treatment for AD, but with limited success. Therefore, potential disease-modifying treatment for AD is still under extensive study, and symptomatic treatments are currently recommended for the disease. One such symptomatic therapy used extensively is to increase BDNF levels to delay the progression of AD in the cerebrum of AD patients. Preclinical studies have proposed various potential pharmacological therapies to increase BDNF levels in transgenic mice [74, 75]. BDNF levels were enhanced when caffeine was administered in AD transgenic mice, and cognitive impairment was also ameliorated during chronic treatment with caffeine. A phase 2 study suggested that pioglitazone, a hypoglycemic drug previously indicated for diabetes, is safe and well-tolerated. It also demonstrated restoration of BDNF concentrations and attenuation of inflammatory markers in an animal model of AD stimulated by administration of Aβ peptide. Fingolimod showed a significant increase in the levels of BDNF and improvement in cognitive impairment in animals when administered via an oral route [76,77,78]. To prevent viral distribution of BDNF into the brain, alternative therapeutic approaches have been proposed for transplantation of neurotrophin-releasing cells [79,80,81]. In recent years, stem cell-based therapy has also shown effectiveness for treatment of AD, as neuronal loss is believed to be the primary cause of cognitive deficit in AD. Mesenchymal stem cells and NSCs are two core cell types that are analyzed for AD, but unfortunately the goal of substituting the damaged nerve cells in the AD cerebrum has yet to be attained. Engrafted mesenchymal stem cells aid in Aβ clearance and inflammation modulation, while engrafted NSCs assist primarily due to BDNF supplementation, although these remain very capable of differentiating into neuronal cells (40). It should be emphasized that these approaches carry a risk of rejection and can cause accidental tumor growth. Another technique for treatment of CNS lesions has employed non-viral vectors for transfer of genes encoding for BDNF and NGF [79, 82].
Other treatments that trigger an increase in BDNF levels include acetylcholinesterase (AChE) inhibitors [83] and antidepressants [84]. AChE inhibitors such as donepezil and galantamine have been successful in treating early AD [85]. However, several conflicting findings were reported. For instance, donepezil is a selective and reversible inhibitor of AChE by increasing the concentration of existing acetylcholine, which compensates for the damage in the functioning of the cholinergic brain cells, and in AD patients chronic medication with donepezil may reverse decreased BDNF concentrations [77]. However, chronic treatment with galantamine (3 mg/kg, intraperitoneally, 14 days) did not induce a modification of hippocampal BDNF concentration in rats, i.e., galantamine failed to have a lasting effect on BDNF concentrations [86]. In cases where the concentration of BDNF is amplified, benzodiazepines are used to reduce levels [87,88,89]. Sakr et al. effectively discovered encouraging promising correlation between central acetylcholine and BDNF expression concentration in the hippocampal area of a rat model for determination of vascular dementia [90] i.e., elevation in acetylcholine concentration observed in the CNS caused by AChE inhibitors might induce an increase in BDNF expression. Considering that one of the major risk factors for AD is depression, antidepressive treatments are believed to contribute in delaying AD progression by targeting BDNF concentrations; therefore, antidepressants such as SSRIs and lithium [91] are often prescribed [84, 85]. Drugs like venlafaxine and valproic acid, which is also well known as a mood stabilizer, have shown positive effects on BDNF [92, 93]. In addition to psychoactive drugs, lipid-lowering drugs [94, 95] and antidiabetics [96] are also extensively prescribed to the elderly and affect BDNF levels [97].
One of the strategies to deliver BDNF in the patient includes nanotechnology. Soft nanotechnology is the process of designing, engineering, and manipulating soft substances and developing structures such as nanoparticles (drug carriers) and devices at nanometer scale. The CNS is the target for drug transport mechanisms by commonly known nanoparticle systems [79, 98, 99]. Various nanocarriers, including dendrimers, nanospheres, solid lipid nanoparticles (SLN), nano-emulsions, polymeric micelles, multifunctional nanoparticles (NPs), and nanoscale systems for imaging [79, 100, 101] are used for mnemonic purposes among people under normal and pathological conditions. Importantly, memory impairment is found to correlate with measurable blood protein concentration in various disorders. The variations found in the transfer and release of BDNF due to the Val66Met polymorphism has also been acknowledged in studies exploring the controlled release of neurotrophins. In searching for innovative therapeutic approaches to treat neurodegenerative syndromes, colloidal nanoparticle carriers have been designed as reservoirs for neurotrophins, ensuring their protection against enzymatic degradation and other destructive stressors. Indeed, the walls of the nanoparticle containers (such as lipid membrane-type vehicles, nanocapsules, and nanospheres) can completely isolate the therapeutic substances from the environment. Hydrophilic polymers (polyethylene glycol) or albumin are used to coat colloidal nanocarriers in order to moderate their opsonization and enhance their in vivo circulation time. Importantly, neurotrophin encapsulation in NPs may aid in the local administration of neuroprotective agents in a concentrated state to target sites, while minimizing subsequent side effects and toxicity. NP delivery systems also offer the prospect of combining different types of active molecules in one nanocarrier. The surface of the nanocarriers can also be modified by anchoring of specific ligands for receptor recognition, and thus targeted delivery can be achieved [79, 102, 103].
Exercise, chronic administration of fluoxetine, and cognitive training are also effective in increasing BDNF levels [73]. Fluoxetine is a selective serotonin reuptake inhibitor (SSRI) antidepressant, which may be effective in reducing depression and anxiety among AD patients. The role of exercise is illustrated by the flowchart in Fig. 5.

Exercise decreasing the risk of AD. Exercise improves vascular health and increases the level of BDNF in the brain, which stimulates neurogenesis, survival of new neurons, and the development of new synaptic connections
A study revealed that factors including synaptic loss and neuronal atrophy controlled neuronal stimulus were all reversed, and the associated mnemonic shortfalls devoid of deviations in the amyloid plaque load were reduced in an amyloid-transgenic mouse due to BDNF delivery to its entorhinal cortex region [97]. This study signifies the potential of BDNF to exert its protective effect by acting through amyloid-independent mechanisms. Moreover, a few drugs are known to reverse memory impairment in animal models of AD or even in preclinical trials, which include 7,8-dihydroxyflavone (7,8 DHF), neotrofin, and neuropep-1 [64, 104,105,106]. 7,8-DHF is an effective small molecule TrkB agonist, which shows high therapeutic value. Additionally, in order to overcome preclinical complications like low oral bioavailability and moderate pharmacokinetic profile, it is suggested to incorporate a prodrug strategy to enhance its oral bioavailability and cerebral exposure, and subsequently it was found that the ideal prodrug R13 demonstrated satisfactory properties along with dose-dependent reversal of cognitive deficiencies in an AD mouse model [107].Neuropep-1 is a novel tripeptide which induces a substantial increase in BDNF mRNA and protein concentration in H19-7 cells, and it was found that this drug could enhance spatial learning and memory (SLM) by stimulating the BDNF/TrkB signaling in the hippocampus of rats [108]. Thus, the protective effect of exercise and diet against neuronal deterioration is facilitated by BDNF [109]. This highlights the need to acquire observable interventions that would prevent the development of dementia or decrease the progression of dementia in patients, a pathway which is presently under study. Such a novel pathway can be primarily achieved by slight lifestyle modifications, antioxidant nutrition, environmental fortification and public interventions to physical or cognitive training as a few of the effective interventions [73].
Conclusion
Substantial evidence supports the important role of BDNF in AD. Changes in the levels of BDNF observed in the brains of AD patients are associated with moderate to severe stages of senile dementia, although there are exceptional cases where the BDNF level remains unchanged. Early-stage AD is marked by the disruption of BDNF signaling through TrkB. Postmortem brain tissue has shown elevated expression of BDNF in the hippocampal and the amygdala regions, which are responsible for memory and emotional functions. Despite the ambiguous findings regarding the association between memory and BDNF, numerous studies have attempted to establish a causal relationship between the two factors by investigating memory functions under specific conditions of upregulation or downregulation of BDNF expression. The association between cerebral BDNF and BDNF concentration has been extensively investigated to determine the significance of BDNF levels with respect to mnemonic performance and anatomical variations in memory-associated areas in both fit and unhealthy individuals. The current symptomatic treatments that target the increase in BDNF levels may be useful for delaying disease progression. In addition, exercise, chronic administration of fluoxetine, and cognitive training is reported to increase BDNF concentration and is associated with improved performance in memory tasks. As discussed earlier, BDNF is responsible for the growth, maturation, and maintenance of nerve cells present in the brain and thus controls functions such as memory, learning, higher thinking, and speech. Consequently, a correlational relationship between memory and BDNF has been inferred. However, because existing practice does not permit the regulation of BDNF expression in humans, in order to understand the dynamics and possible role of BDNF as a surrogate marker of disease progression in individuals with AD, further studies are needed.
Future Perspectives
As life expectancy continues to increase, age-related diseases are of increasing concern. Based on the various scientific studies and experimental findings, the prevalence of AD is predicted to grow exponentially among the elderly in the coming years, and therefore finding an exact treatment for this disease is extremely important. The ambiguous nature of BDNF in AD currently prevents its role as a target therapy or a biomarker. Therefore, increased effort in clinical research focusing on BDNF, including the use of imaging to elucidate the temporal relationship of BDNF in relation to cognitive scores and brain anatomical, functional, and metabolic changes, would be invaluable in achieving deeper insights in this area [110]. As AD pathology is associated with multiple mechanisms, AD therapy must target multiple components of the pathophysiological cascade. The treatment should act on the processing of Aβ, the abnormal accumulation of Aβ, and hyperphosphorylation of tau combined with a neuroprotective neurotrophic factor-like effect [111].
Abbreviations
- Aβ :
-
Amyloid Beta
- AD :
-
Alzheimer’s disease
- APP :
-
Amyloid precursor protein
- BDNF :
-
Brain-derived neurotrophic factor
- NFTs :
-
Neurofibrillary tangles
- NGFs:
-
Nerve growth factors
- TrkB :
-
Tyrosine kinase B
References
Jiao SS, Shen LL, Zhu C et al (2016) Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl Psychiatry. 6(10):e907
Ye X, Tai W, Zhang D (2012) The early events of Alzheimer’s disease pathology: from mitochondrial dysfunction to BDNF axonal transport deficits. Neurobiol Aging. 33(6):1122-e1
Herrmann N, Chau SA, Kircanski I, Lanctot KL (2011) Current and emerging drug treatment options for Alzheimer’s disease. Drugs. 71(15):2031–65
Wang ZH, Xiang J, Liu X et al (2019) Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates δ-Secretase by Upregulating C/EBPβ in Alzheimer’s Disease. Cell Rep. 28(3):655–69
Ninan I (2014) Synaptic regulation of affective behaviors; role of BDNF. Neuropharmacology. 1(76):684–95
Cattaneo A, Cattane N, Begni V, Pariante CM, Riva MA (2016) The human BDNF gene: peripheral gene expression and protein levels as biomarkers for psychiatric disorders. Transl Psychiatry. 6(11):e958
Autry AE, Monteggia LM (2012) Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev. 64(2):238–58
Genius J, Klafki H, Benninghoff J, Esselmann H, Wiltfang J (2012) Current application of neurochemical biomarkers in the prediction and differential diagnosis of Alzheimer’s disease and other neurodegenerative dementias. Eur Arch Psychiatry Clin Neurosci. 262(2):71–7
Fu L, Doreswamy V, Prakash R (2014) The biochemical pathways of central nervous system neural degeneration in niacin deficiency. Neural Regen Res. 9(16):1509
Denham J, Marques FZ, O’Brien BJ, Charchar FJ (2014) Exercise: putting action into our epigenome. Sports Med. 44(2):189–209
Szuhany KL, Bugatti M, Otto MW (2015) A meta-analytic review of the effects of exercise on brain-derived neurotrophic factor. J Psychiatric Res. 1(60):56–64
Bachurin SO, Bovina EV, Ustyugov AA (2017) Drugs in clinical trials for Alzheimer’s disease: the major trends. Med Res Rev. 37(5):1189–225
Broadstock M, Ballard C, Corbett A (2014) Latest treatment options for Alzheimer’s disease, Parkinson’s disease dementia and dementia with Lewy bodies. Expert Opin Pharmacother. 15(13):1797–810
Karch CM, Goate AM (2015) Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 77(1):43–51
Chen X, Yan SD (2006) Mitochondrial Aβ A potential cause of metabolic dysfunction in Alzheimer’s disease. IUBMB Life. 58(12):686–94
Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR et al (2009) Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science. 326(5955):1005–7
Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M et al (2013) Sleep drives metabolite clearance from the adult brain. Science. 342(6156):373–7
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C et al (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature Genet. 45(12):1452–8
Waring SC, Rosenberg RN (2008) Genome-Wide Association Studies in Alzheimer Disease. Arch Neurol. 65(3):329–34
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL et al (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14(4):388–405
Karch CM, Goate AM (2015) Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 77(1):43–51
Hickman SE, El Khoury J (2014) TREM2 and the neuroimmunology of Alzheimer's disease. Biochem Pharmacol 88(4):495–8
Katsumoto A, Takeuchi H, Takahashi K, Tanaka F (2018) Microglia in Alzheimer’s disease: risk factors and inflammation. Front Neurol. 15(9):978
Guerreiro R, Wojtas A, Bras JC, Carrasquillo M, Rogaeva E, Majounie E et al (2013) TREM2 deficiency alters acute macrophage distribution and improves recovery after TBI. N Engl J Med 117–27
Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E (2011) Alzheimer’s disease. Lancet (London, England). 377(9770):1091–31
Kowiański P, Lietzau G, Czuba E, Waśkow M, Steliga A, Moryś J (2018) BDNF: a key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol Neurobiol. 38(3):579–93
Mandel AL, Ozdener H, Utermohlen V (2009) Identification of pro-and mature brain-derived neurotrophic factor in human saliva. Arch Oral Biol 54(7):689–95
Qiu LL, Pan W, Luo D et al (2020) Dysregulation of BDNF/TrkB signaling mediated by NMDAR/Ca 2+/calpain might contribute to postoperative cognitive dysfunction in aging mice. J Neuroinflammation. 17(1):1–5
Zheng F, Wang H (2009) NMDA-mediated and self-induced bdnf exon IV transcriptions are differentially regulated in cultured cortical neurons. Neurochem Int. 54(5–6):385–92
Yamada K, Nabeshima T (2003) Brain-derived neurotrophic factor/TrkB signaling in memory processes. J Pharm Sci. 91(4):267–70
Bath KG, Lee FS (2006) Variant BDNF (Val66Met) impact on brain structure and function. Cogn Affect Behav Neurosci. 6(1):79–85
Lu B, Nagappan G, Guan X, Nathan PJ, Wren P (2013) BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci. 14(6):401–16
Tsai SJ, Hong CJ, Liu HC, Liu TY, Hsu LE, Lin CH (2004) Association analysis of brain-derived neurotrophic factor Val66Met polymorphisms with Alzheimer’s disease and age of onset. Neuropsychobiology. 49(1):10–2
Tsai SJ, Hong CJ, Liu HC, Liu TY, Liou YJ (2006) The brain-derived neurotrophic factor gene as a possible susceptibility candidate for Alzheimer’s disease in a chinese population. Dement Geriatr Cogn Disord. 21(3):139–43
Amidfar M, de Oliveira J, Kucharska E, Budni J, Kim YK (2020) CREB and BDNF: Neurobiology and treatment of Alzheimer’s disease. Life Sci. 27:118020
Tejeda GS, Díaz-Guerra M (2017) Integral characterization of defective BDNF/TrkB signalling in neurological and psychiatric disorders leads the way to new therapies. Int J Mol Sci. 18(2):268
Fahnestock M, Garzon D, Holsinger RM, Michalski B (2002) Neurotrophic factors and Alzheimer’s disease: are we focusing on the wrong molecule? In Ageing and Dementia Current and Future Concepts. Springer, Vienna, pp 241–252
Peng S, Wuu J, Mufson EJ, Fahnestock M (2005) Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J Neurochem. 93(6):1412–21
Wu CC, Lien CC, Hou WH, Chiang PM, Tsai KJ (2016) Gain of BDNF function in engrafted neural stem cells promotes the therapeutic potential for Alzheimer’s disease. Sci Rep 6(27358)
Mitchelmore C, Gede L (2014) Brain derived neurotrophic factor: epigenetic regulation in psychiatric disorders. Brain Res. 24(162–72):1586
Pramanik S, Sulistio YA, Heese K (2017) Neurotrophin signalling and stem cells-implications for neurodegenerative diseases and stem cell therapy. Mol Biol. 54(9):7401–59
Kao PF, Banigan MG, Vanderburg CR et al (2012) Increased expression of TrkB and Capzb2 accompanies preserved cognitive status in early Alzheimer disease pathology. J Neuropathy Exp Neurol 71(7):654–64
Weinstein G, Beiser AS, Choi SH et al (2014) Serum brain-derived neurotrophic factor and the risk for dementia: the Framingham Heart Study. JAMA Neurol 71(1):55–61
Bitner RS (2012) Cyclic AMP response element-binding protein (CREB) phosphorylation: a mechanistic marker in the development of memory enhancing Alzheimer’s disease therapeutics. Biochem Pharmacol. 83(6):705–14
Hu YS, Long N, Pigino G, Brady ST, Lazarov O (2013) Molecular mechanisms of environmental enrichment: impairments in Akt/GSK3β, neurotrophin-3 and CREB signaling. PloS One. 8(5):e64460
Pláteník J, Fišar Z, Buchal R et al (2014) GSK3β, CREB, and BDNF in peripheral blood of patients with Alzheimer’s disease and depression. Prog Neuro-Psychopharmacol Biol Psychiatry. 3(50):83–93
Yoshii A, Constantine-Paton M (2010) Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol. 70(5):304–22
Zhang F, Kang Z, Li W, Xiao Z, Zhou X (2012) Roles of brain-derived neurotrophic factor/tropomyosin-related kinase B (BDNF/TrkB) signalling in Alzheimer’s disease. J Clin Neurosci. 19(7):946–9
Chen Q, Zhou Z, Zhang L et al (2012) Tau protein is involved in morphological plasticity in hippocampal neurons in response to BDNF. Neurochem Int. 60(3):233–42
Rosa E, Mahendram S, Ke YD, Ittner LM, Ginsberg SD, Fahnestock M (2016) Tau downregulates BDNF expression in animal and cellular models of Alzheimer's disease. Neurobiol Aging 135–42
Davis J, Maes M, Andreazza A, McGrath JJ, Tye SJ, Berk M (2015) Towards a classification of biomarkers of neuropsychiatric disease: from encompass to compass. Mol Psychiatry. 20(2):152–3
Pillai A, Buckley PF (2012) Reliable biomarkers and predictors of schizophrenia and its treatment. Psychiatric Clin. 35(3):645–59
O’Bryant SE, Hobson VL, Hall JR et al (2011) Serum brain-derived neurotrophic factor levels are specifically associated with memory performance among Alzheimer’s disease cases. Dement Geriatr Cogn Disord. 31(6):31–6
Angelucci F, Spalletta G, Iulio FD et al (2010) Alzheimer’s disease (AD) and Mild Cognitive Impairment (MCI) patients are characterized by increased BDNF serum levels. Curr Alzheimer Res. 7(1):15–20
Laske C, Stransky E, Leyhe T et al (2006) Stage-dependent BDNF serum concentrations in Alzheimer’s disease. J Neural Transm. 113(9):1217–24
Winblad B, Palmer K, Kivipelto M et al (2004) Mild cognitive impairment–beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med. 256(3):240–6
Gezen-Ak D, Dursun E, Hanağası H et al (2013) BDNF, TNFα, HSP90, CFH, and IL-10 serum levels in patients with early or late onset Alzheimer’s disease or mild cognitive impairment. J Alzheimer’s Dis. 37(1):185–95
O’Bryant SE, Hobson V, Hall JR et al (2009) Brain-derived neurotrophic factor levels in Alzheimer’s disease. J Alzheimer’s Dis. 17(2):337–41
Buchman AS, Yu L, Boyle PA, Schneider JA, De Jager PL, Bennett DA (2016) Higher brain BDNF gene expression is associated with slower cognitive decline in older adults. Neurology. 86(8):735–41
Balietti M (2020) Blood brain-derived neurotrophic factor as a biomarker of Alzheimer's disease. In Diagnosis and Management in Dementia. Academic Press: 281–296
Diniz BS, Teixeira AL (2011) Brain-derived neurotrophic factor and Alzheimer’s disease: physiopathology and beyond. Neuromol Med. 13(4):217–22
Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM (2004) Synaptic changes in Alzheimer’s disease: increased amyloid-β and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 165(5):1809–17
Lee L, Dale E, Staniszewski A et al (2014) Regulation of synaptic plasticity and cognition by SUMO in normal physiology and Alzheimer’s disease. Sci Rep. 2(4):7190
Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature. 429(6993):761–6
McDole B, Isgor C, Pare C, Guthrie K (2015) BDNF over-expression increases olfactory bulb granule cell dendritic spine density in vivo. Neuroscience. 24(304):146–60
Somers C, Struyfs H, Goossens J, Niemantsverdriet E, Luyckx J, De Roeck N et al (2016) A decade of cerebrospinal fluid biomarkers for Alzheimer’s disease in Belgium. J Alzheimer’s Dis. 54(1):383–95
Goossens J, Bjerke M, Struyfs H, Niemantsverdriet E, Somers C, Van den Bossche T et al (2017) No added diagnostic value of non-phosphorylated tau fraction (p-tau rel) in CSF as a biomarker for differential dementia diagnosis. Alzheimer’s Res Ther. 9(1):1–7
Anoop A, Singh PK, Jacob RS, Maji SK (2010) CSF biomarkers for Alzheimer’s disease diagnosis. Int J Alzheimer’s Dis. 23:2010
Blennow K, Hampel H, Weiner M, Zetterberg H (2010) Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 6(3):131–44
Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L (2006) Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 5(3):228–34
Zetterberg H (2006) Neurological Biomarkers. Rev Cell Biol Mol Med. 2(1):62–75
Welge V, Fiege O, Lewczuk P, Mollenhauer B, Esselmann H, Klafki HW et al (2009) Combined CSF tau, p-tau181 and amyloid-β 38/40/42 for diagnosing Alzheimer’s disease. J Neural Transm. 1(11):203–12
Miranda M, Morici JF, Zanoni MB, Bekinschtein P (2019) Brain-derived neurotrophic factor: a key molecule for memory in the healthy and the pathological brain. Front Cell Neurosci. 13:363
Hsiao YH, Hung HC, Chen SH, Gean PW (2014) Social interaction rescues memory deficit in an animal model of Alzheimer’s disease by increasing BDNF-dependent hippocampal neurogenesis. J Neurosci. 34(49):16207–19
Han K, Jia N, Li J, Yang L, Min LQ (2013) Chronic caffeine treatment reverses memory impairment and the expression of brain BNDF and TrkB in the PS1/APP double transgenic mouse model of Alzheimer’s disease. Mol Med Rep. 8(3):737–40
Prakash A, Kumar A (2014) Role of nuclear receptor on regulation of BDNF and neuroinflammation in hippocampus of β-amyloid animal model of Alzheimer’s disease. Neurotox Res. 25(4):335–47
Fukumoto K, Mizoguchi H, Takeuchi H et al (2014) Fingolimod increases brain-derived neurotrophic factor levels and ameliorates amyloid β-induced memory impairment. Behav Brain Res. 15(268):88–93
Budni J, Bellettini-Santos T, Mina F, Garcez ML, Zugno AI (2015) The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. 6(5):331
Géral C, Angelova A, Lesieur S (2013) From molecular to nanotechnology strategies for delivery of neurotrophins: emphasis on brain-derived neurotrophic factor (BDNF). Pharmaceutics. 5(1):127–67
Wahlberg LU, Lind G, Almqvist PM, Kusk P, Tornøe J, Juliusson B, Söderman M, Selldén E, Seiger Å, Eriksdotter-Jönhagen M, Linderoth B (2012) Targeted delivery of nerve growth factor via encapsulated cell biodelivery in Alzheimer disease: a technology platform for restorative neurosurgery. J Neurosurg. 117(2):340–7
Lu P, Jones LL, Tuszynski MH (2005) BDNF-expressing marrow stromal cells support extensive axonal growth at sites of spinal cord injury. Exp Neurol. 191(2):344–60
Lu KW, Chen ZY, Jin DD, Hou TS, Cao L, Fu Q (2002) Cationic liposome-mediated GDNF gene transfer after spinal cord injury. J Neurotrauma. 19(9):1081–90
Leyhe T, Stransky E, Eschweiler GW, Buchkremer G, Laske C (2008) Increase of BDNF serum concentration during donepezil treatment of patients with early Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 258(2):124–8
Polyakova M, Stuke K, Schuemberg K, Mueller K, Schoenknecht P, Schroeter ML (2015) BDNF as a biomarker for successful treatment of mood disorders: a systematic & quantitative meta-analysis. J Affect Disord. 15(174):432–40
Song JH, Yu JT, Tan L (2015) Brain-derived neurotrophic factor in Alzheimer’s disease: risk, mechanisms, and therapy. Mol Neurobiol. 52(3):1477–93
Autio H, Mätlik K, Rantamäki T et al (2011) Acetylcholinesterase inhibitors rapidly activate Trk neurotrophin receptors in the mouse hippocampus. Neuropharmacology. 61(8):1291–6
Ventriglia M, Zanardini R, Bonomini C, Zanetti O, Volpe D, Pasqualetti P et al (2013) Serum brain-derived neurotrophic factor levels in different neurological diseases. BioMed Res Int
Huang TL, Hung YY (2009) Lorazepam reduces the serum brain-derived neurotrophic factor level in schizophrenia patients with catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 1(33):158–9
Balietti M, Giuli C, Conti F (2018) Peripheral blood brain-derived neurotrophic factor as a biomarker of Alzheimer’s disease: are there methodological biases? Mol Neurobiol. 55(8):6661–72
Sakr HF, Khalil KI, Hussein AM, Zaki MS, Eid RA, Alkhateeb M (2014) Effect of dehydroepiandrosterone (DHEA) on memory and brain derived neurotrophic factor (BDNF) in a rat model of vascular dementia. J Physiol Pharmacol. 65(1):41–53
Watanabe K, Hashimoto E, Ukai W et al (2010) Effect of antidepressants on brain-derived neurotrophic factor (BDNF) release from platelets in the rats. Prog Neuro-Psychopharmacol Biol Psychiatry. 34(8):1450–4
Calabrese F, Luoni A, Guidotti G, Racagni G, Fumagalli F, Riva MA (2013) Modulation of neuronal plasticity following chronic concomitant administration of the novel antipsychotic lurasidone with the mood stabilizer valproic acid. Psychopharmacology. 226(1):101–12
Martisova E, Aisa B, Guereñu G, Javier Ramirez M (2013) Effects of early maternal separation on biobehavioral and neuropathological markers of Alzheimer’s disease in adult male rats. Curr Alzheimer Res. 10(4):420–32
Weinstein G, R Preis S, S Beiser A et al (2017) Clinical and environmental correlates of serum BDNF: a descriptive study with plausible implications for AD research. Curr Alzheimer Res 14(7):722–30
Zhang J, Mu X, Breker DA, Li Y, Gao Z, Huang Y (2017) Atorvastatin treatment is associated with increased BDNF level and improved functional recovery after atherothrombotic stroke. Int J Neurosci. 127(1):92–7
Allard JS, Perez EJ, Fukui K, Carpenter P, Ingram DK, de Cabo R (2016) Prolonged metformin treatment leads to reduced transcription of Nrf2 and neurotrophic factors without cognitive impairment in older C57BL/6J mice. Behav Brain Res. 15(301):1–9
Nagahara AH, Merrill DA, Coppola G et al (2009) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 15(3):331–7
Begley DJ (2004) Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacol Ther. 104(1):29–45
Craparo EF, Bondì ML, Pitarresi G, Cavallaro G (2011) Nanoparticulate systems for drug delivery and targeting to the central nervous system. CNS Neurosci Ther. 17(6):670–7
Petros RA, DeSimone JM (2010) Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 9(8):615–27
Zhang S, Uludağ H (2009) Nanoparticulate systems for growth factor delivery. Pharmaceutical research. 26(7):1561
Xie Y, Ye L, Zhang X, Cui W, Lou J, Nagai T, Hou X (2005) Transport of nerve growth factor encapsulated into liposomes across the blood–brain barrier: in vitro and in vivo studies. J Control Release. 105(1–2):106–19
Pang Z, Lu W, Gao H, Hu K, Chen J, Zhang C, Gao X, Jiang X, Zhu C (2008) Preparation and brain delivery property of biodegradable polymersomes conjugated with OX26. J Control Release. 128(2):120–7
Glasky AJ, Melchior CL, Pirzadeh B, Heydari N, Ritzmann RF (1994) Effect of AIT-082, a purine analog, on working memory in normal and aged mice. Pharmacol Biochem Behav. 47(2):325–9
Devi L, Ohno M (2012) 7, 8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 37(2):434–44
Shin MK, Kim HG, Baek SH et al (2014) Neuropep-1 ameliorates learning and memory deficits in an Alzheimer’s disease mouse model, increases brain-derived neurotrophic factor expression in the brain, and causes reduction of amyloid beta plaques. Neurobiol Aging. 35(5):990–1001
Chen C, Wang Z, Zhang Z et al (2018) The prodrug of 7, 8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proc Natl Acad Sci. 115(3):578–83
Shin MK, Kim HG, Kim KL (2011) A novel trimeric peptide, Neuropep-1-stimulating brain-derived neurotrophic factor expression in rat brain improves spatial learning and memory as measured by the Y-maze and Morris water maze. J Neurochem. 116(2):205–16
Vaynman S, Ying Z, Gomez-Pinilla F (2004) Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur J Neurosci. 20(10):2580–90
Ng TK, Ho CS, Tam WW, Kua EH, Ho RC (2019) Decreased serum brain-derived neurotrophic factor (BDNF) levels in patients with Alzheimer’s disease (AD): a systematic review and meta-analysis. Int J Mol Sci. 20(2):257
Ubhi K, Masliah E (2013) Alzheimer’s disease: recent advances and future perspectives. J Alzheimer’s Dis. 33(s1):S185-94
Acknowledgements
The authors would like to thank Chitkara College of Pharmacy, Chitkara University, Punjab, India for providing the basic facilities for completion of the current article.
Funding
The present review article did not receive any funding.
Author information
Authors and Affiliations
Contributions
P.G. and T.B.: Conceived the idea and wrote the first draft; A.S.: Figure work; S.S.: Data compilation; S.B.: Proofreading.
Corresponding author
Ethics declarations
Consent for Publication
All the authors have given consent for publication.
Competing Interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Girotra, P., Behl, T., Sehgal, A. et al. Investigation of the Molecular Role of Brain-Derived Neurotrophic Factor in Alzheimer’s Disease. J Mol Neurosci 72, 173–186 (2022). https://doi.org/10.1007/s12031-021-01824-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-021-01824-8