
Abstract
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the most common cause of dementia among the elderly population. AD is accompanied with the dysregulation of specific neurotrophic factors (NTFs) and their receptors, which plays a critical role in neuronal degeneration. NTFs are small proteins with therapeutic potential for human neurodegenerative diseases. These growth factors are categorized into four families: neurotrophins, neurokines, the glial cell line-derived NTF family of ligands, and the newly discovered cerebral dopamine NTF/mesencephalic astrocyte-derived NTF family. NTFs are capable of preventing cell death in degenerative conditions and can increase the neuronal growth and function in these disorders. Nevertheless, the adverse side effects of NTFs delivery and poor diffusion of these factors in the brain restrict the efficacy of NTFs therapy in clinical situations.
Materials and methods
In this review, we focus on the current advances in the use of NTFs to treat AD and summarize previous experimental and clinical studies for supporting the protective and therapeutic effects of these factors.
Conclusion
Based on reports, NTFs are considered as new and promising candidates for treating AD and AD-associated cognitive impairment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a progressive, multifarious neurodegenerative disease and the most common cause of dementia in older people. The World Alzheimer’s Report informed that 46.8 million people throughout the world were living with dementia in 2015 [1]. Specific diets, physical inactivity, midlife hypertension, depression, obesity, smoking, and low educational achievements can be accounted as some of the risk factors of AD. Likewise, diabetes is a risk factor for AD and neurodegeneration, and one-third of people with AD in the world have diabetes [2]. Impaired memory, loss of directionality, language disorders, and anxiety behaviors are the main early clinical manifestations of AD. Besides declined cognition, loss of body functions, and abnormal mental activities such as emotion and behavior are considered as late-stage manifestations of AD, which eventually lead to disability and death [3]. AD is characterized by three main histopathological features: extracellular senile plaques (SPs), intracellular neurofibrillary tangles (NFTs), and degeneration of basal forebrain cholinergic neurons (BFCNs). SPs are primarily made from β-amyloid (Aβ) peptide aggregation, whereas NFTs are composed of hyperphosphorylation of microtubule-associated protein (MAP) tau [4].
Although this disease was under research for an extended period, available drugs such as N-methyl d-aspartate receptor antagonist (memantine) and acetylcholinesterase inhibitors (rivastigmine, galantamine, donepezil) provide only symptomatic relief [5]. Pharmacological interventions for AD are severely limited and earlier approved drugs only offer symptomatic improvement, but not disease-modifying therapy. Fortunately, On June 7, 2021, aducanumab received a conditional approval form the US Food and Drug Administration (FDA) as the first new drug for the treatment of AD. Although aducanumab showed promise in clearing amyloid plaques from the brain tissue, but its efficacy in improving cognitive deficits remains to be proven [6].
Neurotrophic factors (NTFs) are small secretory proteins that play a pivotal role in the synaptic plasticity, survival, growth, differentiation, and myelination of neuronal cells [7]. It came to the limelight that altered expression of NTFs such as brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), and glial cell-derived neurotrophic factor (GDNF) are correlated with the neurodegeneration process and cognitive impairment in AD [8, 9]. Accordingly, in this review, we attempt to explain some of the recent developments in our understanding of the NTFs dysregulation in AD and the role of these factors in treating AD.
Limitations of neurotrophic factors application in the clinical trial and delivery methods
The earliest studies only considered the feasibility of NTFs therapy for the treatment of neurodegenerative diseases. Nevertheless, the adverse side effects of NTFs delivery in humans limit the efficacy of NTFs therapy in clinical settings. The BBB creates a primary challenge in the clinical application of NTFs, because the proteins do not pass the BBB and need to be delivered intracranially [10]. The other obstacles of clinical usage of NTFs include the short half-life [11], difficulties with administrating proper dose and covering a large brain area, and stage of patients who enrolled in trials and underwent NTF therapy. Thus, a minimally invasive injection method would be optimal [12]. Furthermore, because the NTFs are proteins, chronic delivery of them can be problematic because of aggregation, misfolding, and the development of neutralizing antibodies. Moreover, they typically cannot be given orally or administered systemically because of side effects often induced by systemic exposure to organs and tissues [13]. On the other hand, in humans, some NTFs such as BDNF are present in blood platelets; thus, the levels of these factors are readily measurable in human serum [14].
The main constraint is the difficulty to identify a mode of administration that allows long-term delivery of NTFs in efficient quantities to brain target areas in order to achieve clinical efficacy. Several approaches of the direct delivery of NTFs have been investigated, including direct infusion into the brain [15], intra-nasal injection [16], intra-ventricular transplantation [15], gene therapy, and stem cell-based delivery [17]. Nevertheless, in practice, it is difficult to achieve effective concentrations of NTFs, due to the substantial side-effects and rapid diffusion of a large proportion of them away from the injury site. Accordingly, it is imperative to find a better approach for delivery of NTFs into the target areas [18]. The fundamental solution to this problem is repeated injections of NTFs to the injury site so that the therapeutic concentrations can be obtained. To this end, encapsulated cell bio-delivery of NTFs leads to sustained release into the brain [19]. Another approach is the use of recombinant viral vectors to enable the long-term expression of NTFs in nerve cells [20]. As a novel solution to this issue, various biomaterials have been engineered to provide an effective, and sustained drug-delivery system. This method allows stem cells and/or small drug molecules, including NTFs to bypass the BBB and be delivered directly to the injury site [21].
Classification of neurotrophic Factors
NTFs are growth factors that promote and regulate neuronal growth, survival, differentiation, and functions in the developing and mature nervous systems. Likewise, they are vital for the maintenance of neuronal health and function. These proteins, according to their different cellular mechanisms, are categorized into four following groups: neurotrophins, neuropoietic cytokines, glial cell line-derived NTF (GDNF) family ligands (GFLs), an evolutionary conserved cerebral dopamine neurotrophic factor- mesencephalic astrocyte-derived neurotrophic factor family (CDNF/MANF) [22]. All NTFs that are implicated in AD treatment are shown in Table 1; Fig. 1.

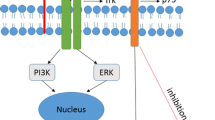
The schematic representation of the neurotrophic factors (NTFs) involvement in Alzheimer’s disease. Nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) bind to two families of tyrosine kinase receptors (TRKA and TRKB). Pro-NGF mediates apoptosis via interacting with p75NTR. Mature NGF has neuroprotective effects on cholinergic neurons and promotes the survival of them. Pro-BDNF binds specifically to p75NTR and mediates cell death, while mature BDNF promotes the survival and differentiation of cholinergic neurons, stimulates the acetylcholine release in cholinergic neurons, and induces the directional differentiation of cortical neural stem cells (NSCs) into the cholinergic neurons. Neurotrophin-3 (NT-3) binds to TRKC, and neurotrophin-4/5 (NT-4/5) binds to TRKB and promotes the survival of neural cells. Binding of glial cell line-derived neurotrophic factor (GDNF) to the GDNFα receptor and tyrosine kinase RET receptor leads to the cell survival and induces neuroprotective effect on nerve cells against atrophy. Ciliary neurotrophic factor (CNTF) is secreted by astrocytes and binds to CNTFRα and two subunits, gp130 and leukemia inhibitory factor (LIFRβ) that are expressed by astrocytes and neurons. It has survival and differentiation-inducing effects on cerebral sensory and motor neurons and hippocampal neurons. CDNF/MANF family may exert neuroprotective effects against Aβ-induced neuronal cell death by suppressing ER stress and modulating protein synthesis and folding
Neurotrophin family
NGF, BDNF, neurotrophin 3 (NT-3), and neurotrophin 4/5 (NT-4/5) constitute a structurally related family named neurotrophins that mediate different cellular processes, including the growth, survival, and death of neurons in the central and peripheral nervous systems [23]. These diverse biological roles are depending on their interactions with one of two receptors: tyrosine receptor kinase (Trk), one member of the tropomyosin-related tyrosine kinase receptors, or p75 neurotrophin receptor (p75NTR), a member of the tumor necrosis factor (TNF) receptor superfamily. When neurotrophins interact with specific Trk receptors (NGF binds to TrkA, BDNF and NT4/5 bind to TrkB, and NT3 binds to TrkC), mediate survival and growth responses. Moreover, their interaction with P75 neurotrophic receptor (p75NTR) leads to the modulation of brain plasticity and apoptosis [24].
In the central nervous system (CNS), the most prominent neurotrophin is BDNF because of the wide TrkB expression in the brain, especially in the cortex, hippocampus, and basal forebrain. However, NGF provides trophic support to the BFCNs, which mainly express TrkA receptors [25].
Nerve growth factor and Alzheimer’s disease
NGF is the first identified neurotrophic factor discovered by Rita Levi-Montalcini and Cohen in the 1950s. They hypothesized that during development, the secretion of NTFs supports the innervation of a sufficient number of neurons in the target tissue. NGF is secreted in the neocortex and the hippocampus and then retrogradely transported to the BFCNs and promotes survival and differentiation of these neurons [26] (Fig. 1). NGF is produced as large precursor proteins named pro-NGF, containing an N-terminal pro-domain and a C-terminal mature domain. Pro-NGF mediates apoptosis via interacting with p75NTR. Cleavage of pro-form and creating non-covalently linked homodimers result in mature NGF generation, which supports survival through binding to TrkA [27]. Mature NGF ultimately degraded by metalloproteases in the extracellular space. In AD brain, pro-NGF levels are increased, while NGF biosynthesis is normal, but unfortunately, the conversion of the pro-NGF molecule to its mature form is reduced in AD and the degradation of the mature form is augmented [28].
Neuropathological studies indicate that memory loss in AD patients is associated with neurodegeneration and dysfunction of the cholinergic neurons. Besides, evidence shows that decreased NGF levels is responsible for cholinergic atrophy in AD. In the AD brain, NGF receptor dysregulation has been found before phenotypic changes of BFCNs, indicating that the absence of neurotrophic support is a causative agent for basal forebrain deterioration [29]. According to these findings, NGF administration has been considered as a therapeutic approach for the regeneration of cholinergic neurons and, consequently treatment of AD.
Additionally, animal studies demonstrated that NGF stimulator drugs with oxidative stress suppressing activity might be a novel therapeutic option to protect and repair cellular damage in AD. For example, the aqueous extract of A. cochinchinesis root (AEAC) shows the NGF stimulatory activity and anti-oxidant effect in Tg2576 mice showing AD phenotypes of humans. AEAC can improve neural cell injuries by enhancing the NGF secretion and suppressing oxidative stress [30].
Based on data from animal models suggesting the neuroprotective effects of NGF on the cholinergic neurons, this NTF has been tested in patients with AD in clinical settings. Several approaches have been applied for NGF delivery in human, including intranasal administration of hNGF-61 (engineered mutein of hNGF) [31], direct injection into the cerebral lateral ventricle of the AD brain [15], gene therapy approaches [32], the cell-based delivery method by using stem-cells [17], and encapsulated cell bio delivery [33].
Intraventricular infusion of NGF in AD patients was one of the earliest neurotrophic programs. In 1992 and 1993, NGF was tested as a possible treatment for AD in two single-patient case reports. Both of these studies reported that protein infusion enhanced cholinergic activity and improved memory in these patients. No side effects were observed in either study [34, 35].
In contrast to before reports, Jönhagen et al. noted that the infusion of NGF protein showed slight improvements in a few neuropsychology tests and could not alleviate cognitive impairments. Also, they reported side effects, including constant back pain and weight reduction during the infusion of NGF in AD patients. Eventually, these researchers suggested that a change of routes or infusion doses may eliminate the side effects [15]. The next delivery method to improve NTFs efficacy in clinical trials is gene therapy. Gene therapy has two main forms, including ex vivo and in vivo. In the ex vivo method, researchers have used bioengineered cultural cells that express therapeutic proteins such as NTFs. In contrast, direct gene delivery into the endogenous cells of the patient is considered as an in vivo form of gene therapy [36].
Tuszynski et al. performed a phase 1 trial of ex vivo gene delivery of autologous fibroblasts that expressed human NGF into the forebrain of eight patients with mild AD. This report was the first gene therapy for AD in a small open-label trial and showed that cholinergic cells extended their projections toward the implant in response to secreted NGF from transplanted cells. Also, no serious side effects were reported as a consequence of gene therapy or NGF expression [37]. Eriksdotter and coworkers performed the next ex vivo gene therapy trial several years later. Their study indicated that encapsulated cell bio-delivery of NGF to the basal forebrain of individuals with AD caused positive effects in cognition, electroencephalography, and nicotinic receptor binding. They confirmed that this delivery method is “safe, well-tolerated, and feasible” [33].
Another in vivo gene therapy trial used genetically engineered vector adeno-associated virus serotype 2 (AAV2) to deliver three ascending doses of NGF to the basal forebrain of AD patients. This open-label clinical trial indicated that AAV2-NGF was safe and well-tolerated, and also it demonstrated the initial preliminary efficacy of this method [32]. Later gene therapy described postmortem findings from 10 AD patients who enrolled in previous ex vivo and in vivo NGF gene transfer trials conducted by Tuszynski et al. and Rafii et al. This study indicated that NGF induced 10 years of persistence axonal sprouting, cell hypertrophy, and functional markers in neurons of the degenerating brain [37, 32]. NGF gene therapy appears safe over extended periods and confirms the neurotrophic effect of NGF on treating AD disease, and opens a new approach for continuing studies. Rafii et al. noted their observation of a Phase II trial of AAV2-NGF in mild to moderate AD that confirmed the feasibility and safety of this approach, but no NGF-related symptomatic improvement was detected [38]. These findings aligned with the results of the later randomized clinical trial, which reported that AAV2-NGF was safe and well-tolerated through 2 years, but no significant difference between the treatment group and placebo was measured [39]. In a recent study, AAV2-NGF was delivered into the nucleus basalis of Meynert in cases with early-to middle-stage AD. NGF could not directly reach cholinergic neurons due to restricted distribution and imprecise stereotactic targeting. Because AAV2-NGF did not directly affect the target cholinergic neurons, it cannot concluded that NTFs gene therapy is inefficient for AD treatment [40]. Accordingly, further studies are required to detect the efficacy of NGF gene therapy in AD treatment and the effective approach for successful gene therapy in patients.
Some researchers practiced the encapsulated cell bio-delivery (ECB) implant to deliver human NGF to the basal forebrain of AD patients in a phase I pilot study. They expected that this delivery technology allows the transplanted factor to concentrate highly at the target area, and has fewer off-target side-effects [33]. Their results demonstrated that the ECB-NGF device was safe and feasible. It also indicated decreased rates of brain atrophy, stable cognitive function, and improvement of the level of cholinergic markers in cerebrospinal fluid (CSF) of AD patients [41]. Inflammatory proteins such as IL-1β may have a negative effect on NGF production by ECB cells. Therefore, the combination of ECB-NGF and anti-inflammatory drugs may be a potential candidate to protect or repair cellular damage and improve behavioral performance in AD [42].
Taken together, the mentioned studies reveal that NGF can partially restore the function of cholinergic neurons and can protect them from degeneration in AD. However, it has exerted less alleviative effects on cognitive performance in AD patients. Further studies and clinical trials should be done to optimize the best delivery method of NGF in AD patients.
Brain-derived neurotrophic factor and Alzheimer’s disease
BDNF, another neurotrophin family member, mediates synaptic plasticity and has a critical role in cognitive processes, learning, and memory formation. This molecule has a vital role in the survival, maintenance, and function of neurons [43]. BDNF gene is highly expressed in the cortex, hippocampus, and basal forebrain regions, that plays a crucial role in memory, learning, and higher cognitive performances. Pro-BDNF form binds specifically to p75NTR and mediates cell death, while mature form acts via binding to TrkB [44]. BDNF promotes neurogenesis and neurotransmission across the synapses, elevates synaptic growth, and modulates synaptic plasticity. BDNF also takes part in memory formation by inducing hippocampal long-term potentiation [45]. A cohort study demonstrated that polymorphism in the gene encoding BDNF leads to the higher vulnerability of hippocampus-frontal connectivity to AD pathology and AD-related cognitive impairment [46]. It seems that the basal forebrain GABAergic neurons express NGF mRNA and can produce minor amount of NGF [47]. Besides, basal forebrain neurons rely on their cortical and hippocampal targets to generate BDNF and NGF that are transported back to cell bodies of BFCNs through retrograde axonal transport. Therefore, retrograde axonal transport of NTFs by BFCNs has an essential role in their appropriate functioning and survival [29]. Accumulating studies declare that BDNF level is decreased during dementia and neurodegenerative diseases such as AD [48]. Recently, BDNF serum levels were decreased in dementia patients than healthy participants, concomitantly dementia cases presented a reduced hippocampal volume compared to healthy contributors [49]. In contrast, Blasko et al. did not find a significant difference in BDNF levels between AD and controls [50]. A recent meta-analysis study examined the aggregate levels of serum BDNF in patients with AD and individuals with mild cognitive impairment (MCI) compared to healthy controls. They indicated that serum BDNF levels were significantly lower in patients with AD but not MCI in comparison with healthy controls [48]. Another recent meta-analysis study evaluated the diagnostic role of peripheral BDNF concentration in AD patients affected by MCI. It revealed that although the BDNF level reduced in patients with AD or MCI, its levels are not useful markers to diagnose AD and MCI [51].
On the other hand, it has been reported that enhanced serum levels of BDNF reduced the risk of dementia and progression of AD, and acute aerobic exercise increases the plasma levels of BDNF in the elderly with AD [52]. Likewise, experimental evidence demonstrates that blood and plasma levels of BDNF reflect the brain BDNF levels [53]. The molecular mechanisms implicated in the downregulation of BDNF in AD patients are still unknown. Xia et al. disclosed that Aβ1−42 oligomers downregulate the peroxisome proliferators-activated receptor γ coactivator 1α (PGC-1α) and fibronectin type III domain-containing 5 (FNDC5) and probably by this way suppress the expression of BDNF and consequently cause a cognitive deficit in AD mice [54].
An experimental study indicated the specific and dose-response protective effect of BDNF on neural toxicity induced by Aβ peptides. BDNF Val66Met polymorphism is associated with altered neuronal integrity and synaptic excitation. This process also moderates Aβ-related changes in cognition and neurodegeneration, memory decline and hippocampal atrophy in AD cases [55]. Lim et al. proposed that these properties of BDNF Val66Met polymorphism are probably due to the enhanced vulnerability of brain to the toxic effects of Aβ peptide [56]. Another experimental study used a transgenic mouse model overexpressing tau and showed that overexpression of wild-type tau downregulates BDNF [57].
Like NGF, BDNF promotes the survival of BFCNs and increases choline acetyl transferase activity in vitro and in vivo. BDNF also can improve the function of cholinergic neurons and increase acetylcholine release in hippocampal synapses [58]. Moreover, forebrain-specific knockout of BDNF receptor (trkB) resulted in severe impairment in learning, indicating that increasing the level of BDNF in the forebrain could improve cognitive deficit and attenuate AD symptoms. Given this evidences, the delivery of exogenous BDNF has been considered a valuable therapeutic approach for AD [59]. However, delivery of exogenous BDNF faces different problems. As BDNF does not readily cross the BBB, the main problem is the amount of BDNF that reaches the affected neurons. The too low amounts might not be sufficient to have the desired effects, whereas too high doses might have paradoxically dangerous consequences such as neuronal excitability [60]. Similarly, Scharfman et al. reported spontaneous limbic seizures following intrahippocampal infusion of BDNF, supporting the hypothesis that BDNF injection can facilitate seizure activity [61]. To overcome such delivery problems and to get more effective results, different methods have been developed, such as ECB, gene therapy and stem cell therapy. In this context, Xuan et al. obtained neural stem cells (NSCs) from the hippocampus and transplanted them into the rat basal forebrain, followed by the transplantation of BDNF into the lateral ventricle. They revealed the differentiation of labeled NSCs into neurons and astrocytes in the basal forebrain. Besides, BDNF enhanced the therapeutic effects of transplanted NSCs after injection into the lesioned brain [62].
An in vitro study reveals that overexpression of BDNF has a neuroprotective effect on Aβ-treated NSCs [63]. Additionally, engrafted neurons derived from BDNF-overexpressing NSCs demonstrated electrophysiological properties of mature neurons. They were capable of integrating into existing brain circuits, and eventually improved the cognitive decline in the transgenic AD mouse model. Likewise, overexpression of BDNF ameliorated the engrafted cells’ viability, neurite complexity, neuronal fate, synaptic plasticity, and maturation of electrical property in AD mice [64]. In line with these findings, when BDNF was knockdown in BDNF-NSCs combination, the therapeutic efficacy of stem cell-based therapy was reduced [63]. Another study showed that BDNF modified human umbilical cord-derived mesenchymal stem cells (hUC-MSCs)-derived cholinergic-like neurons may be a promising therapeutic method for AD [58]. As another delivery method, a recent study examined the effects of ECB device-mediated long-term release of BDNF on epileptic seizures. These devices, which transplanted into the hippocampus of rats, represented a valid therapeutic effect against epilepsy and some of its comorbidities [65].
Leyhe et al. reported that the up-regulation of BDNF might be part of a neuroprotective effect of acetylcholinesterase (AChE)-inhibitors [66]. Also, it has been revealed that donepezil, an AChE-inhibitor protects neurons, improves cognitive function, and ameliorates learning and memory impairment through regulating the cholinergic system, upregulating the expression of BDNF, increasing the phosphorylated level of TrkB, and subsequently activation of the BDNF/TrkB signaling pathway [67]. Besides, it has been reported that lithium may exert its neuroprotective effect in AD patients through the upregulation of BDNF [68]. Alvarez et al. evaluated the synergistic action of cerebrolysin and donepezil on increasing the BDNF serum level and observed that this treatment delayed cognitive decline in AD patients [69]. Recently, the effects of BDNF-modified human amniotic membrane-derived mesenchymal stem cells (AM-MSCs) were examined on the learning and memory performance of AD rats. Human AM-MSCs transplantation improved learning and memory abilities of AD animals, and BDNF-modified cells could further ameliorate this therapeutic potential [70].
Furthermore, pharmacological mimetics or exercise, which enhance BDNF levels, may improve cognition in AD, and their application at the early stages of AD has a protective role against subsequent neuronal cell death [71]. With respect to the above-mentioned results and regarding the conflicting reports on the alteration of BDNF concentration in serum and brain tissue of AD cases, as well as limited delivery methods of BDNF, additional studies are required to achieve the best approach of BDNF delivery for exerting maximum effect on cognitive problems and treatment of AD.
Neurotrophin 3, neurotrophin 4/5, and Alzheimer’s disease
Few studies have evaluated the role of NT-3 or NT-4/5, another neurotrophin family members, in AD. NT-4/5 contributes in promoting survival and differentiation of hippocampal, noradrenergic and dopaminergic neurons [72]. NT-3 is commonly expressed in the dentate gyrus of the hippocampus, a main neurogenic microenvironment. By interacting with Trk-C, NT-3 can regulate neurogenesis and facilitate hippocampal plasticity, indicating the key role of NT-3 in memory function [73]. A study revealed that the deletion of genes for NT-3 and/or its associated receptor (TrkC) results in a loss of about 30% of striatal neurons [74]. Besides, a significant reduction of NT-3 levels in the motor cortex of post-mortem AD patients but not in the dentate gyrus and entorhinal cortex was reported in another study [75]. By contrast, Durany et al. and Hock et al. did not find altered post-mortem brain NT-3 levels in AD patients [76, 77]. Since improvements in white matter pathology are strongly correlated with the recovery of memory deficits, it was recently reported that Electroacupuncture (EA) stimulation promotes oligodendrocyte regeneration and NT4/5-TrkB signaling and the recovery of memory function following white matter injury [9]. One study has used transfected bone marrow-derived NSCs (BM-NSCs) with NT-3 to increase the delivery of NT3. The results of this study indicated that NT-3 gene transfection could promote the in vitro proliferation and differentiation of BM-NSCs into cholinergic neurons by affecting the Notch signaling pathway [78]. A more recent study investigated the effects of NT-3 on the differentiation of bone marrow-derived MSCs (BM-MSCs) into neurons in vitro and in vivo. They revealed that NT-3 overexpression increased the neuronal differentiation of BM-MSCs in vitro and in vivo and improved cognitive performance in rat model of AD. While, silencing NT-3 suppressed the differentiation of BM-MSCs and reduced cognitive abilities in AD rats. They also found that NT-3 gene operated through triggering Wnt/β-catenin signaling pathway [73]. Based on available results, unlike BDNF and NGF, the NT-3 and NT-4/5 may not be related to the pathogenesis of AD. As well, regarding the role of these neurotrophins in the neuronal differentiation of stem cells, additional cell-based studies are needed to further evaluate the role of NT-3 and NT-4/5 in treatment of AD.
Neuropoietic cytokine (neurokine) family and Alzheimer’s disease
The family of neuropoietic cytokines, also known as neurokine, represents the small and structurally secretory proteins signaling via transmembrane gp130 receptor. This family consists of ciliary neurotrophic factor (CNTF), cardiotrophin-1 (CT-1), leukemia inhibitory factor (LIF), neuropoietin (NPN), oncostatin M (OSM), cardiotrophin-like cytokine (CLC), interleukin 6 (IL-6), IL-11 and IL-27. Neurokines use either a two or three-component receptor system and play an important role in the survival of motoneurons [79]. Among neurokines, CNTF is the most studied neurokine for AD treatment and other members of this family have not yet been studied for treating AD and related cognitive decline.
Ciliary neurotrophic factor and Alzheimer’s disease
CNTF, a neural cytokine, is expressed in the peripheral and central nervous system. In the brain, CNTF is secreted by astrocytes and binds to CNTFRα, which is represented by astrocytes and neurons (Fig. 1). The interaction between CNTF and its receptor activates the recruitment of two other transmembrane receptor proteins, LIFRβ and gp130, leading to their dimerization and tyrosine phosphorylation [80]. Previously, CNTF was revealed to operate synergistically with BDNF and encourage the survival of choline acetyl transferase-positive neurons in the cultured rat-derived BFCNs. BDNF alone caused a threefold enhancement in neural survival. However, CNTF along with BDNF led to an eight-fold increase in the number of survived neurons. This synergetic activity was limited only to BFCNs and not to the hippocampus, or cerebellum. CNTF showed neuronal protective activity in synergy with NT-4 but not NGF [81]. CNTF has survival and differentiation-inducing effects on cerebral sensory and motor neurons, parasympathetic neurons, and hippocampal neurons [82]. It additionally enhances neurite outgrowth in the hippocampus and hypothalamus, that are implicated in stress and anxiety responses. Furthermore, CNTF enhances adult neurogenesis, and cognitive performance in rodent models. CNTF levels are also enhanced following brain injury and promote neuronal survival after injury [80]. Given the role of CNTF in cell survival, neurite outgrowth, and hippocampal neurogenesis, its over-expressing in the AD brain may be a promising approach to improve AD-induced cognitive decline. However, its clinical usage is limited because of its inability to automatically cross the bio-membranes and spread in the target region. Accordingly, some novel approaches have been utilized to solve this problem [83]. In a previous study, continuous direct CNTF implantation to the brain of Aβ-received mice had neuroprotective effects against neuronal and behavioral impairments [84]. In the other study, the researchers used P11 linked to CNTF to solve this problem and help fast and efficient delivery of CNTF to the brain in a non-invasive way. P11-CNTF treatment in the AD model of mice significantly shortened the mean latency in the Morris water maze test. The outstanding characteristics of P11-CNTF as a non-invasive way make it a novel efficient therapeutic method for AD [82]. Recently, treatment with P021, a small peptidergic compound derived from CNTF that is the BBB permeable, successfully prevented synaptic and neurogenesis deficits, neurodegeneration, Aβ and tau pathologies, and subsequently cognitive impairment in the transgenic model of mice. These results confirm the potential therapeutic outcome of P021 on several main features of AD [85]. In the other work, the trans activator of transcription (TAT) was connected to truncate CNTF (tCNTF) and generated TAT-tCNTF, to overcome the inability of CNTF to access the brain. TAT-tCNTF could transport automatically through bio-membranes and enter into the cells generally by macro-pinocytosis. TAT-tCNTF promoted cell viability in hippocampal neurons treated with Aβ by interacting with its receptor, CNTFRα, and activating the extracellular regulated protein kinases (Erk) and Protein kinase B/Akt pathways [86]. Overall, according to evidence, CNTF is potentially able to promote neurogenesis and neuronal survival as well as has neuroprotective effects against neuronal and neurobehavioral impairments following AD. However, its clinical application is restricted due to its inability to cross bio-membranes and reach the target region. Despite the findings as mentioned earlier to overcome this problem, additional investigations should be done to provide the supreme solution for this obstacle.
Cardiotrophin-1 and Alzheimer’s disease
This trophic factor displays remarkable neuroprotective effects and delays motor neurons degeneration via promoting neuronal regeneration, extending the neuronal survival, and improving motor function [87]. CT-1 is capable of alleviating high fat diet-induced cognitive impairment by suppressing proinflammatory cytokines in the brain, improving mitochondrial/synaptic dysfunction, and increasing insulin signaling sensibility by AMPK activation [88, 89]. Another study by Wang et al. examined the involvement of CT-1 in AD-related cognitive decline. They indicated the down-regulation of CT-1 in the brain of the APPswe/PS1dE9 transgenic mice. The APPswe/PS1dE9 transgenic mouse model of AD shows generation of senile plaque and amyloid neuropathy. Moreover, in their study, induced expression of CT-1 ameliorated cognitive deficits through GSK-3β inhibition [87].
Glial cell line-derived neurotrophic factor family ligands and Alzheimer’s disease
GFLs are a distinct family within the TGF-β superfamily, which consists of four members including, GDNF, neurturin (NRTN), artemin (ARTN), and persephin (PSPN). Complex signaling made of the receptor tyrosine kinase Ret and one of the four members of the glycosylphosphatidylinositol-linked GDNF family receptor α (GFRα1, GFRα2, GFRα3, and GFRα4) network mediates the biological functions of these proteins. Supportive and neurorestorative effects of these molecules on various neuronal populations such as basal forebrain, hippocampal, dopaminergic, motor, sensory, enteric, sympathetic, and parasympathetic neurons have attracted significant attention as a potential treatment for degenerative diseases [90]. Based on the evidence, GDNF is the best understood and most commonly used GFL for AD treatment and other members of this family have not yet been studied for treating AD and associated cognitive deficits.
Glial cell line-derived neurotrophic factor and Alzheimer’s disease
GDNF, a potent NTF and a member of GFLs, protects and repairs midbrain dopamine neurons. Besides, GDNF can inhibit the degeneration of noradrenergic neurons of the locus coeruleus, which diminished during AD progression. It has shown that a decreased concentration of GDNF leads to excessive glutamate release and excitotoxicity in the CNS, resulting in dopaminergic degeneration [91]. Degeneration of dopaminergic neurons takes part in AD pathogenesis and, therefore, pharmacological approaches focusing on enhancing the cortical and hippocampal dopamine transmission may promote synaptic functions and improve memory impairment in AD patients [92]. A recent study revealed that endogenous GDNF acts as a potential modulator of cholinergic transmission and protects against cognitive impairments in neurodegenerative disorders [93]. Additionally, data of a postmortem investigation displayed GDNF downregulation in the middle temporal gyrus of AD subjects [94]. In the context of altered expression of GDNF in AD serum, a recent study reported the decreased serum GDNF levels in AD patients compared to the control group, indicating the involvement of GDNF in disease pathology [95]. However, an increased concentration of GDNF was detected in the serum and CSF of patients with early stages AD to compensate for the neurotrophic loss in the impaired brain [96]. Moreover, this protein promotes neuronal plasticity, and age-related cognitive deficits. Furthermore, GDNF signaling is mainly affected during both the aging process and AD, leading to cognitive impairment [91]. In hippocampal neuron culture, GDNF seems to enhance the number of synapses. Also, mutant mice with decreased levels of GDNF showed reduced presynaptic maturation during hippocampal synaptogenesis [97]. Only a handful of literature examined the therapeutic potential of GDNF for AD. A study performed by Ghribi et al. showed that the GDNF administration has a protective role against AD-like changes induced by the injection of aluminum complexes in rabbits [98]. Moreover, Revilla et al. have unveiled that GDNF was down-regulated in transgenic 3xTgAD mice and voluntary exercise for 6 months reversed this effect [99]. Besides, 6 months of overexpressing GDNF and BDNF by using engineered-lentiviral vectors in 10-month‐old 3xTg‐AD mice may protect neurons from atrophy and degeneration and by this way protect against AD neuropathology such as learning and memory impairment [100].
In a more recent study, GDNF-overexpressing umbilical cord blood mononuclear cells (UCBMCs) were administered to AD transgenic mice and this gene-cell therapy method stimulated synaptogenesis and promoted long-lasting neuroprotection [101]. A considerable number of preclinical and clinical studies demonstrated the beneficial effects of lithium on cognitive performance in subjects with AD. According to Straten et al. study, GDNF is the main target of lithium in AD patients, especially in early steps. They found a significant negative correlation between lithium concentration in serum and GDNF concentration in serum and CSF. These findings indicate that lithium treatment might reduce the necessity of enhanced GDNF expression in the CNS in early AD [102]. A recent work reported that GDNF-overexpressing UCBMCs transplantation significantly improved spatial memory in transgenic AD model in mice (APP/PS1 mice), which has confirmed by the existence of implanted cells and their GDNF expression in the hippocampus and cortex for several weeks after transplantation [103]. Another recent study indicated the contribution of the Hippo/YAP signaling pathway in the action of GDNF against Aβ-induced inflammatory response in microglia. Accordingly, targeting GDNF or the Hippo/YAP signaling may be a promising treatment for AD [104]. Therefore, based on these results, it can be concluded that the GDNF administration or over-expression can be a therapeutic approach for the treatment of AD and subsequent cognitive impairment.
Cerebral dopamine neurotrophic factor: mesencephalic astrocyte—derived neurotrophic factor family and Alzheimer’s disease
CDNF and MANF proteins are two members of the evolutionary conserved CDNF/MANF family and have varied critical roles in the brain. These proteins are widely expressed in different areas in the mammalian brain, such as the hippocampus, thalamus, cerebral cortex, cerebellum, and striatum [7]. Some neuropathological studies provided evidence that misfolded proteins’ aggregation leads to endoplasmic reticulum (ER) stress, which is a primary feature of AD. The accumulation of misfolded proteins in the ER disrupts its homeostasis and activates a cellular stress response named the unfolded protein response (UPR) [88]. Whereas UPR protects the cell against the formation of toxic misfolded proteins, its long-term activation can induce cell death [105]. Numerous studies have revealed that UPR is activated in the AD brain [106]. MANF and CDNF are two ER stress response proteins that diminish ER stress-induced cell death via interaction with the UPR [107]. A study conducted by Kemppainen and coworkers showed that intrahippocampal CDNF protein or gene-therapy improved long-term memory in both mice models of AD and wild-type controls, but did not influence spontaneous activity, short-term memory, or object neophobia [108]. Synaptic dysfunction is one of the main characterizations of early-stage AD related to soluble oligomers of Aβ. CDNF shows a protective effect on Aβ-induced synaptotoxicity in hippocampal cells, suggesting that CDNF may protect against Aβ-induced synaptotoxicity through decreased the expression of ER stress-related proteins and attenuating ER stress [109]. Similarly, MANF may exert neuroprotective effects against Aβ-induced neuronal cell death by suppressing ER stress [107]. These findings are proposing CDNF and MANF as a therapeutic candidate for the treatment of AD that needs further investigations before application of them in clinical settings.
Neurotrophic factors and other neurodegenerative conditions
NTFs have been revealed to be effective in the protection of neurons and prevention of progressive cell loss in a range of other neurodegenerative disorders. The bulk of these results were obtained from animal studies. In animal models of Parkinson’s disease, many researchers have reported improved motor function and plasticity, and also observed reduced Lewy neurites/bodies accumulation, inflammation, as well as oxidative stress. Some of these NTFs promote the survival of dopaminergic neurons and prevent their degeneration and exert neuroprotective and neurorestorative effects on these cells; in contrast to their robust effects in animal models, NTFs have shown relatively weak results in human clinical trials [107].
In the case of amyotrophic lateral sclerosis (ALS), administration of NTFs in experimental animal models of ALS revealed significant improvement in the survival of degenerating motoneuron and rescue of the axotomized motoneuron. Likewise, several NTFs such as BDNF, CNTF and, GDNT have been tested in clinical settings to treat ALS patients. However, dose-limited side effects, particularly weight loss, anorexia, and inappropriate administration methods cause poor outcomes [110].
Furthermore, several delivery approaches like gene therapy have since been used to distribute NTFs in many different animal models of other neurodegenerative diseases, including Huntington’s disease [111] and stroke [112]; still, these programs have not advanced into clinical trials.
Concluding remarks and future directions
As mentioned above, although aducanumab is efficient in removing amyloid plaques from the brain, but its efficacy in treating AD-related cognitive deficits has not confirmed. Accordingly, another approach may be more applicable in treatment of cognitive decline following AD. In this context, NTFs as new and promising candidates for treating AD and AD-associated cognitive impairment are currently under investigation. Mounting evidence shows that NTFs have varied biological effects such as survival, development, and neurons’ maintenance. Changes in the levels or expression of NTFs may have a critical role in the progression of AD and concomitant cognitive decline. Nevertheless, mounting shreds of evidence have been collected in the last decades; more studies are needed before the application of NTFs as therapeutic promises for AD management. In this context, some limitations for clinical application of NTFs such as their delivery across the BBB, having short half-lives, difficulties with the injection of proper dose, and side effects on organs and tissues should be resolved in the future. However, emergent delivery approaches, such as gene therapy vectors and biomaterial-based drug-delivery system, seem to improve NTF therapy outcomes, decreased side effects, and increased clinical benefit.
Abbreviations
- AD:
-
Alzheimer’s disease
- NTF:
-
Neurotrophic factors
- NFT:
-
Neurofibrillary tangle
- SP:
-
Extracellular senile plaque
- Aβ:
-
β-amyloid
- BDNF:
-
Brain-derived neurotrophic factor
- NGF:
-
Nerve growth factor
- GDNF:
-
Glial cell line-derived neurotrophic factor
- GFL:
-
Glial cell line-derived NTF (GDNF) family ligands
- NT-3:
-
Neurotrophin 3
- NT-4/5:
-
Neurotrophin 4/5
- Trk:
-
Tyrosine receptor kinase
- p75NTR:
-
p75 neurotrophin receptor
- TNF:
-
Tumor necrosis factor
- AAV2:
-
Adeno-associated virus serotype 2
- CSF:
-
Cerebrospinal fluid
- ECB:
-
Encapsulated cell bio-delivery
- BFCNs:
-
Basal forebrain cholinergic neurons
- MCI:
-
Mild cognitive impairment
- MANF:
-
Mesencephalic astrocyte-derived neurotrophic factor
- PGC-1α:
-
Peroxisome proliferators-activated receptor γ coactivator 1α
- FNDC5:
-
Fibronectin type III domain-containing 5
- AM-MSCs:
-
Amniotic membrane-derived mesenchymal stem cells
- BM-NSCs:
-
Bone marrow-derived neural stem cells
- hUC-MSCs:
-
Human umbilical cord-derived mesenchymal stem cells
- CNTF:
-
Ciliary neurotrophic factor
- LIF:
-
Leukemia inhibitory factor
- TAT:
-
Transactivator of transcription
- UCBMCs:
-
Umbilical cord blood mononuclear cells
- UPR:
-
Unfolded protein response
- ER:
-
Endoplasmic reticulum
References
Huang L-K, Chao S-P, Hu C-J (2020) Clinical trials of new drugs for Alzheimer disease. J Biomed Sci 27(1):1–13
Chakraborty A, Diwan A (2020) Alzheimer and it’s possible therapy: a review. J Exp Neurol 1(4):115–122
Bature F et al (2017) Signs and symptoms preceding the diagnosis of Alzheimer’s disease: a systematic scoping review of literature from 1937 to 2016. BMJ Open 7(8):e015746
Li T, Shi H, Zhao Y (2018) Phosphorylation of microtubule-associated protein tau by mitogen-activated protein kinase in Alzheimer’s disease. IOP Publishing, Bristol
Kumar A et al (2016) Current and novel therapeutic molecules and targets in Alzheimer’s disease. J Formos Med Assoc 115(1):3–10
Dunn B, Stein P, Cavazzoni P (2021) Approval of dducanumab for Alzheimer disease—the FDA’s perspective. JAMA Internal Med 181(10):1276–1278
Nasrolahi A et al (2019) Effect of cerebral dopamine neurotrophic factor on endogenous neural progenitor cell migration in a rat model of Parkinson’s disease. EXCLI J 18:139
Sopova K et al (2014) Dysregulation of neurotrophic and haematopoietic growth factors in Alzheimer’s disease: from pathophysiology to novel treatment strategies. Curr Alzheimer Res 11(1):27–39
Ahn SM et al (2016) Electroacupuncture ameliorates memory impairments by enhancing oligodendrocyte regeneration in a mouse model of prolonged cerebral hypoperfusion. Sci Rep 6:28646
Domanskyi A, Saarma M, Airavaara M (2015) Prospects of neurotrophic factors for Parkinson’s disease: comparison of protein and gene therapy. Hum Gene Ther 26(8):550–559
Houlton J et al (2019) Therapeutic potential of neurotrophins for repair after brain injury: a helping hand from biomaterials. Front NeuroSci 13:790
Bartus RT, Johnson EM Jr. (2017) Clinical tests of neurotrophic factors for human neurodegenerative diseases, part 1: where have we been and what have we learned? Neurobiol Dis 97:156–168
Bartus RT et al (2013) Advancing neurotrophic factors as treatments for age-related neurodegenerative diseases: developing and demonstrating “clinical proof-of-concept” for AAV-neurturin (CERE-120) in Parkinson’s disease. Neurobiol Aging 34(1):35–61
Naegelin Y et al (2018) Measuring and validating the levels of brain-derived neurotrophic factor in human serum. Eneuro. https://doi.org/10.1523/ENEURO.0419-17.2018
Jönhagen ME et al (1998) Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement Geriatr Cogn Disord 9(5):246–257
Lauzon M-A et al (2015) Nanoparticle-mediated growth factor delivery systems: A new way to treat Alzheimer’s disease. J Controlled Release 206:187–205
Lee HJ et al (2012) Human neural stem cells genetically modified to express human nerve growth factor (NGF) gene restore cognition in the mouse with ibotenic acid-induced cognitive dysfunction. Cell Transplant 21(11):2487–2496
Han Q et al (2011) The promotion of cerebral ischemia recovery in rats by laminin-binding BDNF. Biomaterials 32(22):5077–5085
Eyjolfsdottir H et al (2016) Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: application of a second-generation encapsulated cell biodelivery device. Alzheimer’s Res Ther 8:1–111
Iwasaki Y et al (2012) Sendai virus vector-mediated brain‐derived neurotrophic factor expression ameliorates memory deficits and synaptic degeneration in a transgenic mouse model of Alzheimer’s disease. J Neurosci Res 90(5):981–989
Shabani Z et al (2021) Transplantation of bioengineered Reelin-loaded PLGA/PEG micelles can accelerate neural tissue regeneration in photothrombotic stroke model of mouse. Bioeng Transl Med. https://doi.org/10.1002/btm2.10264
Skaper SD (2018) Neurotrophic factors: an overview. Neurotrop Factors 1727:1–17
Sánchez-Sánchez J, Arévalo JC (2017) A review on ubiquitination of neurotrophin receptors: facts and perspectives. Int J Mol Sci 18(3):630
Bhardwaj R, Deshmukh R (2018) Neurotrophic factors and Parkinson’s disease. Clin Invest 7(4):53–62
Weissmiller AM, Wu C (2012) Current advances in using neurotrophic factors to treat neurodegenerative disorders. Translational neurodegeneration 1(1):1–9
Zhou L-T et al (2021) Elevated levels of miR-144-3p induce cholinergic degeneration by impairing the maturation of NGF in Alzheimer’s disease. Front Cell Dev Biol. https://doi.org/10.3389/fcell.2021.667412
Keefe KM, Sheikh IS, Smith GM (2017) Targeting neurotrophins to specific populations of neurons: NGF, BDNF, and NT-3 and their relevance for treatment of spinal cord injury. Int J Mol Sci 18(3):548
Cuello AC, Pentz R, Hall H (2019) The brain NGF metabolic pathway in health and in Alzheimer’s pathology. Front NeuroSci 13:62
Shekari A, Fahnestock M (2019) Retrograde axonal transport of BDNF and proNGF diminishes with age in basal forebrain cholinergic neurons. Neurobiol Aging 84:131–140
Lee HA et al (2018) Asparagus cochinchinensis stimulates release of nerve growth factor and abrogates oxidative stress in the Tg2576 model for Alzheimer’s disease. BMC Complement Altern Med 18(1):125
Mitra S et al (2021) A review of techniques for biodelivery of nerve growth factor (NGF) to the brain in relation to Alzheimer’s disease. Recent advances in NGF and related molecules. Springer, Cham, pp 167–191
Rafii MS et al (2014) A phase1 study of stereotactic gene delivery of AAV2-NGF for Alzheimer’s disease. Alzheimer’s Dement 10(5):571–581
Eriksdotter-Jönhagen M et al (2012) Encapsulated cell biodelivery of nerve growth factor to the basal forebrain in patients with Alzheimer’s disease. Dement Geriatr Cogn Disord 33(1):18–28
Olson L et al (1992) Nerve growth factor affects 11 C-nicotine binding, blood flow, EEG, and verbal episodic memory in an Alzheimer patient (case report). Journal of Neural Transmission-Parkinson’s Disease and Dementia Section 4(1):79–95
Seiger à et al (1993) Intracranial infusion of purified nerve growth factor to an Alzheimer patient: the first attempt of a possible future treatment strategy. Behav Brain Res 57(2):255–261
Bartus RT, Johnson EM Jr. (2017) Clinical tests of neurotrophic factors for human neurodegenerative diseases, part 2: Where do we stand and where must we go next? Neurobiol Dis 97:169–178
Tuszynski MH et al (2005) A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med 11(5):551
Rafii M (2015) A Phase II trial of AAV2-NGF in mild to moderate Alzheimer’s disease. J Prev Alzheimers Dis 2015:274–275
Rafii MS et al (2018) Adeno-associated viral vector (serotype 2)–nerve growth factor for patients with Alzheimer disease: a randomized clinical trial. JAMA Neurol 75(7):834–841
Castle MJ et al (2020) Postmortem analysis in a clinical trial of AAV2-NGF gene therapy for Alzheimer’s disease identifies a need for improved vector delivery. Hum Gene Ther 31(7–8):415–422
Ferreira D et al (2015) Brain changes in Alzheimer’s disease patients with implanted encapsulated cells releasing nerve growth factor. J Alzheimers Dis 43(3):1059–1072
Eriksdotter M et al (2018) Cerebrospinal fluid from Alzheimer patients affects cell-mediated nerve growth factor production and cell survival in vitro. Exp Cell Res 371(1):175–184
Silakarma D, Sudewi AAR (2019) The role of brain-derived neurotrophic factor (BDNF) in cognitive functions. Bali Medical Journal 8(2):427–434
Yang B et al (2017) Altered expression of BDNF, BDNF pro-peptide and their precursor proBDNF in brain and liver tissues from psychiatric disorders: rethinking the brain–liver axis. Transl Psychiatry 7(5):e1128–e1128
Colucci-D’Amato L, Speranza L, Volpicelli F (2020) Neurotrophic factor BDNF, physiological functions and therapeutic potential in depression, neurodegeneration and brain cancer. Int J Mol Sci 21(20):7777
Franzmeier N et al (2021) The BDNF Val66Met SNP modulates the association between beta-amyloid and hippocampal disconnection in Alzheimer’s disease. Mol Psychiatry 26(2):614–628
Lauterborn JC et al (1995) NGF mRNA is expressed by GABAergic but not cholinergic neurons in rat basal forebrain. Journal of Comparative Neurology 360(3):454–462
Ng TKS et al (2019) Decreased serum brain-derived neurotrophic factor (BDNF) levels in patients with Alzheimer’s disease (AD): a systematic review and meta-analysis. Int J Mol Sci 20(2):257
Borba EM et al (2016) Brain-derived neurotrophic factor serum levels and hippocampal volume in mild cognitive impairment and dementia due to Alzheimer disease. Dementia and geriatric cognitive disorders extra 6(3):559–567
Blasko I et al (2006) Measurement of thirteen biological markers in CSF of patients with Alzheimer’s disease and other dementias. Dement Geriatr Cogn Disord 21(1):9–15
Xie B et al (2019) Evaluation of the diagnostic value of peripheral BDNF levels for Alzheimer’s disease and mild cognitive impairment: results of a meta-analysis. Int J Neurosci 130(3):218–230
Coelho FGdM et al (2014) Acute aerobic exercise increases brain-derived neurotrophic factor levels in elderly with Alzheimer’s disease. J Alzheimers Dis 39(2):401–408
Klein AB et al (2011) Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int J Neuropsychopharmacol 14(3):347–353
Xia D-Y et al (2017) PGC-1α or FNDC5 is involved in modulating the effects of Aβ1– 42 oligomers on suppressing the expression of BDNF, a beneficial factor for inhibiting neuronal apoptosis, Aβ deposition and cognitive decline of APP/PS1 Tg mice. Front Aging Neurosci 9:65
Lim YY et al (2016) BDNF Val66Met moderates memory impairment, hippocampal function and tau in preclinical autosomal dominant Alzheimer’s disease. Brain 139(10):2766–2777
Lim YY et al (2015) APOE and BDNF polymorphisms moderate amyloid β-related cognitive decline in preclinical Alzheimer’s disease. Mol Psychiatry 20(11):1322
Rosa E et al (2016) Tau downregulates BDNF expression in animal and cellular models of Alzheimer’s disease. Neurobiol Aging 48:135–142
Hu W et al (2019) Brain-derived neurotrophic factor modified human umbilical cord mesenchymal stem cells-derived cholinergic-like neurons improve spatial learning and memory ability in Alzheimer’s disease rats. Brain Res 1710:61–73
Minichiello L et al (1999) Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24(2):401–414
Knusel B et al (1992) Brain-derived neurotrophic factor administration protects basal forebrain cholinergic but not nigral dopaminergic neurons from degenerative changes after axotomy in the adult rat brain. J Neurosci 12(11):4391–4402
Scharfman HE et al (2002) Spontaneous limbic seizures after intrahippocampal infusion of brain-derived neurotrophic factor. Exp Neurol 174(2):201–214
Xuan A et al (2008) BDNF improves the effects of neural stem cells on the rat model of Alzheimer’s disease with unilateral lesion of fimbria-fornix. Neurosci Lett 440(3):331–335
Wu C-C et al (2016) Gain of BDNF function in engrafted neural stem cells promotes the therapeutic potential for Alzheimer’s disease. Sci Rep 6:27358
Wu C-C et al (2016) Gain of BDNF function in engrafted neural stem cells promotes the therapeutic potential for Alzheimer’s disease. Sci Rep 6(1):1–16
Falcicchia C et al (2018) Seizure-suppressant and neuroprotective effects of encapsulated BDNF-producing cells in a rat model of temporal lobe epilepsy. Mol Ther-Methods Clin Dev 9:211–224
Leyhe T et al (2008) Increase of BDNF serum concentration during donepezil treatment of patients with early Alzheimer’s disease. Eur Arch Psychiatry Clin NeuroSci 258(2):124–128
Zheng H et al (2018) Donepezil improves the cognitive impairment in a tree shrew model of Alzheimer’s disease induced by amyloid-β 1–40 via activating the BDNF/TrkB signal pathway. Metab Brain Dis 33(6):1961–1974
Leyhe T et al (2009) Increase of BDNF serum concentration in lithium treated patients with early Alzheimer’s disease. J Alzheimers Dis 16(3):649–656
Alvarez XA et al (2016) Synergistic increase of serum BDNF in Alzheimer patients treated with cerebrolysin and donepezil: association with cognitive improvement in ApoE4 cases. Int J Neuropsychopharmacol 19(6):pyw024
Gao M-l et al (2018) Effects of braln-derived neurotrophic factor-modified human amniotic membrane-derived mesenchymal stem cell transplantation on learning and memory abilities of alzheimer’s disease rats. Chin J Tissue Eng Res 22(9):1419–1424
Choi SH et al (2018) Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science. https://doi.org/10.1126/science.aan8821
Ribeiro D et al (2021) The impact of physical exercise on the circulating levels of BDNF and NT 4/5: a review. Int J Mol Sci 22(16):8814
Yan Z et al (2021) Neurotrophin-3 promotes the neuronal differentiation of BMSCs and improves cognitive function in a rat model of Alzheimer’s disease. Front Cell Neurosci 15:18
Baydyuk M et al (2013) Midbrain-derived neurotrophins support survival of immature striatal projection neurons. J Neurosci 33(8):3363–3369
Narisawa-Saito M et al (1996) Regional specificity of alterations in NGF, BDNF and NT-3 levels in Alzheimer’s disease. NeuroReport 7(18):2925–2928
Durany N et al (2000) Brain-derived neurotrophic factor and neurotrophin-3 levels in Alzheimer’s disease brains. Int J Dev Neurosci 18(8):807–813
Hock C et al (2000) Region-specific neurotrophin imbalances in Alzheimer disease: decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch Neurol 57(6):846–851
Yan Y-h et al (2016) Neurotrophin-3 promotes proliferation and cholinergic neuronal differentiation of bone marrow-derived neural stem cells via notch signaling pathway. Life Sci 166:131–138
Staudt MD et al (2016) Advances in neurotrophic factor and cell-based therapies for Parkinson’s disease: a mini-review. Gerontology 62(3):371–380
Jia C et al (2019) Ciliary neurotrophic factor is a key sex-specific regulator of depressive-like behavior in mice. Psychoneuroendocrinology 100:96–105
Hashimoto Y et al (1999) Synergistic effects of brain-derived neurotrophic factor and ciliary neurotrophic factor on cultured basal forebrain cholinergic neurons from postnatal 2-week-old rats. Dev Brain Res 115(1):25–32
Qu HY et al (2008) Transducible P11-CNTF rescues the learning and memory impairments induced by amyloid-beta peptide in mice. Eur J Pharmacol 594(1–3):93–100
Mitsumoto H, Tsuzaka K (1999) Neurotrophic factors and neuromuscular disease: I. General comments, the neurotrophin family, and neuropoietic cytokines. Muscle Nerve: Off J Am Assoc Electrodiagn Med 22(8):983–999
Garcia P et al (2010) Ciliary neurotrophic factor cell-based delivery prevents synaptic impairment and improves memory in mouse models of Alzheimer’s disease. J Neurosci 30(22):7516–7527
Baazaoui N (2016) Effect of CNTF derived peptide, P021 on cognition and pathology in 3xTG-AD mouse model of Alzheimer’s disease. City University of New York, New York
Bi G et al (2018) Therapeutic effect of transmembrane TAT-tCNTF via Erk and Akt activation using in vitro and in vivo models of Alzheimer’s disease. Int J Clin Exp Pathol 11(4):1855
Wang D et al (2013) Cardiotrophin-1 (CTF1) ameliorates glucose-uptake defects and improves memory and learning deficits in a transgenic mouse model of Alzheimer’s disease. Pharmacol Biochem Behav 107:48–57
Wang D et al (2015) Cardiotrophin-1 (CT-1) improves high fat diet-induced cognitive deficits in mice. Neurochem Res 40(4):843–853
Wang D et al (2017) Treatment effects of Cardiotrophin-1 (CT-1) on streptozotocin-induced memory deficits in mice. Exp Gerontol 92:42–45
Sidorova Y, Saarma M (2016) Glial cell line-derived neurotrophic factor family ligands and their therapeutic potential. Mol Biol 50(4):521–531
Budni J et al (2015) The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis 6(5):331
Krashia P, Nobili A, D’Amelio M (2019) Unifying hypothesis of dopamine neuron loss in neurodegenerative diseases: focusing on Alzheimer’s disease. Front Mol Neurosci 12:123
Mitra S et al (2021) Increased endogenous GDNF in mice protects against age-related decline in neuronal cholinergic markers. Front Aging Neurosci. https://doi.org/10.3389/fnagi.2021.714186
Airavaara M et al (2011) Identification of novel GDNF isoforms and cis-antisense GDNFOS gene and their regulation in human middle temporal gyrus of Alzheimer disease. J Biol Chem 286(52):45093–45102
Sharif M, Noroozian M, Hashemian F (2021) Do serum GDNF levels correlate with severity of Alzheimer’s disease? Neurol Sci 42(7):2865–2872
Marksteiner J et al (2011) Five out of 16 plasma signaling proteins are enhanced in plasma of patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging 32(3):539–540
Ledda F et al (2007) GDNF and GFRα1 promote formation of neuronal synapses by ligand-induced cell adhesion. Nat Neurosci 10(3):293
Ghribi O et al (2001) GDNF protects against aluminum-induced apoptosis in rabbits by upregulating Bcl-2 and Bcl-XL and inhibiting mitochondrial Bax translocation. Neurobiol Dis 8(5):764–773
Revilla S et al (2014) Physical exercise improves synaptic dysfunction and recovers the loss of survival factors in 3xTg-AD mouse brain. Neuropharmacology 81:55–63
Revilla S et al (2014) Lenti-GDNF gene therapy protects against Alzheimer’s disease‐like neuropathology in 3xTg‐AD mice and MC65 cells. CNS Neurosci Ther 20(11):961–972
Petukhova EO et al (2019) Effects of transplanted umbilical cord blood mononuclear cells overexpressing GDNF on spatial memory and hippocampal synaptic proteins in a mouse model of Alzheimer’s disease. J Alzheimer’s Dis 69(2):443–453
Straten G et al (2011) Influence of lithium treatment on GDNF serum and CSF concentrations in patients with early Alzheimer’s disease. Curr Alzheimer Res 8(8):853–859
Petukhova EO et al (2019) Effects of transplanted umbilical cord blood mononuclear cells overexpressing GDNF on spatial memory and hippocampal synaptic proteins in a mouse model of Alzheimer’s disease. J Alzheimers Dis 69(2):443–453
Qing J et al (2020) Hippo/YAP Pathway Plays a Critical Role in Effect of GDNF Against Aβ-Induced Inflammation in Microglial Cells. DNA Cell Biol 39(6):1064–1071
Hiramatsu N et al (2015) Multiple mechanisms of unfolded protein response–induced cell death. Am J Pathol 185(7):1800–1808
Hoozemans J et al (2005) The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol 110(2):165–172
Nasrolahi A et al (2018) Neurotrophic factors hold promise for the future of Parkinson’s disease treatment: is there a light at the end of the tunnel? Rev Neurosci 29(5):475–489
Kemppainen S et al (2015) Cerebral dopamine neurotrophic factor improves long-term memory in APP/PS1 transgenic mice modeling Alzheimer’s disease as well as in wild-type mice. Behav Brain Res 291:1–11
Zhou W et al (2016) Cerebral dopamine neurotrophic factor alleviates Aβ25-35-induced endoplasmic reticulum stress and early synaptotoxicity in rat hippocampal cells. Neurosci Lett 633:40–46
Henriques A, Pitzer C, Schneider A (2010) Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: where do we stand? Front NeuroSci 4:32
McBride JL et al (2003) Structural and functional neuroprotection in a rat model of Huntington’s disease by viral gene transfer of GDNF. Exp Neurol 181(2):213–223
Zhu W et al (2009) Postischemic IGF-1 gene transfer promotes neurovascular regeneration after experimental stroke. J Cereb Blood Flow Metab 29(9):1528–1537
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Literature’s preparation was performed by ZS, AN, FJ, MJ-G, JM, and KDA. The first draft of the manuscript was written by ZS, AN, and KDA. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest to disclose.
Consent to participate
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nasrolahi, A., Javaherforooshzadeh, F., Jafarzadeh-Gharehziaaddin, M. et al. Therapeutic potential of neurotrophic factors in Alzheimer’s Disease. Mol Biol Rep 49, 2345–2357 (2022). https://doi.org/10.1007/s11033-021-06968-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-021-06968-9