
Abstract
Astrocytes play a wide variety of roles in the central nervous system (CNS). Various facets of astrocyte-neuron interplay, investigated for the past few decades, have placed these most abundant and important glial cell types to be of supreme importance for the maintenance of the healthy CNS. Interestingly, glial dysfunctions have proven to be the major contributor to neuronal loss in several CNS disorders and pathologies. Specifically, in the field of neuroAIDS, glial dysfunction–mediated neuronal stress is a major factor contributing to the HIV-1 neuropathogenesis. As there is increasing evidence that astrocytes harbor HIV-1 and serve as “safe haven” for the dormant virus in the brain, the indirect pathway of neuronal damage has taken over the direct neuronal damage in its contribution to HIV-1 neuropathogenesis. In this review, we provide a brief insight into the astrocyte functions and dysfunctions in different CNS conditions with an elaborated insight into neuroAIDS. Detailed understanding of the role of astrocytes in neuroAIDS will help in the better therapeutic management of the neurological problems associated with HIV-1 patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Astrocytes, the star-shaped cells, are the most abundant cell type of the brain. These glial cells overshadow neurons both in numbers as well as the diversity of roles they play in the mammalian brain. Astrocytes gathered due attention of basic and clinical researchers worldwide, majorly as they play a variety of roles in maintaining the central nervous system (CNS) homeostasis. Astrocytes are also implicated in CNS disorders and pathologies that necessitated in-depth research to understand their functioning in normal and diseased conditions. Last two decades witnessed immense interest and diligent research efforts to understand the mechanisms of basic functions of astrocytes. Several anatomical, genetic, and functional investigations on human and other mammalian brains designate astrocytes to be crucial in human brain evolution and critical for the improved cognitive abilities of humans (Robertson 2014). The neuro-centric view has now shifted towards astrocytes, with insights into emerging research areas to understand the intricacies of the dynamic cross talk between glia and non-glia cell types. These have paved way for studies into the glia-neuron interactions that further provided new insights into brain functioning and helped to elevate the hierarchy of astrocytes among other brain cells, especially in terms of their functions and relevance for CNS.
As mentioned earlier, astrocytes were studied extensively in the last 20 years and are now believed to be the most important cell types of CNS. This review is an attempt to provide a brief account of astrocyte functions and their relationships with neurons and list out their most important functions in the healthy and diseased brain with special emphasis on neuroAIDS.
Astrocyte-Neuron Interplay in Healthy CNS
Astrocytes play diverse roles ranging from early brain development to lifelong trophic and metabolic support to neurons. Some of the basic functions of astrocytes are the maintenance of pH, water and ionic homeostasis, release and uptake of neurotransmitters, maintenance of blood flow, and maintaining the integrity of blood-brain barrier. Astrocytes also modulate synaptic transmission, confer neuronal protection, and help in detoxification and repair of the nervous system.
The neurotransmitters released by the neurons trigger astrocytic [Ca2+]i elevations leading to the release of gliotransmitters such as glutamate, D-serine, and ATP, which further cause neuronal excitation (Fellin et al. 2004; Lee et al. 2010; Newman 2003; Parpura et al. 1994; Yang et al. 2003). Such shreds of evidence suggest that astrocytes get influenced and also can influence the flow of information in the neural circuitry, strengthening the evidence for interplay between these two important cells of the brain. The uptake of the excess of glutamate present at the synapse site through the efficient glutamate transporters is a key step in the maintenance of glutamate homeostasis. Glutamate is further converted to glutamine and then transported to the neurons (Bergles and Jahr 1998; Hertz and Zielke 2004).
The astrocytic activity has the potential to affect almost every aspect of neuronal functioning by influencing blood flow, energy, and synapses. Investigations for the past two decades highlight the role of astrocytes in influencing the cerebral blood flow. The neuronal activity–induced dilation of the arterioles is dependent on the glutamate-mediated intracellular Ca2+ oscillations seen in astroglia as the activity-dependent vasodilation was found to be impaired after blocking astrocytic calcium responses, both in vitro and in vivo (Takano et al. 2006; Zonta et al. 2003). Metabolic support provided by astrocytes to neurons in the form of lactate is crucial for events such as long-term memory formation. Increase in lactate release as a result of astrocytic glycogen breakdown during learning is essential for the maintenance of long-term potentiation (LTP) and memory formation. Disruption of astrocytic lactate transporters, monocarboxylate transporter 4 (MCT4) which exports the lactate out of the cell, causes amnesia, which can be rescued by L-lactate (Suzuki et al. 2011).
Astrocytes play an important extrinsic role in controlling spine formation, their density, and maturation of adult-born hippocampal neurons. Sultan et al. demonstrated vesicular release from astrocytes, especially the release of D-serine, which plays a critical role in local dendritic spine maturation, critical for functional integration of newborn neurons to the circuitry (Sultan et al. 2015). Further, astrocytes also control and support synaptogenesis in the CNS through the release of soluble signals. Christopherson et al. observed that members of the thrombospondin family play a significant role in synaptogenesis. Thrombospondins released by the immature astrocytes of the developing brain is necessary and sufficient for inducing synaptogenesis, although post-synaptically silent, in rat retinal ganglion cells (RGCs) (Christopherson et al. 2005). A study focusing on the mechanism of thrombospondin-mediated synaptogenesis revealed that this synaptogenic activity of thrombospondins is mediated through the neuronal thrombospondin receptor a2S-1 (Eroglu et al. 2009). In order to delineate the signal molecules that regulate functional synapse formation, experiments performed on RGCs cultured on astrocytic feeder layer revealed that astrocyte-derived signals enhance the levels and clustering of pre-existing AMPARs on the neuronal surface. Detailed biochemical analysis confirmed that glypicans 4 and 6 are the signal molecules present in the astrocytic-conditioned media that regulate the formation of post-synaptically active synapses (Allen et al. 2012). Other astrocyte-secreted signals, belonging to matricellular proteins, which modulate synapse formation, are Hevin and SPARC. Hevin enhances synaptogenesis whereas SPARC specifically antagonizes the hevin-mediated synaptogenesis both in vitro and in vivo. The balance between the two determines the formation and morphology of the synapses (Kucukdereli et al. 2011). These functions attributed to astrocytes make them almost indispensable for optimal functioning and survival of neurons.
Astrocyte-Neuron Interaction in CNS Disorders and Pathology
Astrocytes undergo various morphological, molecular, and functional alterations following CNS insult. These alterations can be beneficial or detrimental to the neurons in vicinity based on the challenges faced by the astrocytes. Reactive astrogliosis, characterized by upregulated glial fibrillary acidic protein (GFAP), increased cell proliferation, morphological changes, and enhanced release of various cytokines, affects neurons in both beneficial and detrimental ways (Sofroniew 2009). While astrocytes play several important roles for optimal functioning of neurons, deregulated functioning of astrocytes can perturb neuronal functions under diseased conditions and result in irreversible pathologies. A large number of reports support the idea that astrocyte dysfunction can be considered to be one of the major factors responsible for neurodegeneration in many neurological diseases. As it is difficult to discuss all the neurological disorders in this review, authors have limited the discussion to some of the major disorders, in brief.
CNS Trauma
CNS injury leads to tissue damage and often results in barriers to axonal regeneration. One such barrier is the glial scar comprised of reactive astrocytes and proteoglycans. Glial scar is a collective mass of reactive astrocytes beyond which the axons cannot grow resulting in dystrophic appearance. The reactive astrocytes in the glial scar produce inhibitory extracellular matrix molecules, mainly chondroitin and keratan sulfate proteoglycans which are thought to be playing a role in inhibiting axonal regeneration (Silver and Miller 2004).
Apart from acting as a barrier to axonal regeneration, reactive astrocytes also play protective roles in CNS trauma. Ablation of reactive astrocytes adjacent to the forebrain stab injury leads to neuronal degeneration, an increased outgrowth of the nerve fibers in the injured tissue, leukocyte infiltration, and failure of blood-brain barrier repair (Bush et al. 1999). Astrocytes protect tissue and preserve function after mild or moderate spinal cord injury (SCI). Ablation of reactive astrocytes in the close vicinity of the injury causes failure in blood-brain repair, leukocyte infiltration, local tissue disruption, severe demyelination, neuronal and oligodendrocyte death, failure of wound contraction, and motor deficits. Thus, preserving reactive astrocytes and its functions can help to reduce the secondary tissue degeneration in SCI (Faulkner et al. 2004).
Stroke and Cerebrovascular Disease
Astrocytes convert their glycogen to lactate and transport it to the neighboring neurons thus supporting their normal energy requirements in stressful conditions such as ischemia. This process delays the ATP loss and preserves energy status in both astrocytes and neurons during ischemia. In contrast, studies of hyperglycemia during stroke have shown that glycolysis occurring in the absence of oxygen can add to the ischemic damage because of the enhanced lactic acidosis (Lindsberg and Roine 2004).
In general, ischemia promotes the metabolism of astrocytic glycogen which induces astrocytic calcium overload. Increasing calcium overload in astrocytes causes increased ATP release which leads to glutamate release from astrocytes through the opening of P2X receptors culminating into glutamate excitotoxicity (Duan et al. 2003). Astrocytic glutamate vesicle release can also be induced by increased calcium release from the intracellular stores (Hua et al. 2004); this glutamate can cause neuronal excitation through mGluR. Perturbed glutamate transmission is a key contributor to ischemic brain damage. Astrocyte-neuron communication is substantially affected by ischemia as astrocyte is the major contributor to glutamatergic transmission.
A mitochondrial mechanism of neuroglial cross talk can be observed in conditions such as ischemic stroke where astrocytes release functional mitochondria that get transferred to neurons. A study using the rat model system proves that the release of extracellular mitochondrial particles is occurring through a Ca2+-dependent mechanism which involves CD38 and cyclic ADP ribose signaling (Hayakawa et al. 2016).
Alzheimer’s Disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and a leading cause of dementia across the globe. It is characterized by β-amyloid deposition, the formation of neuritic plaque and neurofibrillary tangles in the brain.
The disease progression is accompanied by alterations in astrocyte morphology and activities that make astrocytes reactive (Fu et al. 2015). Various signaling pathways related to astrocyte-neuron interaction, primarily the pathways involving calcium, proteoglycans, transforming growth factor β (TGF-β), nuclear factor-kappa B (NF-κB), and complement, have been reviewed in detail by Lian and Zheng (2016). Astrocyte migration and accumulation occurring at the site of Aβ deposition is a consequence of augmentation of a chemokine, monocyte chemoattractant protein-1 (MCP-1). The migration stops upon interaction with Aβ1–42. Astrocytes associate with Aβ deposits and carry out its degradation. The astroglial clearance of Aβ is impaired in AD, thus contributing to the disease progression (Wyss-Coray et al. 2003). Apolipoprotein E plays an important role in the astroglial clearance of Aβ deposits, as Apoe−/− astrocytes from adult mouse astrocytes are unable to degrade Aβ deposits in brain sections with Aβ plaque (Koistinaho et al. 2004).
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by the death of motor neurons in cortex, brain stem, and spinal cord that causes weakening of skeletal muscles. Rodent astrocytes expressing mutated SOD1 cause the death of motor neurons by releasing toxic factors through a Bax-dependent mechanism. Interestingly, these SOD1-mutated astrocytes are not harmful to other neurons such as spinal GABAergic or dorsal root ganglion or interneurons (Nagai et al. 2007).
Major loss of motor neurons is attributed to the neurotoxic levels of extracellular glutamate, linked to the impaired glutamate transport system in the brain and spinal cord of ALS patients (Rothstein et al. 1992). Spinal motor neurons show reduced expression of major histocompatibility complex class I (MHCI) in ALS. MHCI level in motor neurons determines the susceptibility to ALS astrocyte-induced toxicity (Song et al. 2016). Co-culturing of motor neurons with ALS astrocytes results in reduced expression of MHCI on motor neurons, thereby confirming the implication of astrocytes in ALS. The astroglial population may also respond to the neuronal loss, adding more to the neurodegeneration by influencing the disease progression. miRNA released extracellularly from the dying neurons affect the glial cells present in the arena. miRNA-218, for instance, released from the motor neuron are taken up by the astrocytes, modulates EAAT2 expression, and thus causes astrocyte dysfunction, which further adds to the neurodegenerative environment (Hoye et al. 2018).
Huntington’s Disease
Huntington’s disease (HD) is a disorder caused by the neurodegeneration of the striatal neurons. It is an autosomal dominant disorder, arising due to the expansion of CAG repeats in the gene called Huntingtin (Htt). The cognitive decline and the movement disorders seen in HD occur due to the neuronal loss in cortex and striatum (Kim et al. 2014; Nana et al. 2014).
Neuronal death in HD is caused by increased astrocytic activity seen in the form of enhanced release of proinflammatory cytokines, caused by prolonged NF-kB pathway activation (Hsiao et al. 2013). Using genetically encoded Ca2+ and glutamate indicators to track the astrocyte involvement in the corticostriatal circuit of adult WT and HD model mice, it has been demonstrated that WT striatal astrocytes show prominent spontaneous Ca2+ signals but did not show evoked Ca2+ response upon cortical stimulation. Whereas in HD mice model, the striatal astrocytes show reduced spontaneous Ca2+ signals but enhanced evoked Ca2+ signals upon cortical stimulation. The altered calcium and glutamate signaling are major results of Glt-1 dysfunction. Furthermore, K+ homeostasis is compromised in HD model mice. Restoring Kir4.1 can rescue the dysfunctional Ca2+ and glutamate signaling significantly (Jiang et al. 2016).
CNS Infections
Astrocyte-neuron communications are severely affected by the challenge with infectious agents. Reactive and scar-forming astrocytes play a crucial role in protecting neurons during some of the CNS infections. For example, GFAP+ astrocytes get activated, increase in number, and restrict the spread of invading pathogens such as Toxoplasma gondii into the CNS parenchyma (Drogemuller et al. 2008). Viral infections in the CNS pose a bigger health issue and have gained the attention of researchers worldwide. The vast arena of neurotropic viruses possessing their unique tropism makes the situation even more complex. Different viruses and the type of inflammation caused by them are nicely reviewed (Swanson and McGavern 2015).
HIV-1 Infection in CNS
Once the virus enters the systemic circulation, it seeds itself into the CNS during initial multiplication in the host and at the first peak of viremia. During acute HIV infection in humans, the viral DNA can be detected as early as 8 days post-exposure. Cerebrospinal fluid (CSF) analysis and magnetic resonance spectroscopy (MRS) further confirm the presence of the virus in the CNS. Infected immune cells of the blood such as T cells and monocytes cross the BBB and carry the virus to the brain, following the Trojan horse hypothesis. In the brain, perivascular macrophages and microglia are most susceptible to viral infection as they have both the CD4 receptor and chemokine co-receptors (Gonzalez-Scarano and Martin-Garcia 2005). Increase in microglial cell activity, multinucleated giant cell formation, the formation of myelin pallor, BBB breaching, astrocytic dysfunction, and neurotoxicity are the key outcomes of HIV-1 infection in the CNS (Elbirt et al. 2015).
Extensive investigations exploring the effects of HIV-1 show that the virus and the viral proteins can directly affect the neuronal population and thus causing the cognitive decline in AIDS patients. For instance, human primary neurons when exposed to viral proteins HIV-glycoprotein120 (gp120) and transactivator of transcription (Tat) show significant mitochondrial fragmentation and decreased mitochondrial membrane potential which further leads to deregulated mitochondrial dynamics, hence compromising the neuronal health (Teodorof-Diedrich and Spector 2018). HIV-1-associated neuroinflammation and cognitive decline are also observed in mouse model where the hippocampus is directly infused with viral proteins gp120 or Tat for 14 days. Microglial activation, upregulation of inflammatory mediators, and decrease in survival and proliferation of neural stem cells (NSCs) altogether lead to the cognitive dysfunction observed in the animal. HIV-1 proteins also affect the differentiation ability of the NSCs in the subgranular zone, as observed in the HIV-1 Tg26 transgenic mouse model. Increase in astrocytic differentiation and reduced neuronal differentiation marks the deficits in the early- and late-stage differentiation potential of the NSCs (Hill et al. 2019; Putatunda et al. 2018). Importantly, the time of the infection plays a role in disease progression and the development of cognitive deficits. In rats, exposure to HIV-1 viral proteins, Tat1–86 and gp120, on postnatal day 1 (P1) has severe effects on hippocampus anatomy and cognitive development. In contrast, exposure to P10 results in deficits in spatial learning specifically, without affecting the somatic growth (Fitting et al. 2018). HIV-1 gp120 also affects the striatal neurons by upregulating α7-nicotinic acetylcholine receptor (α7-nAChR) in the gp120IIIB-transgenic mouse (gp120-tgm) model. By mediating calcium overload in the cells, nAChR plays a key role in the gp120-induced neurotoxicity. Moreover, the use of α7-nAChR antagonists ameliorates the deficits observed in the striatum-dependent behavior in the mice (Capo-Velez et al. 2018).
With the advent of highly active antiretroviral therapy (HAART), AIDS patients are living longer, but it is estimated that nearly 50% of AIDS patients experience some degree of HIV-associated neurocognitive disorder (HAND) (Heaton et al. 2010). These motor and cognitive deficits are collectively termed as HAND and studied as a field of neuroAIDS. Although combinatorial antiretroviral therapy (cART) is successful, in most cases, in rapidly reducing HIV RNA copies to < 50 copies/ml, the virus typically rebounds back quickly, sometimes as early as within 2 weeks of cessation of therapy (Davey et al. 1999). To achieve successful treatment of AIDS, it is important to purge the viral reservoirs thoroughly. Hence, a detailed understanding of the neuroAIDS field and the niche areas that harbor the virus will surely help in better management of the disease as well as in improving therapeutic approaches.
Macrophages and microglia exhibit productive infection and are the major reservoirs of HIV in the brain. Microglia being long-lived cells can harbor the virus for a long period of time. They can also be activated by cytokines and viral proteins released by infected cells. Activated microglia may further cause neuronal damage by releasing excitatory amino acids (EAAs) and EAA-like substances such as quinolinate, glutamate, and cysteine, which induce neuronal apoptosis by causing excitotoxicity. Also, microglia-derived inflammatory cytokines, such as IL-1β and TNF-α, arachidonate, and its metabolites and viral proteins, cause indirect neurotoxicity (Kaul et al. 2001). In contrast, HIV infection in astrocytes is of very low level (Takahashi et al. 1996), and it is non-productive, reviewed in Churchill and Nath (2013). The restricted HIV-1 replication in astrocytes is because of insufficient translation of the structural proteins of the virus, such as Gag, Nev, and Nef. However, there occurs an efficient translation of Tat and Rev mRNA (Gorry et al. 1999). An RNA binding protein kinase (PKR) plays a role in restricting this translation. This PKR response is favored by the low levels of its antagonist transactivation response element (TAR) RNA binding protein (TRBP) (Ong et al. 2005). AIDS could have been a devastating CNS disorder if astrocytes were sites for productive infections for HIV-1. Despite the non-productive infection, the latently infected astrocytes produce some of the HIV-1 proteins, which are neurotoxic. HIV-1 Tat is detected even in patients, successfully treated with cART, with low to undetectable viral load in their systemic circulation, which indicates that Tat is sourced through HIV-1 residing in glial cells (Chauhan et al. 2003).
Both microglia and astrocytes may work synergistically and contribute to the indirect neuronal loss observed in HIV-1 neuropathogenesis. But the link between HIV-1 and astrocytes is little underexplored and awaits better exploration. The next section highlights some of the key findings which justify their link and also pave the way to focus on the astrocyte-mediated neuronal loss which may further amplify the neurotoxic effects of activated or infected microglia.
HIV-1 and Astrocytes
Although HIV-1 infection in astrocytes is considered rare, up to 19% of GFAP+ cells, of subjects with HIV-associated dementia, have HIV DNA. The frequency of infection increases when astrocytes are in close proximity to macrophage, especially at the perivascular regions. Also, the extent of infection is correlated with the severity of neuropathological changes observed in such patients (Churchill et al. 2009). Further several recent reviews have emphasized the role of astrocytes in HIV neuropathogenesis (Al-Harti et al. 2018; Barat et al. 2018; Churchill and Nath 2013; Gorry et al. 2003; Gray et al. 2014; Li et al. 2016). Studies focusing on pediatric HIV-1-associated neuropathology describe the presence of viral particles in some astrocytes. Particularly, the astrocytes in the subcortical white matter possess a detectable level of HIV-1 nucleic acid and thus can be considered as a reservoir (Tornatore et al. 1994). An interesting study focusing on the mode of transmission of HIV-1 to astrocytes shows that an efficient cell-to-cell contact and transmission of the virus occurs between T lymphocytes and astrocytes. Co-cultures of astrocytes and HIV-infected lymphocytes reveal that a CXCR4-dependent fusion process occurs between the two cells which result in budding of immature viral particles from lymphocytes to directly on the astrocyte membrane (Li et al. 2015).
Various CD4/CXCR4-independent pathways may also be involved in the entry of the virus into the cell. HIV virions have been observed in clathrin-coated pits suggesting the involvement of receptor-mediated endocytosis in viral entry. A 65-kDa surface protein acts as a receptor and facilitates specific binding to HIV-gp120 protein paving the way for the endocytic pathway. HIV infection decreases in Rab-depleted primary astrocytes, suggesting a CXCR4-independent viral entry by endocytosis. Also, in non-human R5 simian-human immunodeficiency virus (SHIV)–induced encephalitis primate model, experimental studies reveal that the virus can enter the brain in CD4-independent mechanisms and infect microglia and CD4 negative astrocytes as well (Chauhan et al. 2014; Hao and Lyman 1999; Zhuang et al. 2014).
Based on these findings, HIV-1 infection of astrocytes is no longer a matter of debate; however, there is an urgent need to gain an in-depth understanding of cellular and molecular mechanisms for astrocyte-mediated neuronal damage. In fact, the past decade has witnessed significant improvement in our understanding of the indirect pathways of HIV-1-induced astrocyte-mediated neuronal damage. It is hoped that this will help in the better therapeutic management of neuroAIDS patients.
Astrocytes Contribute to Neuronal Damage in NeuroAIDS
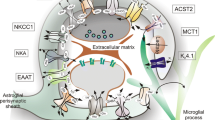
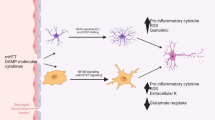
Glial dysfunction is an important contributing factor in HIV-1 neuropathogenesis. Apart from the direct neuronal death caused by HIV-1 and its proteins, a significant amount of neurotoxicity is mediated by astrocytes, in addition to the toxicity caused by activated or infected microglia, that further culminate into the indirect death of neurons. In the post-HAART era, indirect neuronal death is prevalent over direct death pathway, as most of the neuronal damage is through glia, the cell type that safely harbors the HIV-1 virions. There are several pathways that contribute to astrocyte-mediated neuronal damage; some of the important ones are discussed under this section (also see Fig. 1).

A tripartite synapse showing astrocyte-neuron communication
Astrocyte Gene Dysfunctions
HIV and its proteins cause astrogliosis by inducing several modulations in the astrocyte genetic machinery which further leads to astrocyte dysfunctions, most of which happen to be detrimental for the neurons in the vicinity. Simian immunodeficiency virus (SIV)–infected macaques possess decreased expression of aquaporin 4 (AQP4) and excitatory amino acid transporter-2 (EAAT2) in the frontal cortex. Moreover, swelling and vacuolar changes of astrocytic processes are observed especially in the perivascular area in the frontal cortex. Interestingly, areas of frontal cortex with reduced AQP4 tend to show intense gliosis (Xing et al. 2017). HIV-1 protein Tat expression in mouse primary astrocytes results in GFAP upregulation, aggregate formation, and intermediate filament alterations. These events activate ER stress which is further involved in astrocyte-mediated Tat neurotoxicity. In addition to the ER stress, tat also promotes lysosomal exocytosis in astrocytes, thus aggravating the neurotoxicity (Fan and He 2016a, b). Also, HIV-1 gp120 induces apoptosis through ER stress. These ER-mediated effects are IRE1a, JNK, and AP-1 pathway dependent (Shah et al. 2016).
A transgenic (tg) mouse model, GFAP-gp120 tg, expressing gp120 under the control of the promoter of GFAP in astrocytes, manifests several features similar to those observed in brains of AIDS patients. This further provides a tool to study the effects of viral protein on astrocyte functions. The viral proteins also affect the signal transduction pathways and thus contribute to pathogenesis. HIV-1 gp120 exposure to both C6 astrocytoma cells and transgenic mouse astrocytes leads to upregulation of protein kinase C (PKC). Also, the brain tissue of HIV-1 encephalitis patients shows enhanced PKC immunoreactivity which further contributes to astrogliosis observed in HIV-1 neuropathogenesis (Thaney et al. 2018; Wyss-Coray et al. 1996).
Altered transcriptional regulation of several inflammatory molecules in astrocytes in the presence of the virus or viral proteins is a major factor in HIV-1 neuropathogenesis. Histone deacetylase 6 (HDAC6), a member of class IIb HDACs, plays key roles in such regulations. Importantly, HIV-1 Tat induces HDAC6 expression in primary mouse astrocytes. HDAC6 activates mitogen-activated protein kinases which further regulate NF-kB/AP-1 pathways, thereby inducing the expression of neurotoxic chemokines and adhesion molecules (Soo Youn et al. 2015). A correlation study focusing on the cross talk between HDAC6 and NADPH oxidase has shown that knocking down one out of the two reduces the HIV-Tat induced expression of the other and reduces the generation of ROS and proinflammatory chemokines such as CCL2, C-X-C motif chemokine ligand 8 (CXCL8), and CXCL10 (Youn et al. 2017). Few pieces of evidence point towards the fact that deregulation of the complement system may be a contributing factor in disease pathogenesis. Complement components may contribute to astrocyte dysfunction in the CNS as well. A recent study reported the higher expression of complement components in brain tissue of HAND patients. Specifically, HIV indirectly induces C3 expression through NF-kB–driven IL-6 induction that further activates C3 promoter (Nitkiewicz et al. 2017).
Surface receptors or transmembrane proteins may also play a role in mediating the molecular mechanisms which further may contribute astrocyte dysfunction seen in HIV-1 neuropathogenesis. CD38, a type II single-chain transmembrane glycoprotein expression has been reported to be upregulated in primary human fetal astrocytes co-stimulated with HIV-1 and IL-1β when compared with untreated or HIV-1 alone–treated group. Altered CD38 activity and signaling then contribute to the astrocyte reactivity. Increased CD38 expression has also been validated on brain tissue specimens from HIVE patients. Also, HIV-1 gp120 enhances CD38 expression, in a dose-dependent manner, and upregulates the enzyme activity (Banerjee et al. 2008; Kou et al. 2009). Immunohistochemical assessment of tissues from HAD cases also reveals the accumulation of nuclear p53 protein in glial cells. A subpopulation of both astrocytes and microglia shows p53 immunoreactivity in the HIV-infected brain. This p53 activation promotes upregulation of proapoptotic genes such as Bax and also cyclin-dependent kinase inhibitor, p21WAF1 which promotes cell cycle arrest (Jayadev et al. 2007).
Energy metabolism of glial cells in the neuroAIDS is an area which has not received due attention by the researchers of the field. However, there are few reports that discuss the altered levels of the brain metabolites observed in neuroAIDS. Using 1H MRS on macaque models of neuroAIDS, decrease in N-acetyl aspartate (NAA) and increase in creatine concentration have been reported. Neuronal loss, as indicated by increased NAA, has been further validated by an experiment which shows downregulated expression of synaptophysin, microtubule-associated protein-2, as well as the reduced density of the neuronal population. This study also attributes the high levels of creatine to the enhanced metabolism of the activated glial cell types in neuroAIDS conditions (Ratai et al. 2011).
Studying the spatial relationship between astrocytes and neurons in inflammatory conditions may provide us with even more information about their interactions. Alterations, if any, in spatial relationships may affect the brain function. In an investigation done on superior frontal gyrus from 29 patients who died of AIDS, it was observed that a unique spatial pattern exists between astrocytes and neurons. As dementia progresses, astrocytes showed a clustered pattern. Moreover, bivariate spatial pattern analysis reveals that as dementia progresses, astrocytes and neurons tend to stay away from each other whereas astrocytes and interneurons showed evidence of staying in close proximity to each other (Roberts et al. 2013). Detailed investigations are needed in support of such observations and also to investigate the mechanisms responsible for such spatial relationships in HAND pathogenesis.
Glutamate Excitotoxicity
The excess of glutamate at the synapse leads to sustained opening and thus heavy inflow of ions across the channels which further activate Ca2+ signaling and also Ca2+-dependent downstream effectors, leading to neuronal damage. One of the major contributions of astrocyte in the maintenance of neuronal physiology is the uptake of an excess of glutamate that occurs through glutamate transporters EAAT1 and EAAT2. HIV-1 infection or the viral proteins (gp120 and Tat) reduce the expression of these glutamate transporters present on the astrocyte membrane (Wang et al. 2003; Zhou et al. 2004). This results in poor clearance and ultimately the excess of glutamate concentrations, which causes excitotoxicity to the neurons and hence the very cells that were supposed to be friends of neurons, turn foe.
A study, which directly links HIV-1 Tat to astrocytosis and subsequent neuronal death, shows Tat significantly increases GFAP, a marker of astrocytic activity. Moreover, Tat disturbs the glutamate uptake functioning of astrocytes. Further, cell supernatants from these deregulated astrocytes cause significant neuronal death (Zhou et al. 2004). Glutamate disbalance and neurotoxicity are also associated with reduced levels of heme oxygenase-1 (HO-1), a phase II detoxifying enzyme, in brains of HAND patients. A major portion of HO-1 in the brain is contributed by the astrocytes. In prolonged HIV infection, IFNγ downregulates the protein levels of HO-1 without reducing its RNA levels. The reduced expression of HO-1 is linked to the enhanced immunoproteasome subunits (Kovacsics et al. 2017). The impaired glutamate uptake by astrocytes can also be a secondary outcome of the effects of HIV protein gp120 on macrophages. Under the effect of gp120, macrophages release arachidonic acid which further inhibits the glutamate uptake by astrocytes (Dreyer and Lipton 1995).
Release of Proinflammatory and Neurotoxic Substances
The cytokines released by the infected and/or activated microglia and macrophages further activate the astroglial cells which lead to enhanced release of proinflammatory cytokines from the astroglial cells which culminate to neurotoxicity. Some of the signaling molecules released by astrocytes along with pathways mediating their release are discussed in this section.
Extracellular signaling molecule such as adenosine-5′-triphosphate (ATP) is released by both astrocytes and neurons; its levels are found to be high in several pathological conditions including neuroAIDS. Experiments performed on primary progenitor-derived astrocytes and neurons suggest that voltage-dependent anion channel 1 (VDAC1) expression increases in response to HIV-1 Tat leading to the enhanced release of ATP. VDAC1 expression was further found to be regulated by miR-320a. Enhanced ATP release mediated by miR320a-VDAC1 axis is an important contributing factor in astrocyte-mediated neuronal loss seen in neuroAIDS (Fatima et al. 2017). ATP also serves as a ligand for a family of receptors known as purinergic receptors. It has been demonstrated that P2X7 receptors (a type of purinergic receptor) play a significant role in HIV-1 Tat-induced astrocyte-mediated neurotoxicity. By enhancing P2X7R expression, HIV-1 Tat mediates enhanced release of chemokine (C-C motif) ligand 2 (CCL2) in Ca2+- and extracellular signal-regulated kinase (ERK)1/2-dependent manner, thereby augmenting infected monocyte infiltration into the brain (Tewari et al. 2015). Also, HIV-1 protein Nef-expressing human astrocytic U251MG cells show upregulated expression of CCL2/MCP-1 which is a chemoattractant for monocytes/macrophages and T cells. This study also showed that calmodulin mediates the process of Nef-induced CCL2/MCP-1 expression in astrocytes (Lehmann et al. 2006). Other chemokines such as chemokine (C-C motif) ligand 5 (CCL5) production in astrocytes is induced by HIV-1 viral proteins such as Tat, viral protein R (Vpr), Nef, and gp120. In astrocytes, these HIV-1 proteins activate PI3K-Akt and p38 MAPK pathways which further activate downstream transcription factors such as NF-kB, C/EBPα, C/EBPγ, and AP-1 to regulate the expression of CCL5 (Gangwani et al. 2013; Liu et al. 2014; Nookala et al. 2013; Shah et al. 2011). Co-stimulation of primary human astrocytes with HIV-1 Tat, IFN-γ, and TNF-α shows increased induction of neurotoxic chemokine CXCL10. This increase is regulated by p38, Jnk, and Akt signaling pathways and the transcription factors downstream to these pathways (Williams et al. 2009).
The deleterious effects of reactive astrocytes can also be conferred upon the neuronal population by nitric oxide (NO) production by the astroglial cells infected with HIV-1. HIV-1 Tat gene expressing human astrocytes show enhanced expression of inducible nitric oxide synthase (iNOS) and also NO production. These processes occur under the effect of NF-kB and C/EBPβ activation. Also, HIV-1 gp120 exposure to astrocytes induces iNOS and further causes astrogliosis. Ascorbate incubation attenuates the astrocyte hyperactivity and prevents neuronal injury, thus confirming the NO-mediated toxicity seen in neuroAIDS (Liu et al. 2002; Walsh et al. 2004). By modulating the levels of arachidonic acid as well, HIV-1 gp120 inhibits neuronal NOS with subsequent activation of IL-1b and iNOS transcription in astrocytes, ultimately contributing to neuronal stress (Persichini et al. 2014).
Astrocytes once infected with HIV-1 not only release an excess of signaling molecules but also can release viral proteins encapsulated in vesicles which bring about their effects after entering neurons. HIV-1 Nef is released in such extracellular vesicles from primary human fetal astrocytes expressing HIV-1 Nef. These vesicles carrying Nef cause neuronal toxicity that is manifested in the form of axonal and neurite degeneration, and they also alter the neuronal action potential, contributing to neurotoxicity seen in HAND patients (Sami Saribas et al. 2017).
WNT/β-Catenin Signaling Deregulation
A significant number of studies show that Wnt, a glycoprotein, acting independently or via β-catenin, plays important roles in regulating CNS activities. Experiments performed on astrocytic cell lines U87MG, U251MG, and primary progenitor-derived astrocytes reveal that both endogenously expressed and exogenously added HIV-Tat downregulate β-catenin/Wnt signaling that was dependent on the intact core and cysteine-rich domains of Tat. The downregulation of the β-catenin signaling by Tat is mediated through miRNAs (Henderson et al. 2012; Sardo et al. 2016). Tat binding to a subset of miRNAs may alter the cellular miRNA profile and thereby affect the CNS.
In the context of studying the interaction between peripheral blood mononuclear cells (PBMCs) and astrocytes, it has been observed that human progenitor-derived astrocytes have a distinct profile of released Wnt ligands. Out of all the released ligands, Wnt 2b and Wnt 10b expressions increase post-HIV infection. Using astrocytic-conditioned media over PBMCs, it has been observed that Wnts inhibit HIV replication in PBMCs with Wnt 1 and Wnt 2b having the highest inhibitory effect. In contrast, conditioned media from the activated CD8+ T cells enhance HLA-DR and IFNγ in astrocytes. Also, it enhances the HIV infection in astrocytes in an IFNγ/Stat-3–dependent manner (Richards et al. 2015).
Hemichannel or Gap Junction–Mediated Toxicity
Junction proteins form hemichannels and/or gap junctions. These contribute to the signal transmission and intercellular trafficking in astrocytes and modulate cell activity affecting neuronal physiology. The gap junction communication amplifies the adverse effects of even a very small fraction of astrocytes infected with HIV-1. For instance, a study has demonstrated that the expression of a gap junction protein connexin 43 (Cx43) gets upregulated under the effect of HIV-1 Tat. This leads to an active communication among astrocytes including the transfer of toxic signals from infected to uninfected astrocytes, causing bystander killing (Berman et al. 2016). The gap junctions mediate the adverse effects of HIV-1-infected astrocytes on the blood-brain barrier (BBB) integrity (Eugenin et al. 2011). Alterations in these proteins in astrocytes also affect the neuronal population. HIV infection of human astrocytes leads to an increased opening of Cx43 hemichannels which mediates an increase in expression and secretion of a soluble Wnt signaling inhibitor Dickkopf-1 (DKK1). HIV-mediated upregulation of DKK1 and exogenous addition of DKK1 both lead to the collapse of neuronal processes (Orellana et al. 2014).
Conclusion
Recent research works on astrocyte biology have proved the importance of this cell type in maintaining the health of the brain. The dynamic interaction of astroglia with the other cell types in contexts such as astrocyte-neuron communication, synaptic transmission, plasticity, neural protection, and repair have made these cells indispensable for the maintenance of healthy CNS. In fact, the cross talk is dynamic and bidirectional; recently, few studies have also suggested that neurons modulate astrocytic functions (Hasel et al. 2017), which has opened a new avenue of research. Discoveries of new roles for astrocytes are continuing even today, while this review was being compiled; a study performed in rats suggests that astrocytes help the brain to tune breathing rhythms (Sheikhbahaei et al. 2018). Astrocyte deregulations have emerged to be one of the important contributing factors of the several CNS disorders and pathology. Specifically, in neuroAIDS, despite the low productive infections, astrocytes amplify the neurotoxic effects of microglia to contribute to neuronal damage through different mechanisms. Although dissecting out those mechanisms demand much more exploration, some of them include the release of proinflammatory or neurotoxic substances, glutamate misbalance, wnt/β-catenin signaling deregulations, the spread of toxicants via hemichannels, and gap junctions. Adding new observations to the existing pool of information will lead to a better understanding of astrocyte physiology and its altered behavior in neuroAIDS and other CNS disorders and pathology.
References
Al-Harti L, Joseph J, Nath A (2018) Astrocytes as an HIV CNS reservoir: highlights and reflections of an NIMH-sponsored symposium. J Neurovirol 24:665–669
Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, Barres BA (2012) Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486:410–414
Banerjee S, Walseth TF, Borgmann K, Wu L, Bidasee KR, Kannan MS, Ghorpade A (2008) CD38/cyclic ADP-ribose regulates astrocyte calcium signaling: implications for neuroinflammation and HIV-1-associated dementia. J NeuroImmune Pharmacol 3:154–164
Barat C, Proust A, Deshiere A, Leboeuf M, Drouin J, Tremblay MJ (2018) Astrocytes sustain long-term productive HIV-1 infection without establishment of reactivable viral latency. Glia 66:1363–1381
Bergles DE, Jahr CE (1998) Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci 18:7709–7716
Berman JW, Carvallo L, Buckner CM, Luers A, Prevedel L, Bennett MV, Eugenin EA (2016) HIV-tat alters Connexin43 expression and trafficking in human astrocytes: role in NeuroAIDS. J Neuroinflammation 13:54
Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV (1999) Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23:297–308
Capo-Velez CM et al (2018) The alpha7-nicotinic receptor contributes to gp120-induced neurotoxicity: implications in HIV-associated neurocognitive disorders. Sci Rep 8:1829
Chauhan A, Mehla R, Vijayakumar TS, Handy I (2014) Endocytosis-mediated HIV-1 entry and its significance in the elusive behavior of the virus in astrocytes. Virology 456-457:1–19
Chauhan A, Turchan J, Pocernich C, Bruce-Keller A, Roth S, Butterfield DA, Major EO, Nath A (2003) Intracellular human immunodeficiency virus Tat expression in astrocytes promotes astrocyte survival but induces potent neurotoxicity at distant sites via axonal transport. J Biol Chem 278:13512–13519
Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120:421–433
Churchill M, Nath A (2013) Where does HIV hide? A focus on the central nervous system. Curr Opin HIV AIDS 8:165–169
Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, Gorry PR (2009) Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol 66:253–258
Davey RT Jr et al (1999) HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci U S A 96:15109–15114
Dreyer EB, Lipton SA (1995) The coat protein gp120 of HIV-1 inhibits astrocyte uptake of excitatory amino acids via macrophage arachidonic acid. Eur J Neurosci 7:2502–2507
Drogemuller K, Helmuth U, Brunn A, Sakowicz-Burkiewicz M, Gutmann DH, Mueller W, Deckert M, Schluter D (2008) Astrocyte gp130 expression is critical for the control of toxoplasma encephalitis. J Immunol 181:2683–2693
Duan S, Anderson CM, Keung EC, Chen Y, Chen Y, Swanson RA (2003) P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J Neurosci 23:1320–1328
Elbirt D, Mahlab-Guri K, Bezalel-Rosenberg S, Gill H, Attali M, Asher I (2015) HIV-associated neurocognitive disorders (HAND). Isr Med Assoc J 17:54–59
Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Özkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA (2009) Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139:380–392
Eugenin EA, Clements JE, Zink MC, Berman JW (2011) Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J Neurosci 31:9456–9465
Fan Y, He JJ (2016a) HIV-1 tat induces unfolded protein response and endoplasmic reticulum stress in astrocytes and causes neurotoxicity through glial fibrillary acidic protein (GFAP) activation and aggregation. J Biol Chem 291:22819–22829
Fan Y, He JJ (2016b) HIV-1 tat promotes lysosomal exocytosis in astrocytes and contributes to astrocyte-mediated tat neurotoxicity. J Biol Chem 291:22830–22840
Fatima M, Prajapati B, Saleem K, Kumari R, Mohindar Singh Singal C, Seth P (2017) Novel insights into role of miR-320a-VDAC1 axis in astrocyte-mediated neuronal damage in neuroAIDS. Glia 65:250–263
Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV (2004) Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci 24:2143–2155
Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G (2004) Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43:729–743
Fitting S, McLaurin KA, Booze RM, Mactutus CF (2018) Dose-dependent neurocognitive deficits following postnatal day 10 HIV-1 viral protein exposure: relationship to hippocampal anatomy parameters. Int J Dev Neurosci 65:66–82
Fu W, Shi D, Westaway D, Jhamandas JH (2015) Bioenergetic mechanisms in astrocytes may contribute to amyloid plaque deposition and toxicity. J Biol Chem 290:12504–12513
Gangwani MR, Noel RJ Jr, Shah A, Rivera-Amill V, Kumar A (2013) Human immunodeficiency virus type 1 viral protein R (Vpr) induces CCL5 expression in astrocytes via PI3K and MAPK signaling pathways. J Neuroinflammation 10:136
Gonzalez-Scarano F, Martin-Garcia J (2005) The neuropathogenesis of AIDS. Nat Rev Immunol 5:69–81
Gorry PR, Howard JL, Churchill MJ, Anderson JL, Cunningham A, Adrian D, McPhee D, Purcell DF (1999) Diminished production of human immunodeficiency virus type 1 in astrocytes results from inefficient translation of gag, env, and nef mRNAs despite efficient expression of. Tat Rev J Virol 73:352–361
Gorry PR, Ong C, Thorpe J, Bannwarth S, Thompson K, Gatignol A, Wesselingh S, Purcell D (2003) Astrocyte infection by HIV-1: mechanisms of restricted virus replication, and role in the pathogenesis of HIV-1-associated dementia. Curr HIV Res 1:463–473
Gray LR, Roche M, Flynn JK, Wesselingh SL, Gorry PR, Churchill MJ (2014) Is the central nervous system a reservoir of HIV-1? Curr Opin HIV AIDS 9:552–558
Hao HN, Lyman WD (1999) HIV infection of fetal human astrocytes: the potential role of a receptor-mediated endocytic pathway. Brain Res 823:24–32
Hasel P, Dando O, Jiwaji Z, Baxter P, Todd AC, Heron S, Márkus NM, McQueen J, Hampton DW, Torvell M, Tiwari SS, McKay S, Eraso-Pichot A, Zorzano A, Masgrau R, Galea E, Chandran S, Wyllie DJA, Simpson TI, Hardingham GE (2017) Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat Commun 8:15132
Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535:551–555
Heaton RK, Clifford DB, Franklin DR, Woods SP, Ake C, Vaida F, Ellis RJ, Letendre SL, Marcotte TD, Atkinson JH, Rivera-Mindt M, Vigil OR, Taylor MJ, Collier AC, Marra CM, Gelman BB, McArthur JC, Morgello S, Simpson DM, McCutchan JA, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, For the CHARTER Group (2010) HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER study. Neurology 75:2087–2096
Henderson LJ, Sharma A, Monaco MC, Major EO, Al-Harthi L (2012) Human immunodeficiency virus type 1 (HIV-1) transactivator of transcription through its intact core and cysteine-rich domains inhibits Wnt/beta-catenin signaling in astrocytes: relevance to HIV neuropathogenesis. J Neurosci 32:16306–16313
Hertz L, Zielke HR (2004) Astrocytic control of glutamatergic activity: astrocytes as stars of the show. Trends Neurosci 27:735–743
Hill JD, Zuluaga-Ramirez V, Gajghate S, Winfield M, Persidsky Y (2019) Chronic intrahippocampal infusion of HIV-1 neurotoxic proteins: a novel mouse model of HIV-1 associated inflammation and neural stem cell dysfunction J Neuroimmune Pharmacol
Hoye ML, Regan MR, Jensen LA, Lake AM, Reddy LV, Vidensky S, Richard JP, Maragakis NJ, Rothstein JD, Dougherty JD, Miller TM (2018) Motor neuron-derived microRNAs cause astrocyte dysfunction in amyotrophic lateral sclerosis. Brain 141:2561–2575
Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y (2013) A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington’s disease. Hum Mol Genet 22:1826–1842
Hua X, Malarkey EB, Sunjara V, Rosenwald SE, Li WH, Parpura V (2004) Ca(2+)-dependent glutamate release involves two classes of endoplasmic reticulum Ca(2+) stores in astrocytes. J Neurosci Res 76:86–97
Jayadev S, Yun B, Nguyen H, Yokoo H, Morrison RS, Garden GA (2007) The glial response to CNS HIV infection includes p53 activation and increased expression of p53 target genes. J NeuroImmune Pharmacol 2:359–370
Jiang R, Diaz-Castro B, Looger LL, Khakh BS (2016) Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J Neurosci 36:3453–3470
Kaul M, Garden GA, Lipton SA (2001) Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410:988–994
Kim HA, Jiang L, Madsen H, Parish CL, Massalas J, Smardencas A, O’Leary C, Gantois I, O’Tuathaigh C, Waddington JL, Ehrlich ME, Lawrence AJ, Drago J (2014) Resolving pathobiological mechanisms relating to Huntington disease: gait, balance, and involuntary movements in mice with targeted ablation of striatal D1 dopamine receptor cells. Neurobiol Dis 62:323–337
Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM (2004) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 10:719–726
Kou W, Banerjee S, Eudy J, Smith LM, Persidsky R, Borgmann K, Wu L, Sakhuja N, Deshpande MS, Walseth TF, Ghorpade A (2009) CD38 regulation in activated astrocytes: implications for neuroinflammation and HIV-1 brain infection. J Neurosci Res 87:2326–2339
Kovacsics CE, Gill AJ, Ambegaokar SS, Gelman BB, Kolson DL (2017) Degradation of heme oxygenase-1 by the immunoproteasome in astrocytes: a potential interferon-gamma-dependent mechanism contributing to HIV neuropathogenesis. Glia 65:1264–1277
Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA, Eroglu C (2011) Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A 108:E440–E449
Lee S et al (2010) Channel-mediated tonic GABA release from glia science, vol 330, New York, NY, pp 790–796
Lehmann MH, Masanetz S, Kramer S, Erfle V (2006) HIV-1 Nef upregulates CCL2/MCP-1 expression in astrocytes in a myristoylation- and calmodulin-dependent manner. J Cell Sci 119:4520–4530
Li GH, Anderson C, Jaeger L, Do T, Major EO, Nath A (2015) Cell-to-cell contact facilitates HIV transmission from lymphocytes to astrocytes via CXCR4. Aids 29:755–766
Li GH, Henderson L, Nath A (2016) Astrocytes as an HIV reservoir: mechanism of HIV infection. Curr HIV Res 14:373–381
Lian H, Zheng H (2016) Signaling pathways regulating neuron-glia interaction and their implications in Alzheimer’s disease. J Neurochem 136:475–491
Lindsberg PJ, Roine RO (2004) Hyperglycemia in acute stroke. Stroke 35:363–364
Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K (2002) Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J Biol Chem 277:39312–39319
Liu X, Shah A, Gangwani MR, Silverstein PS, Fu M, Kumar A (2014) HIV-1 Nef induces CCL5 production in astrocytes through p38-MAPK and PI3K/Akt pathway and utilizes NF-kB, CEBP and AP-1 transcription factors. Sci Rep 4:4450
Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S (2007) Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 10:615–622
Nana AL, Kim EH, Thu DC, Oorschot DE, Tippett LJ, Hogg VM, Synek BJ, Roxburgh R, Waldvogel HJ, Faull RL (2014) Widespread heterogeneous neuronal loss across the cerebral cortex in Huntington’s disease. J Huntingtons Dis 3:45–64
Newman EA (2003) Glial cell inhibition of neurons by release of ATP. J Neurosci 23:1659–1666
Nitkiewicz J, Borjabad A, Morgello S, Murray J, Chao W, Emdad L, Fisher PB, Potash MJ, Volsky DJ (2017) HIV induces expression of complement component C3 in astrocytes by NF-kappaB-dependent activation of interleukin-6 synthesis. J Neuroinflammation 14:23
Nookala AR, Shah A, Noel RJ, Kumar A (2013) HIV-1 Tat-mediated induction of CCL5 in astrocytes involves NF-kappaB, AP-1, C/EBPalpha and C/EBPgamma transcription factors and JAK, PI3K/Akt and p38 MAPK signaling pathways. PLoS One 8:e78855
Ong CL, Thorpe JC, Gorry PR, Bannwarth S, Jaworowski A, Howard JL, Chung S, Campbell S, Christensen HS, Clerzius G, Mouland AJ, Gatignol A, Purcell DFJ (2005) Low TRBP levels support an innate human immunodeficiency virus type 1 resistance in astrocytes by enhancing the PKR antiviral response. J Virol 79:12763–12772
Orellana JA, Saez JC, Bennett MV, Berman JW, Morgello S, Eugenin EA (2014) HIV increases the release of dickkopf-1 protein from human astrocytes by a Cx43 hemichannel-dependent mechanism. J Neurochem 128:752–763
Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG (1994) Glutamate-mediated astrocyte-neuron signalling. Nature 369:744–747
Persichini T, Mastrantonio R, Del Matto S, Palomba L, Cantoni O, Colasanti M (2014) The role of arachidonic acid in the regulation of nitric oxide synthase isoforms by HIV gp120 protein in astroglial cells. Free Radic Biol Med 74:14–20
Putatunda R, Zhang Y, Li F, Yang XF, Barbe MF, Hu W (2018) Adult neurogenic deficits in HIV-1 Tg26 transgenic mice. J Neuroinflammation 15:287
Ratai EM, Annamalai L, Burdo T, Joo CG, Bombardier JP, Fell R, Hakimelahi R, He J, Lentz MR, Campbell J, Curran E, Halpern EF, Masliah E, Westmoreland SV, Williams KC, González RG (2011) Brain creatine elevation and N-Acetylaspartate reduction indicates neuronal dysfunction in the setting of enhanced glial energy metabolism in a macaque model of neuroAIDS. Magn Reson Med 66:625–634
Richards MH, Narasipura SD, Kim S, Seaton MS, Lutgen V, Al-Harthi L (2015) Dynamic interaction between astrocytes and infiltrating PBMCs in context of neuroAIDS. Glia 63:441–451
Roberts ES, Chana G, Nguyen TB, Perera G, Landau S, Rabe-Hesketh S, Glass JD, McArthur J, Everall IP (2013) The spatial relationship between neurons and astrocytes in HIV-associated dementia. J Neurovirol 19:123–130
Robertson JM (2014) Astrocytes and the evolution of the human brain. Med Hypotheses 82:236–239
Rothstein JD, Martin LJ, Kuncl RW (1992) Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med 326:1464–1468
Sami Saribas A, Cicalese S, Ahooyi TM, Khalili K, Amini S, Sariyer IK (2017) HIV-1 Nef is released in extracellular vesicles derived from astrocytes: evidence for Nef-mediated neurotoxicity. Cell Death Dis 8:e2542
Sardo L, Vakil PR, Elbezanti W, El-Sayed A, Klase Z (2016) The inhibition of microRNAs by HIV-1 tat suppresses beta catenin activity in astrocytes. Retrovirology 13:25
Shah A, Singh DP, Buch S, Kumar A (2011) HIV-1 envelope protein gp120 up regulates CCL5 production in astrocytes which can be circumvented by inhibitors of NF-kappaB pathway. Biochem Biophys Res Commun 414:112–117
Shah A, Vaidya NK, Bhat HK, Kumar A (2016) HIV-1 gp120 induces type-1 programmed cell death through ER stress employing IRE1alpha, JNK and AP-1 pathway. Sci Rep 6:18929
Sheikhbahaei S, Turovsky EA, Hosford PS, Hadjihambi A, Theparambil SM, Liu B, Marina N, Teschemacher AG, Kasparov S, Smith JC, Gourine AV (2018) Astrocytes modulate brainstem respiratory rhythm-generating circuits and determine exercise capacity. Nat Commun 9:370
Silver J, Miller JH (2004) Regeneration beyond the glial scar. Nat Rev Neurosci 5:146–156
Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32:638–647
Song S, Miranda CJ, Braun L, Meyer K, Frakes AE, Ferraiuolo L, Likhite S, Bevan AK, Foust KD, McConnell MJ, Walker CM, Kaspar BK (2016) Major histocompatibility complex class I molecules protect motor neurons from astrocyte-induced toxicity in amyotrophic lateral sclerosis. Nat Med 22:397–403
Soo Youn G, Ju SM, Choi SY, Park J (2015) HDAC6 mediates HIV-1 tat-induced proinflammatory responses by regulating MAPK-NF-kappaB/AP-1 pathways in astrocytes glia
Sultan S, Li L, Moss J, Petrelli F, Cassé F, Gebara E, Lopatar J, Pfrieger FW, Bezzi P, Bischofberger J, Toni N (2015) Synaptic integration of adult-born hippocampal neurons is locally controlled by astrocytes. Neuron 88:957–972
Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM (2011) Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144:810–823
Swanson PA 2nd, McGavern DB (2015) Viral diseases of the central nervous system. Curr Opin Virol 11:44–54
Takahashi K, Wesselingh SL, Griffin DE, McArthur JC, Johnson RT, Glass JD (1996) Localization of HIV-1 in human brain using polymerase chain reaction/in situ hybridization and immunocytochemistry. Ann Neurol 39:705–711
Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M (2006) Astrocyte-mediated control of cerebral blood flow. Nat Neurosci 9:260–267
Teodorof-Diedrich C, Spector SA (2018) Human immunodeficiency virus type 1 gp120 and tat induce mitochondrial fragmentation and incomplete mitophagy in human neurons. J Virol 92
Tewari M, Monika VRK, Menon M, Seth P (2015) Astrocytes mediate HIV-1 Tat-induced neuronal damage via ligand-gated ion channel P2X7R. J Neurochem 132:464–476
Thaney VE, Sanchez AB, Fields JA, Minassian A, Young JW, Maung R, Kaul M (2018) Transgenic mice expressing HIV-1 envelope protein gp120 in the brain as an animal model in neuroAIDS research. J Neurovirol 24:156–167
Tornatore C, Chandra R, Berger JR, Major EO (1994) HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurology 44:481–487
Walsh KA, Megyesi JF, Wilson JX, Crukley J, Laubach VE, Hammond RR (2004) Antioxidant protection from HIV-1 gp120-induced neuroglial toxicity. J Neuroinflammation 1:8
Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, Rothstein JD, Volsky DJ (2003) Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology 312:60–73
Williams R, Yao H, Dhillon NK, Buch SJ (2009) HIV-1 Tat co-operates with IFN-gamma and TNF-alpha to increase CXCL10 in human astrocytes. PLoS One 4:e5709
Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J (2003) Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 9:453–457
Wyss-Coray T, Masliah E, Toggas SM, Rockenstein EM, Brooker MJ, Lee HS, Mucke L (1996) Dysregulation of signal transduction pathways as a potential mechanism of nervous system alterations in HIV-1 gp120 transgenic mice and humans with HIV-1 encephalitis. J Clin Invest 97:789–798
Xing HQ, Zhang Y, Izumo K, Arishima S, Kubota R, Ye X, Xu Q, Mori K, Izumo S (2017) Decrease of aquaporin-4 and excitatory amino acid transporter-2 indicate astrocyte dysfunction for pathogenesis of cortical degeneration in HIV-associated neurocognitive disorders. Neuropathology 37:25–34
Yang Y, Ge W, Chen Y, Zhang Z, Shen W, Wu C, Poo M, Duan S (2003) Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc Natl Acad Sci U S A 100:15194–15199
Youn GS, Cho H, Kim D, Choi SY, Park J (2017) Crosstalk between HDAC6 and Nox2-based NADPH oxidase mediates HIV-1 Tat-induced pro-inflammatory responses in astrocytes. Redox Biol 12:978–986
Zhou BY, Liu Y, Kim B, Xiao Y, He JJ (2004) Astrocyte activation and dysfunction and neuron death by HIV-1 Tat expression in astrocytes. Mol Cell Neurosci 27:296–305
Zhuang K, Leda AR, Tsai L, Knight H, Harbison C, Gettie A, Blanchard J, Westmoreland S, Cheng-Mayer C (2014) Emergence of CD4 independence envelopes and astrocyte infection in R5 simian-human immunodeficiency virus model of encephalitis. J Virol 88:8407–8420
Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G (2003) Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 6:43–50
Acknowledgments
The authors wish to acknowledge the support of the facilities provided under the Biotechnology Information System Network (BTISNET) grant, Department of Biotechnology, India and Distributed Information Centre at NBRC, Manesar, India.
Funding
The study was supported by a Research Fellowship to Hriday S. Pandey from CSIR, New Delhi, India, and financial support from Department of Biotechnology (DBT) and Department of Science and Technology (DST), New Delhi, and NBRC core funds to Prof. Pankaj Seth.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pandey, H.S., Seth, P. Friends Turn Foe—Astrocytes Contribute to Neuronal Damage in NeuroAIDS. J Mol Neurosci 69, 286–297 (2019). https://doi.org/10.1007/s12031-019-01357-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-019-01357-1