
Abstract
Erythropoietin (EPO) may protect the nervous system of animals against aging damage, making it a potential anti-aging drug for the nervous system. However, experimental evidence from natural aging nerve cell models is lacking, and the efficacy of EPO and underlying mechanism of this effect warrant further study. Thus, the present study used long-term cultured primary nerve cells to successfully mimic the natural aging process of nerve cells. Starting on the 11th day of culture, cells were treated with different concentrations of recombinant human erythropoietin (rhEPO). Using double immunofluorescence labeling, we found that rhEPO significantly improved the morphology of long-term cultured primary nerve cells and increased the total number of long-term cultured primary cells. However, rhEPO did not improve the ratio of nerve cells. A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to measure nerve cell activity and showed that rhEPO significantly improved the activity of long-term cultured primary nerve cells. Moreover, Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) double immunofluorescence labeling flow cytometry revealed that rhEPO reduced the apoptotic rate of long-term cultured primary nerve cells. Senescence-associated β-galactosidase (SA-β-gal) immunohistochemistry staining showed that rhEPO significantly reduced the aging rate of long-term cultured primary nerve cells. Immunochemistry revealed that rhEPO enhanced intracellular superoxide dismutase (SOD) activity and glutathione (GSH) abundance and reduced the intracellular malondialdehyde (MDA) level. In addition, this effect depended on the dose, was maximized at a dose of 100 U/ml and was more pronounced than that of vitamin E. In summary, this study finds that rhEPO protects long-term cultured primary nerve cells from aging in a dose-dependent manner. The mechanism of this effect may be associated with the enhancement of the intracellular anti-oxidant capacity. These findings provide a theoretical basis to further the anti-aging mechanism of EPO in the nervous system, and they provide experimental evidence at the cellular level for the clinical application of EPO to protect the nervous system from aging.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The nervous system is central to bodily function, and its aging affects all systems of the body (Alcedo et al. 2013). In recent years, studies that examine the aging of the nervous system have attracted increasing attention (Carrasco-Garcia et al. 2015), and most of these studies have focused on anti-oxidants (Rikans and Hornbrook 1997; Cutler 1991). Anti-oxidants scavenge free radicals to prevent excessive amounts of these free radicals from damaging the nerve cells. At present, studies examining the use of anti-oxidant to protect nerves from aging have focused on the following three classes of molecules: intracellular anti-oxidants, such as superoxide dismutase (SOD) and glutathione (GSH) (Klichko et al. 1999); natural anti-oxidants, such as vitamin E (Vatassery 1998); and related small molecules that can regulate intracellular anti-oxidase (Smith et al. 2002).
Erythropoietin (EPO) is an important glycoprotein hormone in the body, and previous studies have found that it plays a protective role in a variety of nerve injuries (Juul 2002). The aging of and damage to the nervous system is the result of a concerted action of various factors, but the role EPO in the progression of the aging of the nervous system remains unclear. The expression of EPO in the nervous system has been shown to inversely correlate with age in animals and humans (Chung et al. 2004), and supplementation with exogenous EPO delays the pathogenesis and progression of neurodegenerative disorders (Chong et al. 2013). Specifically, our previous study (Wu et al. 2017) found that recombinant human erythropoietin (rhEPO) upregulates anti-oxidase activity in the nervous system via the extracellular signal-regulated kinase/nuclear factor-like 2 (ERK/Nrf2) pathway, reduces oxidative stress injuries to aged rat brain tissue induced by d-galactose, and improves the learning and memory functions of rats. We and others also found that EPO upregulated intracellular anti-oxidase activity in nerve cell models (Yoo et al. 2016). These results suggest that EPO plays a role in regulating anti-oxidase activity in nerve cells, and it may protect against aging-induced damage in animals. Therefore, EPO may be an anti-aging drug for the nervous system. However, this use requires further experimental verification in natural aging nerve cell models, and the efficacy of EPO and mechanism underlying its effect warrant further exploration.
Aging differs by cell type. Specifically, nerve cells are terminally differentiated and consequently no longer divide and replicate. Thus, they undergo non-replicative aging, which is mainly characterized by the retention of β-galactosidase (β-gal) and other aging-related substances, a decrease in the number of neuronal synapses, reduced neuronal synapse activity, and an increase in apoptotic cells (Ermini and Jucker 2000). Early nerve cell aging models primarily consisted of induced aging models (Dong et al. 2014; Andrews 1996), including d-galactose-induced aging models, ozone-induced aging models, and β-amyloid-induced aging models. In recent years, the introduction of “Neurobasal” medium and “B27 growth factor” has facilitated the long-term culture of primary nerve cells. During long-term culture, protein carbonyls and Aβ proteins accumulate in primary nerve cells and cell mitochondrial membrane potential and anti-oxidant capacity decrease, which ultimately increases the number of apoptotic cells. These changes approximate the changes of nerve cells in the central nervous system during aging. A number of studies have used long-term cultured primary nerve cells to study naturally aging-associated mechanisms in the nervous system at the cellular level (Lesuisse and Martin 2002).
In this study, we aimed to use long-term cultured primary nerve cells to establish a nerve cell model of natural aging. Before the cells entered the aging period, they were treated with various concentrations of rhEPO, and changes in nerve cell aging-associated indicators were then observed to assess the anti-aging effect of rhEPO in nerves at the cellular level. The relevant mechanism was explored to provide a theoretical basis for the clinical application of EPO.
Materials and Methods
Experimental Animals
Neonatal Sprague-Dawley (SD) rats aged 1 day, without a specific requirement for sex, were purchased from the Animal Experiment Center of the Biofavor Biotech Service Co., Ltd., Wuhan, China (No. 42000600012249). The animals were housed at 22 ± 2 °C and a relative humidity of 55 ± 5% under a 12-h light cycle. All experiments were completed at the Experimental Center of the Second Affiliated Hospital of Xi’an Jiaotong University, China. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal use protocol was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Xi’an Jiaotong University.
Long-Term Primary Culture of Neurons
One-day-old neonatal SD rats were sacrificed by cervical dislocation, and the brain cortex was isolated and digested with 0.125% trypsin (Lesuisse and Martin 2002). The digested mixture was filtered with a 200-mesh filter, and the filtrate was centrifuged for 5 min (800 rpm) to harvest the cells. Neurobasal (Invitrogen Gibco, New York, USA) + B27 medium (Invitrogen Gibco, New York, USA) was used to prepare the cell suspension solution, which was seeded into a polylysine-coated culture plate and incubated overnight at 37 °C with 5% CO2 in a saturated humidity atmosphere. Half the medium was changed every 2 days, and the size and morphology of the cells were observed under an inverted microscope.
Experimental Grouping
The control group (group C): the control group was divided into the mature cell group (C-DIV10) and aged cell group (C-DIV22). Cells in the mature group were cultured for a total of 10 days and represented mature nerve cells; cells in the aged group were cultured for a total of 22 days and represented aged nerve cells. EPO treatment group (group E): starting on day 11, the culture medium was supplemented with EPO (PeproTech, New Jersey, USA), and half the medium was changed every 2 days. The group was divided into the following four subgroups according to the final concentration of EPO: E(1) group, 1 U/ml; E(10) group, 10 U/ml; E(100) group, 100 U/ml; and E(200) group, 200 U/ml. The cells were cultured for a total of 22 days. Vitamin E treatment group (Vit E group): starting on day 11, the culture medium was supplemented with vitamin E (Sigma, Santa Clara, USA), and the final concentration of Vit E was 2 mM (Wang et al. 2012). Half the medium was changed every 2 days, and the cells were cultured for a total of 22 days (DIV22).
Double Immunofluorescence Labeling of Nerve Cells to Observe the Purity of the Cells
Isolated cells were seeded into polylysine-treated 12-well plates at a density of 1.0 × 106 cells/ml, and 1 ml of medium was added to each well. The cells were cultured overnight at 37 °C in 5% CO2 and saturated humidity. The cover slip covered with cells was washed with PBS, and the cells were fixed with 4% paraformaldehyde. The cells were permeabilized with 0.5% Triton X-100 at room temperature for 20 min. After being washed with PBS, the cells were blocked with normal goat serum at room temperature for 30 min. The cells were then incubated with the following microtubule-associated protein (MAP)-2 antibody (1:100, species: rabbit, Proteintech Group Inc., Wuhan, China) at 4 °C overnight, followed by incubation with fluorescent (Cy3) goat anti-rabbit immunoglobulin (IgG) secondary antibody (1:200, Boster Biological Technology co. ltd, Wuhan, China) for 1 h at 37 °C in a humidified atmosphere. Glial fibrillary acidic protein (GFAP) antibody (1:100, species: mouse, Santa Cruz Biotechnology, Santa Cruz, USA) was incubated with the cells in the dark at 4 °C overnight, and fluorescein isothiocyanate (FITC) goat anti-mouse IgG (1:200, Boster Biological Technology Co. Ltd, Wuhan, China) was then added, and the slides were incubated at 37 °C for 1 h in a humidified atmosphere. Phosphate-buffered saline containing Tween (PBST) was used to wash the slides three times for 3 min each. Subsequently, 4′6-diamidine-2′-phenylindole dihydrochloride (DAPI) was added dropwise and incubated for 5 min in the dark to counterstain the nuclei. The slides were sealed with mounting medium containing anti-quench agent and then observed under a fluorescence microscope to capture images. Cy3 produced red fluorescence at an excitation wavelength of 550 nm and an emission wavelength of 570 nm; FITC produced green fluorescence at an excitation wavelength of 494 nm and an emission wavelength of 518 nm. All manipulation steps were conducted in the dark.
Determination of Cell Activity by MTT Assay
Isolated neuronal cells were seeded in 96-well plates at a density of 5 × 104 cells/well, with 200 μl of medium in each well, and a blank control group was established simultaneously. The cells were cultured at 37 °C in 5% CO2 and saturated humidity. Half the medium was exchanged with fresh medium every 2 days. At the time of assay, 20 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma, Louisiana, USA) was added to each well, the cells were cultured at 37 °C for 4 h, and the medium was aspirated. Subsequently, 150 μl of dimethyl sulfoxide (DMSO) was added to each well, and the plate was shaken for 10 min. A plate reader was used to determine the absorbance value of each well (Filter: 568).
Determination of Apoptotic Cell Ratio with a Flow Cytometer
Newly isolated neuronal cells were seeded in T25 flasks at a density of 2 × 105 cells/ml, with 5 ml of medium in each flask, and the cells were cultured at 37 °C in 5% CO2 and saturated humidity. Half the medium was replaced with fresh medium every 2 days. At the time of measurement, 0.25% trypsin without ethylenediaminetetraacetic acid (EDTA) was used to digest the cells. After the digestion was terminated, cells were harvested by centrifugation (1500 rpm) for 5 min. The supernatant was removed, and PBS was added to re-suspend the cells. The cells were rinsed twice with PBS and centrifuged (1500 rpm) again for 5 min. An Annexin V-FITC/propidium iodide (PI) apoptosis detection kit (Nanjing KeyGen Biotech Co. Ltd., Nanjing, China) was used for detection.
Observation of the Ratio of Aged Cells by SA-β-gal Staining
Cover slips coated with cells were washed three times with PBS for 3 min each. The cells were fixed for 15 min with 4% paraformaldehyde, and the cover slip was washed again three times with PBS for 3 min each. Subsequently, 100 μl of senescence-associated (SA)-β-gal staining solution (Nanjing KeyGen Biotech Co. Ltd., Nanjing, China) was added to each cover slip, which was then incubated in the dark at 37 °C in a CO2 incubator for 10 h to allow the reaction to proceed. The cover slip was then washed three times with PBS, the solution on the slip was absorbed with a piece of absorbent paper, and the slide was sealed with aqueous mounting medium. The cells were observed under a light microscope, and cells in different fields were counted. The ratio of SA-β-gal-positive cells represented the ratio of aged cells.
Determination of SOD Activity, GSH, and MDA Content
At the measured time points, cell lysis buffer was added to lyse the cells and obtain protein solution. An SOD assay kit (Jiancheng Bioengineering, Nanjing, China) was used to determine SOD activity. A trace-reduced GSH detection kit (Jiancheng Bioengineering, Nanjing, China) was used to determine GSH content. A malondialdehyde (MDA) assay kit (Jiancheng Bioengineering, Nanjing, China) was used to determine MDA content.
Statistical Analysis
All data were analyzed using SPSS (ver. 20.0) software. All the experiments were performed in triplicate, and the data are shown as the mean ± SD. The data were analyzed using a one-way analysis of variance (ANOVA). Probability values of P < 0.05 were considered statistically significant.
Results
rhEPO Improved the Morphology of Long-Term Cultured Primary Nerve Cells
Under an inverted phase-contrast microscope, cells appeared homogeneous and round immediately after seeding, and most cells were floating in medium (Fig. 1). At 3–4 h, the cells began to adhere, and almost all cells were adherent 12 h later. After culture for 1 day (C-DIV1), some cells developed small protrusions, and the cell body was somewhat enlarged. On the 10th day (C-DIV10), the cell body of the neuron was mature; the protrusions were thick and intertwined. On the 22nd day (C-DIV22), the nerve cells degenerated, nuclear pyknosis and lysis occurred, small vacuoles were observed in the cytoplasm, the protrusions decreased in size, the cells shrank, and cell grid became sparse. Twenty-two days after rhEPO treatment, the nerve cell morphology and nerve cell network had significantly improved compared with the cells of the control group at the same time point. This improvement was especially pronounced for EPO concentrations of 100 U/ml (E (100)) and 200 U/ml (E (200)). Vitamin E treatment had a similar effect.

Changes in the morphology of long-term cultured primary nerve cells after different treatments, as observed under an inverted microscope: A ×100, scale bar = 50 μm; B enlarged view of (A), take the arrow as the center, ×400. C-DIV1 morphology of cells in the control group after being cultured for 1 day (very small protrusions grew out from some cells, and cell bodies were enlarged), C-DIV10 morphology of cells in the control group on day 10 (the soma of the neuron was mature, and the protrusions were thick and intertwined into a network), C-DIV22 morphology of cells in the control group after being cultured for 22 days (nerve cells had degenerated, nuclear pyknosis and lysis were evident, small vacuoles were visible in the cytoplasm, the protrusion decreased in size, cells shrank, and the cell grid became sparse), E(1), E(10), E(100), and E(200) the morphology on day 22 treatment with different concentrations (1, 10, 100, or 200 U/ml) of rhEPO starting on day 11. The morphology of nerve cells and the nerve cell network had significantly improved compared with those of group C-DIV22; these improvements were especially significant for groups E(100) and E(200). Vit E on day 22, the effect of vitamin E treatment starting on day 11 was similar to that of E(100) and E(200). Representative images of each group
rhEPO Increased the Total Number of Long-Term Cultured Primary Cells but Failed to Increase the Ratio of Nerve Cells
Double immunofluorescence labeling was used to observe cell morphology and the number of long-term cultured primary cells (Fig. 2). The cytoplasm of cells that stained positive for the neuronal marker MAP-2 was red, whereas the cytoplasm of cells positive for the glial cell marker GFAP was green; DAPI stained the nuclei blue.


a Changes in the morphology of long-term cultured primary nerve cells in various groups as observed by immunofluorescence microscopy (×400, scale = 20 μm). C-DIV10 morphology of cells in the control group on day 10, C-DIV22 morphology of cells in the control group after being cultured for 22 days, E(1), E(10), E(100), and E(200) morphology on day 22 after treatment with different concentrations (1, 10, 100, or 200 U/ml) of rhEPO starting on day 11, Vit E cell morphology on day 22 after treatment with vitamin E starting on day 11, MAP-2 neuronal marker MAP-2 staining (the cytoplasm of positively stained cells fluoresced red), GFAP glial cell marker GFAP staining (the cytoplasm of positively stained cells fluoresced green), DAPI DAPI counterstained the nuclei fluoresced blue and represented the total number of cells. Representative images of each group. b Statistical analysis of the long-term cultured primary nerve cells in the various groups. The data are shown as the mean ± SD values obtained from three separate experiments. The data were analyzed using a one-way ANOVA analysis. ★ P < 0.05 vs. C-DIV10; ▲ P < 0.05 vs. C-DIV22; ◆ P < 0.05 vs. E(1); ■ P < 0.05 vs. E(10)
The number of blue nuclei was counted to determine the number of long-term cultured primary cell, which revealed significantly fewer C-DIV22 cells than C-DIV10 cells (P = 0.0001). After rhEPO or vitamin E treatment, each group contained significantly fewer total cells than the C-DIV10 group (P = 0.0001 vs. E(1), P = 0.0003 vs. E(10), P = 0.001 vs. E(100), P = 0.015 vs. E(200), and P = 0.011 vs. Vit E), but the C-DIV22 showed an increase in cells. Specifically, the increases in groups E(10) (P = 0.005), E(100) (P = 0.0004), E(200) (P = 0.0004), and Vit E (P = 0.0006) were significant, whereas groups E(100), E(200), and Vit E did not significantly differ (P > 0.05).
The number of MAP-2-positive cells represented the number of nerve cells. The results showed that the number of nerve cells in the C-DIV22 group was significantly less than the of C-DIV10 group (P < 0.0001), After rhEPO or vitamin E treatment, each group contained significantly fewer nerve cells than the C-DIV10 group (P < 0.0001 vs. E(1), P = 0.0001 vs. E(10), P = 0.002 vs. E(100), P = 0.003 vs. E(200), and P = 0.002 vs. Vit E). When compared with the C-DIV22 group, the groups all showed an increase in nerve cells. Specifically, the increases in groups E(10) (P = 0.003), E(100) (P = 0.0005), E(200) (P = 0.0006), and Vit E (P = 0.0007) were significant, whereas groups E(100), E(200), and Vit E did not significantly differ (P > 0.05).
The number of GFAP-positive cells represented the number of glial cells. The results showed that the number of glial cells in the C-DIV22 group was significantly higher than the of C-DIV10 group (P = 0.029). After rhEPO or vitamin E treatment, there was a trend toward an increase in the number of glial cells, with group Vit E as the vertex. Specifically, compared with the C-DIV22 group, the increases in groups E(100) (P = 0.015), E(200) (P = 0.002), and Vit E (P = 0.002) were significant, and the increases in groups E(200) (P = 0.029) and Vit E (P = 0.016) were more significant than that in the E(100) group.
The proportion of the number of MAP-2-positive cells to the number of DAPI-positive cells represented the ratio of nerve cells. The results showed that 95% of the cells in the C-DIV10 group were nerve cells, and 77% of the cells in the C-DIV22 group were nerve cells, but this proportion was significantly lower than that of the C-DIV10 group (P = 0.005). Moreover, rhEPO or vitamin E treatment slightly increased the proportion of nerve cells to 70–85%, but this proportion remained significantly lower than that of the C-DIV10 group (P = 0.005 vs. E(1), P = 0.013 vs. E(10), P = 0.008 vs. E(100), P = 0.016 vs. E(200), P = 0.002 vs. Vit E) and did not significantly differ from that of the C-DIV22 group (P > 0.05).
The proportion of the number of GFAP-positive cells to the number of DAPI-positive cells represented the ratio of glial cells. The results showed that less than 1% of the cells in the C-DIV10 group were glial cells, and more than 5% of the cells in the C-DIV22 group were glial cells, and this proportion was significantly higher than that of C-DIV10 group (P = 0.0008). Moreover, rhEPO or vitamin E treatment slightly decreased the proportion of glial cells to 3–5.5%, but this proportion remained significantly higher than that of the C-DIV10 group (P = 0.001 vs. E(1), P = 0.002 vs. E(10), P = 0.001 vs. E(100), P = 0.0007 vs. E(200), and P = 0.002 vs. Vit E) and did not significantly differ from that of the C-DIV22 group (P > 0.05).
rhEPO Improved the Activity of Long-Term Cultured Primary Nerve Cells
The relative optical density (OD) value determined by the MTT assay represented cell activity (Fig. 3a). Specifically, the OD value of the C-DIV22 group was significantly lower than that of the C-DIV10 group (P < 0.0001), and the OD value of each group remained lower than that of the C-DIV10 group after rhEPO or vitamin E treatment (P < 0.0001 vs. E(1), P = 0.0003 vs. E(10), P = 0.004 vs. E(100), P = 0.006 vs. E(200), and P = 0.001 vs. Vit E) but was slightly higher than that of the C-DIV22 group. Of these increases, the increases in the E(10) (P = 0.023), E(100) (P = 0.0008), E(200) (P = 0.002), and Vit E (P = 0.0008) groups were significant. The comparison between various concentrations of rhEPO treatment showed that the OD value of the E(10) group was significantly higher than that of the E(1) group (P = 0.043) and that of the E(100) was significantly higher than that of the E(10) group (P = 0.01). However, groups E(200) and E(100) did not significantly differ (P > 0.05). In addition, the OD values of groups E(100), E(200), and Vit E also did not significantly differ (P > 0.05).

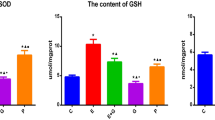
Changes in cell viability, SOD, GSH, and MDA in long-term cultured primary nerve cells in each group. An MTT assay was used to measure the OD value of the long-term cultured nerve cells in the control group on day 10, which was used as the reference value. Cell viability on day 22 (C-DIV22) of cells in the control group after treatment with different concentrations of rhEPO (1, 10, 100, or 200 U/ml) or vitamin E (2 mM). The data are shown as the mean ± SD of values obtained from three separate experiments. The data were analyzed using a one-way ANOVA. ★ P < 0.05 vs. C-DIV10; ▲ P < 0.05 vs. C-DIV22; ◆ P < 0.05 vs. E(1); ■ P < 0.05 vs. E(10); ● P < 0.05 vs. E(100); ╋ P < 0.05 vs. E(200)
rhEPO Reduced Apoptosis in Long-Term Cultured Primary Nerve Cells
Annexin V-FITC/PI double labeling flow cytometry was used to assess apoptosis, which showed that the apoptotic rate of the C-DIV10 group was approximately 7%, and the apoptotic rate of the C-DIV22 group was approximately 61%, which was significantly higher than that of the C-DIV10 group (P = 0.0001) (Fig. 4). After treatment with rhEPO or vitamin E, the apoptotic rate remained higher than that of the C-DIV10 group (P = 0.0001 vs. E(1), P = 0.0002 vs. E(10), P = 0.0004 vs. E(100), P = 0.0005 vs. E(200), and P = 0.0004 vs. Vit E) but lower than that of the C-DIV22 group. Specifically, the decreases in the apoptotic rates of groups E(10) (P = 0.019), E(100) (P = 0.0008), E(200) (P = 0.002), and Vit E (P = 0.003) were significant. Moreover, a comparison of the different rhEPO doses showed that the apoptotic rate of group E(10) was significantly lower than that of group E(1) (P = 0.035) and that of group E(100) was significantly lower than that of group E(10) (P = 0.004). However, groups E(200) and E(100) did not significantly differ (P > 0.05). Furthermore, vitamin E treatment significantly reduced apoptosis compared with group C-DIV22, but this effect was significantly less pronounced than that of group E(100) (P = 0.04).

Changes in the apoptotic rate of long-term cultured primary nerve cells in each group. Flow cytometry was used (Annexin V-FITC/PI double immunofluorescence labeling) to assess apoptosis in response to different treatments. Representative images of each group. St the statistical analysis of the apoptosis rate of each group. The data are shown as the mean ± SD of values obtained from three separate experiments. The data were analyzed using a one-way ANOVA. ★ P < 0.01 vs. C-DIV10; ▲ P < 0.05 vs. C-DIV22; ◆ P < 0.05 vs. E(1); ■ P < 0.05 vs. E(10); ● P < 0.05 vs. E(100)
rhEPO Reduced the Aging Rate of Long-Term Cultured Primary Nerve Cells
SA-β-gal staining was used to observe the ratio of aged cells to total cells, which showed that the proportion of aged cells in group C-DIV10 was only approximately 2%, whereas the ratio of aged cells in group C-DIV22 exceeded 85%, and this difference was significant (P < 0.0001) (Fig. 5). After treatment with rhEPO or vitamin E, the proportion of aged cells remained significantly higher than that of the C-DIV10 group (P = 0.0001 vs. E(1), P = 0.0001 vs. E(10), P = 0.01 vs. E(100), P = 0.002 vs. E(200), and P = 0.001 vs. Vit E) but was lower than that of the C-DIV22 group. Specifically, the decreases in the proportions of aged cells were significant in groups E(10) (P = 0.0004), E(100) (P = 0.0001), E(200) (P = 0.0001), and Vit E (P = 0.0002). The comparison between various concentrations of rhEPO treatment showed that the proportion of aged cells was significantly lower in group E(10) than in group E(1) (P = 0.001) and that of group E(100) was significantly lower than that of group E(10) (P = 0.003). However, groups E(200) and E(100) did not significantly differ (P > 0.05). Moreover, vitamin E treatment significantly reduced the proportion of aged cells compared with group C-DIV22, but this effect was significantly less pronounced than that of group E(100) (P = 0.046).

The proportion of aged cells in long-term cultured primary nerve cells in each group as observed under an ordinary light microscope (×200, scale = 50 μm). SA-β-gal immunohistochemical staining, SA-β-gal-positive cells that were stained blue represent aged cells. Representative images of each group. St the statistical analysis of the proportion of SA-β-gal-positive cells in each group. The data are shown as the mean ± SD of values obtained from three separate experiments. The data were analyzed using a one-way ANOVA. ★ P < 0.01 vs. C-DIV10; ▲ P < 0.01 vs. C-DIV22; ◆ P < 0.01 vs. E(1); ■ P < 0.05 vs. E(10); ● P < 0.05 vs. E(100)
rhEPO Upregulated the Anti-oxidant Capacity of Long-Term Cultured Primary Nerve Cells
Immunochemistry was used to determine the activity of the anti-oxidant SOD, the content of GSH, and the content of the cell peroxidation product of MDA (Fig. 3b–d). The SOD activity and GSH content were significantly lower in the C-DIV22 group than the C-DIV10 group (SOD: P = 0.0004; GSH: P = 0.0002), whereas the MDA content was significantly increased (P = 0.0001). Treatment with rhEPO significantly increased SOD activity and GSH content compared with the C-DIV22 group and decreased the MDA content. Specifically, these changes were significant for groups E(10) (SOD: P = 0.039; GSH: P = 0.0007; MDA: P = 0.007), E(100) (SOD: P = 0.001; GSH: P = 0.0004; MDA: P = 0.0004), and E(200) (SOD: P = 0.001; GSH: P = 0.0001; MDA: P = 0.0004) compared with group C-DIV22. Comparing the various concentrations of rhEPO treatment showed that cell the SOD activity and GSH content were significantly different in group E(10) compared with group E(1) (SOD: P = 0.039; GSH: P = 0.003), whereas the MDA content was significantly decreased (P = 0.048). These parameters also significantly differed between groups E(100) and E(10) (SOD: P = 0.01; GSH: P = 0.004; MDA: P = 0.004) but not between groups E(200) and E(100) (P > 0.05). Moreover, vitamin E treatment did not significantly change the SOD activity and GSH content (P > 0.05) but did significantly decrease the MDA content compared with the C-DIV22 group (P = 0.008). Nevertheless, the MDA content remained significantly higher than those of the E(100) (P = 0.016) and E(200) groups (P = 0.011).
Discussion
At the cellular level, the aging of the nervous system primarily manifests as soma atrophy, a decrease in the disappearance of dendrites and dendritic spines, axonal swelling, and demyelination (Omelyanchuk et al. 2015; Hervonen 1990; Pannese 2011). However, there is controversy about the aging-related changes of nerve cells. Early studies have shown that nerve cell counts are significantly reduced with aging (Brody 1955), but in recent years, an increasing number of studies have shown that the nerve cell counts in the naturally aging of brain tissue were unchanged (Roberts et al. 2012; Uylings and de Brabander 2002). This may be ascribed to neural stem cell transdifferentiation mechanism within the natural aging process in the brain tissue (Limke and Rao 2002). Nerve cells are highly differentiated non-divisible cells, and studies based on the divisible cells cannot completely reflect the characteristics of the aging of nervous system. Therefore, recent aging studies have focused on “neuronal culture,” and significant progress has been made in primary neuronal culture in the past two decades. Specifically, the introduction of “Neurobasal” medium and “B27 growth factor” has significantly prolonged the survival time of neurons in vitro and maintained higher neuronal purity throughout the culture process (Brewer 1995). A number of previous studies used the aforementioned culture system for the primary culture of nerve cells from fetal mice and found that more than 50% of cells had survived until the 4th week of culture, and the proportion of astrocytes was lower than 20% (Bertrand et al. 2011; Dong et al. 2011). In the present study, we used cerebral cortical neurons from 1-day-old neonatal SD rats to perform long-term primary neuronal culture using this culture system; the results showed that the nerve cells in the primary culture had the best cell morphology and the most extensive neural network on day 10 (C-DIV10), and the purity of nerve cells reached 95%. On day 22 (C-DIV22), the nerve cells and the neural network had significantly degenerated, cell viability had decreased to 50%, and nerve cell purity had decreased to 77%. This result is consistent with those of previous studies, suggesting that this culture system is not only suitable for the primary culture of nerve cells from fetal mice but is also suitable for the primary culture of the cerebral cortical neurons from 1-day-old neonatal SD rats. Neurons experience the following three stages during long-term culture: development, maturation and stabilization, and aging and degeneration. As aging progresses, the mitochondrial membrane potential of the primary neurons is reduced, anti-oxidant capacity is decreased, protein carbonyls and Aβ protein accumulate, the expression of apoptosis-associated proteins changes, and the sensitivity of neurons to various stresses that cause neuronal death is eventually increased (Dong et al. 2011). This process is similar to the age-related changes observed in nerve cells in the central nervous system, and it may be a good cell model to study the natural aging of the nervous system. Researchers previously roughly divided the growth process of neurons cultured in vitro into the following stages based on cell morphology (Lesuisse and Martin 2002): the resting stage, the growth stage, the maturation stage, and the aging stage. However, the reports of the timing of cells entering the aging stage are not consistent (Aksenova et al. 1999; Belrose et al. 2012). In the present study, long-term cultured nerve cells were the most stable and the neural network was extensive on day 10 (C-DIV10), whereas the morphology of nerve cells and the neural network had significantly degenerated by day 22 (C-DIV22), showing various typical aging-associated morphological changes. Moreover, the viability of nerve cells had decreased to nearly 50% by C-DIV22, the apoptotic rate exceeded 60%, and the ratio of aged cells exceeded 80%. Therefore, we suggest that long-term cultured primary nerve cells have entered the aging stage by day 22 of culture and show typical signs of aging, allowing them to be used as a model to study aging this time point. Of course, the type of cell that can be used to prepare the aging nerve cell model is not limited to primary nerve cells. PC12 cells, among others, can also be prepared via d-galactose induction, ischemia and hypoxia-related induction, aging-related gene regulation, etc. However, those models are all similar; therefore, it would be more scientific to choose different models when studying different aging mechanisms.
EPO is an important glycoprotein hormone in the body that is widely distributed in the central and peripheral nervous systems. Some researchers suggested that EPO is “a brand new concept for neuroprotection” (Subiros et al. 2012; Castañeda-Arellano et al. 2014; Ugurluer et al. 2016). The aging of and damage to the nervous system is the result of the concerted action of various factors (Brierley et al. 1998), and the correlation between EPO and this damage remains unclear. A number of previous studies showed that the expression of EPO in the nervous system inversely correlated with age in animals and humans (Widl et al. 2007) and supplementation with exogenous EPO delays the pathogenesis and progression of Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD), among other neurodegenerative disorders (Jia et al. 2014; Cervellini et al. 2013). The expression of EPO also reduced oxidative stress injuries in aged rat brain tissue induced by d-galactose and improved the learning and memory functions of rats (Wu et al. 2017). These studies suggest that EPO may protect the nervous system against aging damage, but the experimental evidence at the cellular level is lacking. In this study, we used long-term cultured primary nerve cells to establish a natural aging model, and these cells were treated with exogenous EPO before they entered the aging stage. Although some evidences suggest that the amino acid sequence of the human EPO and the murine EPO is 80% homologous (Shoemaker and Mitsock 1986), the use of human-derived EPO in rat vivo experiments may still have an immune rejection. In this study, we used rhEPO to intervene long-term cultured primary nerve cells in vitro experiments. Therefore, there is no big problem in homology. Our results showed that in addition to significantly improving the morphology of naturally aging nerve cells, exogenous EPO also significantly affected other aging-associated indicators: cell viability was significantly improved, apoptosis was dramatically reduced, and the proportion of aged cells was notably decreased. These changes are similar to and, for some parameters, better than the effects of the widely accepted anti-aging drug vitamin E, which suggests that EPO significantly protects nerve cells against natural aging damage to delay the aging of nerve cells. Thus, EPO has the potential to become an anti-aging drug for the nervous system. Moreover, the concentration of EPO positively correlated with this protective effect on aging nerve cells: increasing the EPO concentration 1 to 100 U/ml improved various aging-associated indicators, but further increases from 100 to 200 U/ml did not further improve these indicators. This finding suggests that the protective effect of EPO on aging nerve cells correlates with dose but is also associated with a saturation concentration. In addition, this study found that rhEPO significantly increased the nerve cell count and glial cell count in aging nerve cell models but changes in the proportion of total cells to nerve cells were not obvious. Although the proportion of glial cells was not significantly different between the groups, the proportion of nerve cells was only 70–85%, and the proportion of glial cells was only 3–5.5%. Each group contained approximately 15% MAP-2-negative cells/GFAP-negative cells. We speculated that the reason for this phenomenon was that in addition to astrocytes, oligodendrocytes and microglia also exist in the central nervous system, but our study only observed the proportion of activated astrocytes. In addition, the observed nerve cells were immunostained using the neuronal dendritic marker MAP-2 in the study. Although this staining allows for the observation of nerve cell morphology and neural network, it has limitations in terms of nerve cell counting. MAP-2-positive cells had relatively complete structures, and their staining of microtubule-damaged nerve cells during partial aging was poor, resulting in partial false negative results. It may be more beneficial to use NeuN staining when determining the nerve cell counts.
The theory of free radicals suggests that aging-related damage to cells is mainly caused by the retention of MDA produced by oxidative stress, which results from the weakening of the cellular anti-oxidant capacity provided by SOD and GSH (Sims-Robinson et al. 2013; Ji 1993; Patel and Sesti 2016). The present study confirmed that EPO significantly protects nerve cells against aging damage, but the mechanism underlying this protection remains unclear. Previously, we used long-term cultured primary nerve cell models to show that rhEPO enhances the anti-oxidant capacity of the nerve cells by regulating the Nrf2 pathway (Wang et al. 2016). We hypothesized that EPO protects nerve cells from aging damage by also improving the cellular anti-oxidant capacity. To verify this hypothesis, we herein observed changes in the anti-oxidant capacity of naturally aging nerve cells after treatment with various concentrations of rhEPO, which showed that the SOD activity and GSH content of naturally aging nerve cells significantly decreased, whereas MDA content significantly increased as the long-term culture of primary nerve cells was extended. However, treatment with rhEPO significantly increased the SOD activity and GSH content of naturally aging nerve cells and decreased the retention of MDA. Moreover, this effect depended on dose and was maximized at a dose of 100 U/ml. This change was also consistent with changes in aging-associated indicators, such as cell viability, apoptotic rate, and the proportion of aged cells. In addition, the present study also found that treatment with the exogenous anti-oxidant vitamin E did not significantly change SOD activity and GSH content in naturally aging nerve cells, whereas the MDA content was significantly decreased, but this decrease was less pronounced than that associated with rhEPO treatment. These results suggest that EPO may protect long-term cultured primary nerve cells from aging damage by improving the endogenous anti-oxidant capacity of aging nerve cells, but the specific cellular pathway underlying this effect requires further investigation.
In summary, we cultured cortical neurons from 1-day-old neonatal rats long term. By observing changes in the morphology of nerve cells, the nerve fiber network, the total number of cells, the viability of cells, the apoptotic rate, and the ratio of aged cells, we found that this model successfully mimicked the natural aging process of nerve cells and identified DIV22 as critical time point of the aging stage. Furthermore, we found that exogenous EPO significantly improved various aging indicators of nerve cells, and this effect depended on dose and was maximized at a dose of 100 U/ml. In addition, this effect was more pronounced than that of vitamin E. Finally, changes in SOD, GSH, and MDA demonstrated that EPO may protect nerve cells from aging damage by improving the endogenous anti-oxidant capacity of aging nerve cells, but the specific cellular pathway underlying this effect requires further study.
References
Aksenova MV, Aksenov MY, Markesbery WR, Butterfield DA (1999) Aging in a dish: age-dependent changes of neuronal survival, protein oxidation, and creatine kinase BB expression in long-term hippocampal cell culture. J Neurosci Res 58:308–317
Alcedo J, Flatt T, Pasyukova EG (2013) The role of the nervous system in aging and longevity. Front Genet 4:124
Andrews TJ (1996) Autonomic nervous system as a model of neuronal aging: the role of target tissues and neurotrophic factors. Microsc Res Tech 35:2–19
Belrose JC, Xie YF, Gierszewski LJ, MacDonald JF, Jackson MF (2012) Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Mol Brain 5:11
Bertrand SJ, Aksenova MV, Aksenov MY, Charles FM, Rosemarie MB (2011) Endogenous amyloidogenesis in long-term rat hippocampal cell cultures. BMC Neurosci 12:38
Brewer GJ (1995) Serum-free B27/Neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res 42:674–683
Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM (1998) Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Ann Neurol 43:217–223
Brody H (1955) Organization of the cerebral cortex. III. A study of aging in the human cerebral cortex. J Comp Neurol 102:511–516
Carrasco-Garcia E, Arrizabalaga O, Serrano M, Lovell-Badge R, Matheu A (2015) Increased gene dosage of Ink4/Arf and p53 delays age-associated central nervous system functional decline. Aging Cell 14:710–714
Castañeda-Arellano R, Feria-Velasco AI, Rivera-Cervantes MC (2014) Activity increase in EpoR and Epo expression by intranasal recombinant human erythropoietin (rhEPO) administration in ischemic hippocampi of adult rats. Neurosci Lett 583:16–20
Cervellini I, Ghezzi P, Mengozzi M (2013) Therapeutic efficacy of erythropoietin in experimental autoimmune encephalomyelitis in mice, a model of multiple sclerosis. Methods Mol Biol 982:163–173
Chong ZZ, Shang YC, Mu YL, Cui SX, Yao QQ, Maiese K (2013) Targeting erythropoietin for chronic neurodegenerative diseases. Expert Opin Ther Targets 17:707–720
Chung YH, Kim SI, Joo KM, Kim YS, Lee WB, Yun KW, Cha CI (2004) Age-related changes in erythropoietin immunoreactivity in the cerebral cortex and hippocampus of rats. Brain Res 1018:141–146
Cutler RG (1991) Antioxidant and aging. Am J Clin Nutr 53:373S–379S
Dong W, Cheng S, Huang F, Fan W, Chen Y, Shi H, He H (2011) Mitochondrial dysfunction in long-term neuronal cultures mimics changes with aging. Med Sci Monit 17:BR91–BR96
Dong CM, Wang XL, Wang GM, Zhang WJ, Zhu L, Gao S, Yang DJ, Qin Y, Liang QJ, Chen YL, Deng HT, Ning K, Liang AB, Gao ZL, Xu J (2014) A stress-induced cellular aging model with postnatal neural stem cells. Cell Death Dis 5:e1116
Ermini F, Jucker M (2000) Nerve cell loss and neurogenesis in the aging brain. Praxis (Bern 1994) 89:1605–1608
Hervonen A (1990) The aging nerve cell. Duodecim 106:1497–1500
Ji LL (1993) Antioxidant enzyme response to exercise and aging[J]. Med Sci Sports Exerc 25:225–231
Jia Y, Mo SJ, Feng QQ, Zhan ML, OuYang LS, Chen JC, Ma YX, Wu JJ, Lei WL (2014) EPO-dependent activation of PI3K/Akt/FoxO3a signalling mediates neuroprotection in in vitro and in vivo models of Parkinson’s disease. J Mol Neurosci 53:117–124
Juul S (2002) Erythropoietin in the central nervous system, and its use to prevent hypoxic-ischemic brain damage. Acta Paediatr Suppl 91:36–42
Klichko VI, Radyuk SN, Orr WC (1999) CuZn-SOD promoter-driven expression in the drosophila central nervous system. Neurobiol Aging 20:537–543
Lesuisse C, Martin LJ (2002) Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J Neurobiol 51:9–23
Limke TL, Rao MS (2002) Neural stem cells in aging and disease. J Cell Mol Med 6:475–496
Omelyanchuk LV, Shaposhnikov MV, Moskalev AA (2015) Drosophila nervous system as a target of aging and anti-aging interventions. Front Genet 6:89
Pannese E (2011) Morphological changes in nerve cells during normal aging. Brain Struct Funct 216:85–89
Patel R, Sesti F (2016) Oxidation of ion channels in the aging nervous system. Brain Res 1639:174
Rikans LE, Hornbrook KR (1997) Lipid peroxidation, antioxidant protection and aging. Biochim Biophys Acta 1362:116–127
Roberts DE, Killiany RJ, Rosene DL (2012) Neuron numbers in the hypothalamus of the normal aging rhesus monkey: stability across the adult lifespan and between the sexes. J Comp Neurol 520:1181–1197
Shoemaker CB, Mitsock LD (1986) Murine erythropoietin gene: cloning, expression, and human gene homology. Mol Cell Biol 6:849–858
Sims-Robinson C, Hur J, Hayes JM, Dauch JR, Keller PJ, Brooks SV, Feldman EL (2013) The role of oxidative stress in nervous system aging. PLoS One 8:e68011
Smith MA, Casadesus G, Joseph JA, Perry G (2002) Amyloid-β and τ serve antioxidant functions in the aging and Alzheimer brain. Free Radic Biol Med 33:1194–1199
Subiros N, Del Barco DG, Coro-Antich RM (2012) Erythropoietin: still on the neuroprotection road. Ther Adv Neurol Disord 5:161–173
Ugurluer G, Cebi A, Mert H, Mert N, Serin M, Erkal HS (2016) Neuroprotective effects of erythropoietin against oxidant injury following brain irradiation: an experimental study. Arch Med Sci 6:1348–1353
Uylings HB, de Brabander JM (2002) Neuronal changes in normal human aging and Alzheimer’s disease. Brain Cogn 49:268–276
Vatassery GT (1998) Vitamin E and other endogenous antioxidants in the central nervous system. Geriatrics 53(Suppl 1):S25–S27
Wang J, Sun P, Bao Y, Dou B, Song D, Li Y (2012) Vitamin E renders protection to PC12 cells against oxidative damage and apoptosis induced by single-walled carbon nanotubes. Toxicol in Vitro 26:32–41
Wang HQ, Wu HQ, Zhang XF, Liu X, Gao Z, Bu N, Sun H, Yu XR (2016) Antioxidant capacities of long-range cultured neurons enhanced by erythropoietin (EPO)-regulated Nrf2 pathway. Int J Clin Exp Pathol 9:662–672
Widl K, Brettschneider J, Schattauer D, Süssmuth S, Huber R, Ludolph AC, Tumani H (2007) Erythropoietin in cerebrospinal fluid: age-related reference values and relevance in neurological disease. Neurochem Res 32:1163–1168
Wu H, Zhao J, Chen M, Wang H, Yao Q, Fan J, Zhang M (2017) The anti-aging effect of erythropoietin via the ERK/Nrf2-ARE pathway in aging rats. J Mol Neurosci 61:449–458
Yoo SJ, Cho B, Moon C, Yu SW, Moon C (2016) Neuroprotective effects of an erythropoietin-derived peptide in PC12 cells under oxidative stress. CNS Neurol Disord Drug Targets
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 81170330).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Wang, H., Fan, J., Chen, M. et al. rhEPO Enhances Cellular Anti-oxidant Capacity to Protect Long-Term Cultured Aging Primary Nerve Cells. J Mol Neurosci 62, 291–303 (2017). https://doi.org/10.1007/s12031-017-0937-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-017-0937-6