
Abstract
Autophagy, or type II programmed cell death, plays a crucial role in many nervous system diseases. However, few studies have examined the role of autophagy in post-traumatic stress disorder (PTSD), and the mechanisms underlying PTSD are poorly understood. The objective of this research was to explore the expression of three important autophagy-related proteins, Beclin-1, microtubule-associated protein 1 light chain 3 (LC3), and p62/SQSTM1 (p62), in the medial prefrontal cortex (mPFC) of an animal model of PTSD to identify changes in autophagic activity during PTSD pathogenesis. PTSD was induced in rats by exposure to a single-prolonged stress (SPS). The Morris water maze was used to assess cognitive changes in rats from the SPS and control groups. Transmission electron microscopy (TEM) was employed to observe mPFC morphological changes. Immunohistochemistry, immunofluorescence, and Western blotting techniques were used to detect expression of Beclin-1, LC3, and p62 in the mPFC. The Morris water maze test results showed that the escape latency time was increased and that the percent time in the target quadrant was decreased in the SPS group compared with that in the control group. Numerous visible autolysosomes in mPFC neurons were observed using TEM after SPS stimulation. Compared with that in the control group, the expression of Beclin-1 and the LC3-II/I ratio significantly decreased at 1 day, then increased and peaked at 7 days, and slightly decreased at 14 days after SPS stimulation, whereas the converse was found for p62 expression. In conclusion, dysregulation of autophagic activity in the mPFC may play a crucial role in PTSD pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Post-traumatic stress disorder (PTSD) is a serious mental and behavioral disorder caused by exposure to catastrophic or life-threatening trauma. PTSD is characterized by a number of neuropathological features including hypothalamic–pituitary–adrenal (HPA) axis dysregulation, abnormal serotonin levels, and noradrenergic dysfunction (Bailey et al. 2013; Pratchett et al. 2011; Mellman et al. 2009; Hughes and Shin 2011). Typical symptoms of PTSD include re-experiencing trauma, avoidance, numbing, and negative mood (American Psychiatric Association 2013; Pollice et al. 2012). These symptoms may appear a few days, months, or even several years after the trauma, which seriously affects the patient’s quality of life (Sherin and Nemeroff 2011). PTSD has attracted increasing attention in the fields of psychiatry and psychology due to the complex physical and behavioral outcomes (Meng et al. 2016). Over the last few decades, researchers worldwide have been committed to studying the pathogenesis of PTSD. However, the precise cellular and molecular mechanisms underlying PTSD remain unclear.
Programmed cell death is classified as apoptosis or autophagy based on morphological characteristics. Autophagy is characterized by the presence of membrane bilayers that wrap around cytoplasmic material and organelles to form autophagosomes, which then fuse with lysosomes to form autolysosomes that mediate degradation (Longatti et al. 2010). Basal levels of autophagy have pleiotropic functions, generally function during cell growth and development and, most importantly, regulate cellular homeostasis in all eukaryotes by degrading long-lived proteins, misfolded proteins, and subcellular organelles (Yamamoto and Yue 2014; Xie and Klionsky 2007; Fleming et al. 2011). Autophagy is involved in the pathophysiology of many diseases including cancer, heart disease, aging, immune disorders, and neurodegenerative diseases (Lee et al. 2009; Rangaraju et al. 2010; Yang et al. 2015). Presently, the autophagic process is known to be mediated by a series of autophagy-related (Atg) proteins, including Beclin-1 and microtubule-associated protein 1 light chain 3 (LC3), which play crucial roles in the autophagic process from yeast to mammals (Chen and Klionsky 2011). Beclin-1 is the mammalian homolog of yeast Atg6 and is essential for recruitment of other Atg proteins during the early stages of autophagy (Rami et al. 2008; Carloni et al. 2008). LC3 is the mammalian homolog of yeast Atg8 that forms LC3-I (cytosolic form) and LC3-II (membrane-bound form). During the autophagic process, cytosolic LC3-I is formed by newly synthesized LC3β, followed by combination with phosphatidylethanolamine to produce membrane-bound LC3-II. Membrane anchoring of LC3-II is necessary for autophagosome formation, and levels of LC3-II closely correlate with the number of autophagosomes (Jiang and Mizushima 2014; Narendra et al. 2010). Autophagy is a fast and dynamic process that is also referred to as autophagic flux. An increase or decrease in the expression of Atg proteins might indicate that autophagic activity increased or decreased at a specific point in time. However, the absence of autophagosome lysosomal fusion or changes in lysosomal activity might also result in abnormal expression of Atg, whereas autophagic flux might not completely be activated or inhibited at this point (Sahani et al. 2014). p62/SQSTM1 (p62), a multiubiquitin-chain-binding protein encoded by SQSTM1, participates in two protein degradation pathways: the ubiquitin–proteasome system (UPS) and autophagy (Weidberg et al. 2010). p62 directly interacts with LC3 through the LC3-interacting region (LIR) and is sequestered into autophagosomes for degradation; thus, the most well-known autophagic substrate p62 can be used to further evaluate autophagic flux by measuring the declining levels (Sahani et al. 2014). Recent studies have shown that autophagy plays an important role in many nervous system diseases such as ischemic brain damage, Alzheimer’s disease (AD), Parkinson’s disease (PD), and cerebral trauma (Sheng et al. 2010; Nixon et al. 2005; Clark et al. 2008). However, there are few studies regarding autophagy in PTSD, and the mechanisms underlying PTSD are not clearly understood.
The medial prefrontal cortex (mPFC) is an important part of the limbic system, which includes the amygdala, hippocampus, and hypothalamus. Numerous studies have shown that the mPFC directly interacts with and forms strong synaptic interconnections with the amygdaloid region, hippocampus, and other brain regions and is closely associated with regulation of emotion, behavior, and especially memory (Roozendaal et al. 2009; Giustino and Maren 2015). Recent studies have also shown that ventral hippocampus (vHPC)–mPFC afferent pathways are critical for the encoding and updating of spatial cues during spatial working memory (Spellman et al. 2015; Gibson and Mair 2016). mPFC neurons are highly sensitive to stress- and anxiety-like behaviors; dysfunction and abnormal activity of the mPFC appears in many stress-related mental illnesses such as depression and anxiety disorders (Moghaddam and Jackson 2004; Chaudhury et al. 2013). Many magnetic resonance imaging studies have indicated that mPFC volume is significantly decreased and that gray matter density is also decreased in PTSD patients compared with that of unaffected individuals; furthermore, these clinical studies showed a significant correlation between mPFC volume and the severity of PTSD symptomatology (Williams et al. 2006; Wang et al. 2016; Boccia et al. 2016). Single-prolonged-stress (SPS)-exposed rats, an established animal model that has been extensively applied in PTSD studies, exhibit HPA-axis dysfunction and abnormal behavior (Ding et al. 2010; Yehuda 2005). In previous studies, we discovered that neuronal apoptosis induced mPFC atrophy in SPS-exposed rats. These results indicate that changes in the structure and function of mPFC neurons are closely related to the neuropathology of PTSD (Zhao et al. 2014; Yu et al. 2014). Research has also provided evidence that autophagy and apoptosis utilize a number of common cellular and molecular regulatory mechanisms or signaling pathways (Oral et al. 2016). We, therefore, suspect that neuronal autophagy may be associated with changes in the morphology and function of the mPFC and may play an important role in the pathogenesis of PTSD.
In the present study, we sought to monitor ultrastructural changes and changes in the expression of Beclin-1, LC3, and p62 in the mPFC of SPS-exposed rats using transmission electron microscopy (TEM), immunohistochemistry, immunofluorescence, and Western blotting analysis to investigate the dysregulation of autophagic activity in the mPFC as a pathogenic mechanism for PTSD.
Materials and Methods
Animals and SPS Model Establishment
Seventy-five healthy male Wistar rats (180–200 g body weight) were provided by the Department of Laboratory Animals at China Medical University. Rats were housed with free access to food and water (23 ± 2 °C, 55 ± 5% humidity, and 12-h light/12-h dark cycle). All rats were randomly divided into either the control group or one of the SPS groups (1, 4, 7, and 14 days), with 15 rats in each group. Rats of the control group remained in home cages with no handling. On the first day, rats of the SPS groups were restrained for 2 h with a plastic container (immobilization) and exposed to a forced swim stress (24 ± 1 °C) for 20 min with a 15-min rest, followed by drying and ether anesthesia (consciousness was lost). Following SPS, the rats were fed routinely. The study protocol was reviewed and approved by the Ethics Committee of China Medical University.
Morris Water Maze Test
The Morris water maze was used to assess the learning and spatial memory performance of rats in the control group and the 7 days after the SPS. Briefly, a test lasting 5 consecutive days with four trials on each day was performed. The rat was placed in the water facing the wall of the pool, with the drop location changing for each trial, and allowed 120 s to locate the submerged platform, where it was allowed to remain for 20 s. If the animal failed to find the platform within 120 s, it was guided gently onto the platform and allowed to stay there for 20 s. The time taken to locate the platform (escape latency time, ELT) was noted as the acquisition index or an index of their learning capabilities. The spatial probe test was used to evaluate spatial memory on the sixth day. In the spatial probe test, the platform was removed, and the rats were given a probe test of the spatial location. The movement track of each rat was recorded by the computer. The percentage of time spent in the target quadrant was calculated as an index for spatial memory capabilities.
Section Preparation
Under anesthesia, six rats per group were transcardially perfused with pre-cooled saline and then perfused with 4% paraformaldehyde (PFA) in 0.1 M phosphate-buffered saline (PBS) through the left ventricle. The whole brain was removed and immersed in 30% sucrose solution after post-fixation in 4% PFA at 4 °C and then frozen in liquid nitrogen. Serial frontal sections of the brain tissue were cut at 15-μm thickness on a Leica CM3050S cryostat and stored at −20 °C in preparation for immunohistochemistry and immunofluorescence staining.
Transmission Electron Microscope
Under anesthesia, four rats per group were transcardially perfused with 4% PFA and 2.5% glutaraldehyde in 0.1 M PBS through the left ventricle, following which the mPFC (interaural 5.90 to 5.14 mm, between bregma 2.1 and 1.34 mm) was fixed in 2.5% glutaraldehyde in 0.1 M PBS. After washing with 0.1 M PBS, the mPFC was cut into blocks measuring approximately 1 mm wide, 5 mm long, and 1 mm thick. The blocks were post-fixed in 1% osmium tetroxide, rinsed in distilled water several times, dehydrated, then placed in propylene oxide, and finally polymerized in pure Epon 812. Then, 70-nm-thick ultrathin sections were cut on an ultra-microtome using diamond knives, collected on copper grids, and stained with 4% uranyl acetate and Reynolds lead citrate. A minimum of 10 sections from each mPFC were observed under TEM (JEM-1200EX; JEOL, Japan).
Double Immunofluorescence Staining
After being washed with PBS three times, the sections were treated with 5% bovine serum albumin (BSA) and 0.3% Triton X-100 in PBS for 30 min to block nonspecific staining at room temperature, followed by washing with PBS three times. For double immunofluorescence staining of Beclin-1 with glial fibrillary acid protein (GFAP) or NeuN and LC3 with GFAP or NeuN, sections were incubated with a mixture of a rabbit anti-Beclin-1 polyclonal antibody (Santa Cruz, USA; 1:500) and either a mouse anti-GFAP polyclonal antibody (Santa Cruz, USA; 1:500) or a mouse anti-NeuN polyclonal antibody (Abcam, British; 1:1000) or incubated with a rabbit anti-LC3 polyclonal antibody (Santa Cruz, USA; 1:500) and either a mouse anti-GFAP polyclonal antibody (Santa Cruz, USA; 1:500) or a mouse anti-NeuN polyclonal antibody (Abcam, British; 1:1000) at 4 °C overnight, respectively. After washing with PBS three times, the sections were incubated with a Cy3-conjugated secondary antibody (Boster Biotechnology, China; 1:200) and fluorescein isothiocyanate (FITC) secondary antibody (Boster Biotechnology, China; 1:100) for 2 h at room temperature, washed, and observed using a Nikon 80i fluorescence confocal microscope. A few sections were incubated in PBS instead of a primary antibody as the negative control in every experiment. The fluorescent intensity was analyzed using the MetaMorph/DPIO/BX41 morphology image analysis system. The LC3 punctate was analyzed as previously described (Wang et al. 2013); the number of green punctate in each field of view was observed under the confocal microscope and was analyzed using the MetaMorph/DPIO/BX41 morphology image analysis system. Five slides were randomly selected from each group, and five random fields were included in each slide.
Immunohistochemical Staining
Immunohistochemical staining was performed using the immunohistochemical SABC method. Sections were treated with 0.3% Triton X-100 in PBS and 5% BSA to block nonspecific staining, blocked with dripped 10% goat serum for 30 min, and incubated with a rabbit anti-p62 polyclonal antibody (Santa Cruz, USA; 1:500) at 4 °C overnight. Following washing with PBS, the sections were incubated with a secondary antibody anti-rabbit IgG-HRP (Boster Biotechnology, China; 1:1000) for 2 h at room temperature. Finally, DAB was used as a chromogen for approximately 10 min until the brown color appeared. Slices were then dehydrated and mounted with neutral balsam. Five slides were randomly selected from each group, and five random fields were included in each slide. The optical density (OD) of positive cells in each field was recorded, and the average of the OD in each field was recorded and analyzed using the MetaMorph/DPIO/BX41 morphology image analysis system.
Western Blotting
Five rats per group were anesthetized and then decapitated, and the whole brain was quickly removed and placed in an ice-cold dish. The mPFC (interaural 5.90 to 5.14 mm, between bregma 2.1 and 1.34 mm) was dissected according to the atlas by cutting perpendicularly, immediately frozen over liquid nitrogen and stored at −80 °C for Western blot analysis. After thawing, the mPFC was lysed using radio immunoprecipitation assay (RIPA) lysis buffer with protease inhibitor in an ice-cold dish, and the protein concentration of the lysate was measured using the Coomassie brilliant blue method. Each sample was separated by using 12% (w/v) gradient sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and transferred to a PVDF membrane. The membrane was blocked with 5% dried skim milk for 2 h and incubated with a rabbit anti-Beclin-1 polyclonal antibody (Santa Cruz, USA; 1:500), rabbit anti-LC3 polyclonal antibody (Santa Cruz, USA; 1:500), and rabbit anti-p62 polyclonal antibody (Santa Cruz, USA; 1:500) at 4 °C overnight. The membranes were then incubated with the secondary antibody, anti-rabbit IgG-HRP (Boster Biotechnology, China; 1:2000), for 2 h at room temperature. The signals on the membranes were detected with enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech, UK). To confirm equal protein loading, the same blots were incubated with antibodies specific for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Santa Cruz, USA; 1:1000). The OD of Beclin-1, LC3-I, LC3-II, p62, and GAPDH was analyzed on the Gel Image Analysis System (Tanon 2500R, China).
Statistical Analysis
All statistical analyses were performed using SPSS 17.0 software. The results were expressed as the mean ± SD. The Morris water maze test was analyzed by two-tailed independent Student’s t test. The differences between the control group and the SPS groups were compared and analyzed by one-way analysis of variance followed by Tukey’s post hoc test. A level of P < 0.05 was considered to be statistically significant.
Results
Morris Water Maze Test Results
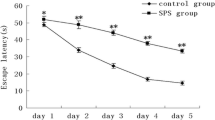
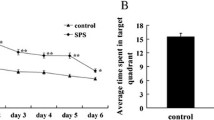
The ELT decreased from day 1 to day 5 in the control and SPS groups; however, day 1–5 ELTs were significantly higher in the SPS group than in the control group (Fig. 1). Spatial memory testing showed that the percent time in the target quadrant in the control and SPS groups was 52.38 ± 5.22 and 24.65 ± 4.15%, respectively. There was a statistically significant difference between the control and SPS groups.

ELT of control and SPS group rats on the 5 test days. n = 5 for each group. Asterisk, compared with the control group, denotes P < 0.05
TEM Analysis of the Morphological Changes in the mPFC Neurons
TEM was used to confirm neuronal autophagy and was performed in both the control and SPS groups. As depicted in Fig. 2, some mPFC neurons exhibited varying degrees of ultrastructural changes at 7 days after stimulation. A few visible autolysosomes appeared in the neuronal cytoplasm in the group tested 7 days after SPS (the SPS 7-day group) but were nearly absent in the control group.

Representative TEM graphs showing morphological changes in mPFC neurons. Bar = 200 nm, Bar = 100 nm. a Control group. b ,c SPS 7-day group. White arrowhead denotes autolysosomes (autophagosomes that have fused with lysosomes, which are characterized by membranous structures that wrap around heterogeneously dense materials)
Immunofluorescent Staining Results
Representative graphs of Beclin-1 immunofluorescent staining are shown in Fig. 3a–e. Beclin-1 was observed in the cytoplasm. In comparison with that of the control group, the fluorescence intensity of Beclin-1 in the mPFC was decreased at 1 day, increased at 4 days, and peaked at 7 days after SPS stimulation. Quantitative analysis revealed significant differences in the fluorescence intensity among the five groups. Moreover, to determine the types of cells that expressed Beclin-1 in the mPFC after SPS stimulation, we compared the localization of Beclin-1 immunoreactivity (ir) with the localization of markers of different cell types. NeuN is a marker of mature neurons; GFAP is a marker of glial cells. Confocal microscopy analysis showed that Beclin-1-ir was primarily found in neurons, and only a few GFAP-ir cells exhibited Beclin-1-ir in the control group and the SPS 7-day group. Representative graphs are shown in Fig. 4.

Beclin-1 expression in the mPFC by immunofluorescence staining (magnification, ×400). Bar = 20 μm. a Control group. b SPS 1-day group. c SPS 4-day group. d SPS 7-day group. e SPS 14-day group. The quantitative analysis of Beclin-1 expression is shown in (f). Asterisk, compared with the control group, denotes P < 0.05. White triangle, compared with the SPS 1-day group, denotes P < 0.05. Black triangle, compared with the SPS 7-day group, denotes P < 0.05

Double immunofluorescence staining for Beclin-1 and NeuN/GFAP in the mPFC in the control and the SPS 7-day group (magnification, ×400). Bar = 20 μm. a–f control group. g–l SPS 7-day group. Expression of Beclin-1 (in green) and NeuN/GFAP (in red) is observed at the mPFC. The yellow color is produced due to the overlapping of red and green, indicating that Beclin-1-ir was primarily found in neurons, and only a few GFAP-ir cells exhibited Beclin-1-ir in the control group and the SPS 7-day group
Previous studies have described LC3 punctate structures that represent isolated membranes and/or autophagosomes by immunofluorescent staining. In this study, the distribution of LC3 puncta in the mPFC was monitored by confocal microscopy. As revealed in Fig. 5a–b, LC3 was mainly distributed in the cytoplasm, and the distribution of specific LC3 puncta was observed in the cytoplasm. The mean fluorescence intensity of LC3 increased, and more LC3 punctate appeared in the SPS 7-day group compared to that in the control group (Fig. 5c–d). We also explored colocalization of LC3-ir and NeuN/GFAP-ir using confocal microscopy. The results showed that LC3-ir was mainly found in neurons of the control and the SPS 7-day group. Colocalization of LC3 and NeuN increased as the number of LC3-ir-positive cells increased. By contrast, we found only a few GFAP-ir cells that exhibited LC3-ir in the control and the SPS 7-day group. Representative graphs are shown in Fig. 6.

The expression of LC3 in mPFC was monitored by immunofluorescence staining (magnification, ×400). Bar = 20 μm. a Control group. b SPS 7-day group. LC3 staining in green and nucleus staining in blue, and LC3 punctate are visible (white arrow). c The quantitative analysis of LC3 expression. d The proportion of specific LC3 punctate per field of view was quantified. Asterisk, compared with the control group, denotes P < 0.05. White triangle, compared with the SPS 1-day group, denotes P < 0.05. Black triangle, compared with the SPS 7-day group, denotes P < 0.05

Double immunofluorescence staining for LC3 and NeuN/GFAP in the mPFC in the control and the SPS 7-day group (magnification, ×400). Bar = 20 μm. a–f control group. g–l SPS 7-day group. High expression of LC3 (in green) and NeuN or GFAP (in red) is observed in the mPFC. The yellow color is produced due to the overlapping of red and green, indicating that LC3-ir was primarily found in neurons, and only a few GFAP-ir cells exhibited LC3-ir in the control group and the SPS 7-day group
Immunohistochemical Staining Results
p62-positive structures were brown and granular and were mainly distributed in the cytoplasm (Fig. 7a–e). One day after SPS stimulation, expression of p62 significantly increased compared with that in the control group. Immunoreactivity slowly decreased from 4 to 14 days after SPS stimulation and remained lower than that in the control group. The mean optical densities of p62 expression are shown in Fig. 7f.

Representative immunohistochemistry of p62 expression in the mPFC in each group (magnification, ×400). Bar = 50 μm. a Control group. b SPS 1-day group. c SPS 4-day group. d SPS 7-day group. e SPS 14-day group. f The quantitative analysis of p62 expression. Asterisk, compared with the control group, denotes P < 0.05. White triangle, compared with the SPS 1-day group, denotes P < 0.05. Black triangle, compared with the SPS 7-day group, denotes P < 0.05
Western Blotting Analysis Results
Immunofluorescent staining identified changes in LC3 expression, but it detected expression of full-length LC3 and reacted with both LC3-I and LC3-II. We used Western blotting analysis to detect the different molecular weight bands of LC3-I and LC3-II and to definitively demonstrate autophagy. The molecular weights of Beclin-1, LC3-I, LC3-II, p62, and GAPDH were 60, 18, 16, 62, and 36 KD, respectively. Compared with that of the control group, the protein expression of Beclin-1 and the LC3-II/LC3-I ratio (LC3-I/GAPDH and LC3-II/GAPDH) decreased at 1 day, increased at 4 and 7 days, and slightly decreased at 14 days after SPS stimulation. Simultaneously, we observed that changes in the p62 expression levels were inversely related to Beclin-1 and the LC3-II/LC3-I ratio (LC3-I/GAPDH and LC3-II/GAPDH) during the experiment period (Fig. 8).

Western blot analysis for Beclin-1, LC3-I, LC3-II, and p62 in the mPFC. The quantification of the Western blot analysis for LC3-I/GAPDH, LC3-II/GAPDH, Beclin-1/GAPDH, LC3-II/LC3-I, and p62/GAPDH is shown. Asterisk, compared with the control group, denotes P < 0.05. White triangle, compared with the SPS 1-day group, denotes P < 0.05. Black triangle, compared with the SPS 7-day group, denotes P < 0.05
Discussion
Use of a standard animal model is the foundation for performing experimental research on PTSD. The 2005 International PTSD Scientific Conference identified rats induced by SPS stimulation as one of the standard PTSD models, and numerous experimental studies have verified that symptoms in the SPS-stimulated rat model correlate well with clinical manifestations in human PTSD (Kohda et al. 2007). Briefly, rats of the SPS rat model have HPA-axis dysfunction and exhibit abnormal behavior, memory, and environmental adaptability (Yehuda 2005; Khan and Liberzon 2004). In the present study, we found that, in SPS-stimulated rats, the ELT was increased, and the percent time in the target quadrant was decreased in the Morris water maze test. All of these results reflect an impairment in learning ability and spatial memory, indicating that SPS stimulation successfully induced PTSD in a rat model.
Autophagy is also known as type II programmed cell death, which is characterized by autophagosome formation and fusion with lysosomes to form autolysosomes. The results of the TEM experiments in this study showed that some mPFC neurons had varying degrees of ultrastructural changes and that a few visible autolysosomes appeared in the neuronal cytoplasm, indicative of neuronal autophagy in the mPFC 7 days after SPS stimulation. By contrast, these changes were nearly absent in the control group. The process of autophagy is rapid, which allows the autophagosomes to be observed a few minutes after induction, and the autolysosomes disappear after 2 h (Wu et al. 2014). A few autophagosomes or autolysosomes were observed by TEM in this study, which may be related to the transient nature of autophagy and the autophagic level. In a previous study by us, autophagosomes/autolysosomes with a double-membrane vesicle were also observed in hippocampal cells 7 days after SPS, and they are important for causing functional damage to neurons (Wan et al. 2016). Therefore, abnormal neuronal autophagy in the mPFC may be one of the reasons why mPFC atrophy and dysfunction are present in SPS-exposed rats. Visualization of autophagosomes (or membrane bilayers, autolysosomes) by TEM is the gold standard for the demonstration of autophagy, whereas levels of Beclin-1 and LC3 are still regarded as reliable and potential indicators of autophagic activity (Jiang and Mizushima 2014; Wang et al. 2015). A brain injury study showed that Beclin-1 levels increased at 4 h post-injury and peaked at 1 week at the site of injury mainly in neurons and astrocytes, suggesting that overexpression of Beclin-1 was important for autophagy (Diskin et al. 2005). A cerebral hypoxia–ischemia (HI) experimental study showed that Beclin-1 levels increased from 4 to 72 h post-injury and decreased after 72 h in both Beclin-1-positive neurons and glial cells at the later HI time points (Carloni et al. 2008). However, many studies concerning neurodegenerative diseases (such as AD, Parkinson’s disease, and Huntington’s disease) have shown autophagy dysregulation and a significant reduction in Beclin-1 levels (Lucin et al. 2013; Son et al. 2012). Interestingly, our study showed that Beclin-1 levels decreased at 1 day after SPS stimulation, increased from 4 to 7 days, and slightly declined at 14 days after SPS using immunofluorescence and Western blotting techniques. Differences in intracellular Atg levels and cell types observed in this study compared to that observed in other studies may be due to differences in the types of diseases or the use of different time points (Rangaraju and Notterpek 2011).
As a Bcl-2 homology-3 (BH3)-only protein, Beclin-1 can directly interact with other antiapoptotic multidomain proteins such as Bcl-2 or Bcl-XL via the BH3 domain (Maiuri et al. 2007). The biological effects of Bcl-2 have largely been attributed to its effect on apoptosis; however, Bcl-2 functions not only as an antiapoptotic protein but also as an anti-autophagy protein (Pattingre et al. 2005). Under normal physiological conditions, Beclin-1 binds to Bcl-2 (or potentially to other Bcl-2 family members) to form a Beclin-1–Bcl-2 complex, which acts as a regulator to ensure that autophagic levels remain within a homeostatic range. Beclin-1 separates from the Beclin-1–Bcl-2 complex and binds to class III phosphatidylinositol kinase or Vps34 (which is associated with the myristoylated, membrane-anchored kinase, Vps15) during amino acid deficiency, injury, stress, etc. (Levine et al. 2008). The lipid kinase activity of the multiprotein Beclin-1/Vps34 complex converts phosphatidylinositol into PI3K, which is involved in the nucleation of pre-autophagosomal structures, activates the lipid kinase Vps34, and thereby induces autophagy (Pattingre et al. 2005). Accordingly, one of the mechanisms by which Bcl-2 inhibits autophagy involves either antagonism of another undefined action of Beclin-1 that is necessary for autophagy or disruption of Beclin-1/hVps34 complex formation (Shimizu et al. 2004). Our research team previously described the relationship between Bcl-2 expression and apoptosis in mPFC neurons in PTSD rats. The previous study showed that expression of Bcl-2 in the mPFC gradually increased at 1 day after SPS stimulation, peaked at 4 days, and began to decline at 7 days through immunohistochemistry, Western blotting, and RT-PCR analysis (data not shown) (Li et al. 2013). We speculate that the abnormal expression of Beclin-1 in the mPFC was correlated with the Bcl-2 levels after SPS stimulation. In addition, Beclin-1 expression can also be influenced by other factors such as PI3K, TSC1/2, and PTEN (Dodson et al. 2013).
Beclin-1 plays a central role in the early stage of autophagy (membrane source formation), and membrane-bound LC3-II is essential for the extension of the autophagosome membrane. LC3 punctate structures are consistent with the presence of autophagic vesicles visualized by TEM (Itakura and Mizushima 2011). In the present study, the distribution of LC3 puncta in the mPFC was observed using immunofluorescent staining, and the LC3-II level was measured by Western blotting. The proportion of LC3 puncta and the LC3-II/I ratio (LC3-I/GAPDH and LC3-II/GAPDH) changed over time in this study, which is consistent with changes in Beclin-1 expression during the experimental period. Changes in Beclin-1 levels, LC3 puncta, and the LC3-II/I ratio indicated that levels of autophagy were decreased in the mPFC at 1 day after SPS stimulation and increased at 4, 7, and 14 days. Our confocal microscopy results showed that Beclin-1-ir and LC3-ir were primarily found in neurons, and only a few GFAP-ir cells exhibited Beclin-1-ir and LC3-ir in the control group and the SPS 7-day group. These results suggested that autophagy was mainly concentrated in mPFC neuronal cells under normal physiological conditions and was sensitive to SPS stimulation.
p62 is involved in autophagolysosome-mediated degradation in the autophagy–lysosomal pathway, and p62 expression negatively correlates with autophagic activity (Kim et al. 2014). Therefore, p62 is an important autophagic substrate, and the expression levels are closely related to autophagic flux. p62 has been reported to be able to directly bind to Bcl-2 to disturb the Bcl-2 and Beclin-1 interaction, thereby affecting autophagy through the Bcl-2- and Beclin-1-mediated pathway (Zhou et al. 2013). Immunohistochemistry and Western blotting analysis showed an increase in p62 levels at 1 day that gradually decreased at 4, 7, and 14 days after SPS stimulation, suggesting that the p62 levels synchronously change with alterations in the expression of Beclin-1 and the LC3-II/I ratio. These results further confirm that autophagic flux in the mPFC was inhibited at 1 day after SPS stimulation and was then activated from 4 to 14 days. Moreover, the multifunctional protein p62 is associated with several neurodegenerative disorders, such as frontotemporal lobar degeneration and AD, and is frequently found in protein inclusions or neurofibrillary tangles (Caccamo et al. 2016). The p62 expression correlates with the level of total tau protein and the phosphorylation of tau protein, which could influence cognitive function (Kuusisto et al. 2002). In this study, p62-positive structures were mainly distributed in the cytoplasm, and the expression changed during the experimental period. An abnormal p62 level might be one of the potential molecular mechanisms for impaired learning and memory function in PTSD.
As mentioned previously, basal levels of autophagy are crucial for maintaining intracellular homeostasis under physiological conditions. Theoretically, autophagy can function during both protective and threatening effects under various physiological or pathological conditions (Larsen and Sulzer 2002). The present study demonstrates that changes in autophagic activity in the mPFC appear to be a response to SPS stimulation and may play an important role in the pathogenesis of PTSD. Apoptosis and autophagy all play crucial roles in mPFC atrophy and dysfunction in SPS-exposed rats. The processes of the regulation of apoptosis and autophagy are very complex and are modulated by some of the same molecular mechanisms or factors. Cross-talk between autophagy and apoptosis during the process of programmed cell death has recently attracted more attention and deserves further investigation in PTSD research.
In conclusion, dysregulation of autophagic activity in the mPFC might be one of the pathologic mechanisms leading to PTSD and might also explain why morphological changes and functional disorders occur in the mPFC of PTSD patients.
References
American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders, 5th edn. American Psychiatric Association, Washington
Bailey CR, Cordell E, Sobin SM et al (2013) Recent progress in understanding the pathophysiology of post-traumatic stress disorder. CNS Drugs 27(3):221–232
Boccia M, D’Amico S, Bianchini F et al (2016) Different neural modifications underpin PTSD after different traumatic events: an fMRI meta-analytic study. Brain Imaging Behav 10(1):226–237
Caccamo A, Ferreira E, Branca C et al (2016) p62 improves AD-like pathology by increasing autophagy. Mol Psychiatry
Carloni S, Buonocore G, Balduini W (2008) Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis 32(3):329–339
Chaudhury D, Walsh JJ, Friedman AK et al (2013) Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature 493(7433):532–536
Chen Y, Klionsky DJ (2011) The regulation of autophagy—unanswered questions. J Cell Sci 124(Pt 2):161–170
Clark RS, Bayir H, Chu CT et al (2008) Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy 4(1):88–90
Ding J, Han F, Shi Y (2010) Single-prolonged stress induces apoptosis in the amygdala in a rat model of post-traumatic stress disorder. J Psychiatr Res 44(1):48–55
Diskin T, Tal-Or P, Erlich S et al (2005) Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J Neurotrauma 22(7):750–762
Dodson M, Darley-Usmar V, Zhang J (2013) Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic Biol Med 63:207–221
Fleming A, Noda T, Yoshimori T et al (2011) Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol 7(1):9–17
Gibson BM, Mair R (2016) A pathway for spatial memory encoding. Learn Behav 44(2):97–98
Giustino TF, Maren S (2015) The role of the medial prefrontal cortex in the conditioning and extinction of fear. Front Behav Neurosci 9:298
Hughes KC, Shin LM (2011) Functional neuroimaging studies of posttraumatic stress disorder. Expert Rev Neurother 11(2):275–285
Itakura E, Mizushima N (2011) p62 targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol 192(1):17–27
Jiang P, Mizushima N (2014) Autophagy and human diseases. Cell Res 24(1):69–79
Khan S, Liberzon I (2004) Topiramate attenuates exaggerated acoustic startle in an animal model of PTSD. Psychopharmacology 172(2):225–229
Kim JH, Hong SK, Wu PK et al (2014) Raf/MEK/ERK can regulate cellular levels of LC3B and SQSTM1/p62 at expression levels. Exp Cell Res 327(2):340–352
Kohda K, Harada K, Kato K et al (2007) Glucocorticoid receptor activation is involved in producing abnormal phenotypes of single-prolonged stress rats: a putative post-traumatic stress disorder model. Neuroscience 148(1):22–33
Kuusisto E, Salminen A, Alafuzoff I (2002) Early accumulation of p62 in neurofibrillary tangles in Alzheimer's disease: possible role in tangle formation. Neuropathol Appl Neurobiol 28(3):228–237
Larsen KE, Sulzer D (2002) Autophagy in neurons: a review. Histol Histopathol 17(3):897–908
Lee JY, He Y, Sagher O et al (2009) Activated autophagy pathway in experimental subarachnoid hemorrhage. Brain Res 1287:126–135
Levine B, Sinha S, Kroemer G (2008) Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 4(5):600–606
Li Y, Han F, Shi Y (2013) Increased neuronal apoptosis in medial prefrontal cortex is accompanied with changes of Bcl-2 and Bax in a rat model of post-traumatic stress disorder. J Mol Neurosci 51(1):127–137
Longatti A, Orsi A, Tooze SA (2010) Autophagosome formation: not necessarily an inside job. Cell Res 20(11):1181–1184
Lucin KM, O'Brien CE, Bieri G et al (2013) Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron 79(5):873–886
Maiuri MC, Le Toumelin G, Criollo A et al (2007) Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 26(10):2527–2539
Mellman TA, Alim T, Brown DD et al (2009) Serotonin polymorphisms and posttraumatic stress disorder in a trauma exposed African American population. Depress Anxiety 26(11):993–997
Meng L, Jiang J, Jin C et al (2016) Trauma-specific grey matter alterations in PTSD. Sci Rep 6:33748
Moghaddam B, Jackson M (2004) Effect of stress on prefrontal cortex function. Neurotox Res 6(1):73–78
Narendra D, Kane LA, Hauser DN et al (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6(8):1090–1106
Nixon RA, Wegiel J, Kumar A et al (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64(2):113–122
Oral O, Akkoc Y, Bayraktar O et al (2016) Physiological and pathological significance of the molecular cross-talk between autophagy and apoptosis. Histol Histopathol 31(5):479–498
Pattingre S, Tassa A, Qu X et al (2005) Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell 122(6):927–939
Pollice R, Bianchini V, Roncone R et al (2012) Psychological distress and post-traumatic stress disorder (PTSD) in young survivors of L'Aquila earthquake. Riv Psichiatr 47(1):59–64
Pratchett LC, Daly K, Bierer LM et al (2011) New approaches to combining pharmacotherapy and psychotherapy for posttraumatic stress disorder. Expert Opin Pharmacother 12(15):2339–2354
Rami A, Langhagen A, Steiger S (2008) Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis 29(1):132–141
Rangaraju S, Notterpek L (2011) Autophagy aids membrane expansion by neuropathic Schwann cells. Autophagy 7(2):238–239
Rangaraju S, Verrier JD, Madorsky I et al (2010) Rapamycin activates autophagy and improves myelination in explant cultures from neuropathic mice. J Neurosci 30(34):11388–11397
Roozendaal B, McReynolds JR, Van der Zee EA et al (2009) Glucocorticoid effects on memory consolidation depend on functional interactions between the medial prefrontal cortex and basolateral amygdala. J Neurosci 29(45):14299–14308
Sahani MH, Itakura E, Mizushima N (2014) Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending ontranscriptional upregulation and autophagy-derived amino acids. Autophagy 10(3):431–441
Sheng R, Zhang LS, Han R et al (2010) Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 6(4):482–494
Sherin JE, Nemeroff CB (2011) Post-traumatic stress disorder: the neurobiological impact of psychological trauma. Dialogues Clin Neurosci 13(3):263–278
Shimizu S, Kanaseki T, Mizushima N et al (2004) Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 6(12):1221–1228
Son JH, Shim JH, Kim KH et al (2012) Neuronal autophagy and neurodegenerative diseases. Exp Mol Med 44(2):89–98
Spellman T, Rigotti M, Ahmari SE et al (2015) Hippocampal-prefrontal input supports spatial encoding in working memory. Nature 522(7556):309–314
Wan J, Liu D, Zhang J et al (2016) Single-prolonged stress induce different change in the cell organelle of the hippocampal cells: a study of ultrastructure. Acta Histochem 118(1):10–19
Wang Q, Zhu J, Zhang K et al (2013) Induction of cytoprotective autophagy in PC-12 cells by cadmium. Biochem Biophys Res Commun 438(1):186–192
Wang D, Tian M, Qi Y et al (2015) Jinlida granule inhibits palmitic acid induced-intracellular lipid accumulation and enhances autophagy in NIT-1 pancreatic β cells through AMPK activation. J Ethnopharmacol 161:99–107
Wang T, Liu J, Zhang J et al (2016) Altered resting-state functional activity in posttraumatic stress disorder: a quantitative meta-analysis. Sci Rep 6:27131
Weidberg H, Shvets E, Shpilka T et al (2010) LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 29(11):1792–1802
Williams LM, Kemp AH, Felmingham K et al (2006) Trauma modulates amygdala and medial prefrontal responses to consciously attended fear. NeuroImage 29(2):347–357
Wu C, Zheng J, Yao X et al (2014) Defective autophagy in chondrocytes with Kashin-Beck disease but higher than osteoarthritis. Osteoarthr Cartil 22(11):1936–1946
Xie Z, Klionsky DJ (2007) Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9(10):1102–1109
Yamamoto A, Yue Z (2014) Autophagy and its normal and pathogenic states in the brain. Annu Rev Neurosci 37:55–78
Yang Z, Goronzy JJ, Weyand CM (2015) Autophagy in autoimmune disease. J Mol Med (Berl) 93(7):707–717
Yehuda R (2005) Neuroendocrine aspects of PTSD. Handb Exp Pharmacol 169:371–403
Yu B, Wen L, Xiao B et al (2014) Single prolonged stress induces ATF6 alpha-dependent endoplasmic reticulum stress and the apoptotic process in medial frontal cortex neurons. BMC Neurosci 15:115
Zhao D, Han F, Shi Y (2014) Effect of glucose-regulated protein 94 and endoplasmic reticulum modulator caspase-12 in medial prefrontal cortex in a rat model of posttraumatic stress disorder. J Mol Neurosci 54(2):147–155
Zhou L, Wang HF, Ren HG (2013) Bcl-2-dependent upregulation of autophagy by sequestosome 1/p62 in vitro. Acta Pharmacol Sin 34(5):651–656
Acknowledgments
This research was supported by grants from the National Natural Science Foundation of China (No. 81571324) and Specialized Research Fund for the Doctoral Program of Higher Education (No. 20132104110021). The authors would like to thank the reviewers for their valuable comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study protocol was reviewed and approved by the Ethics Committee of China Medical University.
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Zheng, S., Han, F., Shi, Y. et al. Single-Prolonged-Stress-Induced Changes in Autophagy-Related Proteins Beclin-1, LC3, and p62 in the Medial Prefrontal Cortex of Rats with Post-traumatic Stress Disorder. J Mol Neurosci 62, 43–54 (2017). https://doi.org/10.1007/s12031-017-0909-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-017-0909-x