
Abstract
An imbalance in metal homeostasis is a prominent feature of Alzheimer’s disease (AD). A wealth of evidence from independent studies over the past two and half decades has found changes to the distribution of brain iron, zinc, and copper in AD patients and animal models of the disease. Early research focused on the association of these metals with amyloid beta (Aβ), particularly extraneuronal Aβ plaque pathology. In contrast, there are numerous studies that have demonstrated a loss of iron-, zinc-, or copper-dependent cellular functions in AD animal and cell models, highlighting the importance of metal homeostasis in maintaining healthy brain function. Characterizing the molecular pathways that are impacted by iron, zinc, or copper will shed light on how these metals affect neuoroprotection, and conversely, neurodegeneration. Of particular interest is the role that the AD-associated presenilins have on protein trafficking and degradation, as metal homeostasis is dependent on the efficient trafficking and recycling of specific metal transporters. This review summarizes what is currently known about presenilin-dependent protein trafficking and the role of presenilin in protein turnover, particularly via the autophagy-lysosomal system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Presenilin (PSEN, PS) is an aspartyl protease. It is most well characterized as the catalytic subunit of γ-secretase, a multiprotein complex minimally comprised of APH-1, nicastrin, PEN-2, and PSEN, which cleave numerous type-1 transmembrane substrates. In mammals, there are two PSEN isoforms, presenilin 1 (PSEN1) and presenilin 2 (PSEN2), that possess nine transmembrane domains, a cytosolic loop domain, and two critical aspartate residues that provide catalytic function to γ-secretase. Most attention has focused on understanding its role in the proteolytic cleavage of the amyloid precursor protein (APP) as it is the final step in the release of Aβ peptides from the parent molecule, one of two major pathological hallmarks associated with AD. Presenilin affects a wide variety of cellular processes including protein trafficking (Naruse et al. 1998), signal transduction and signaling (Boonen et al. 2009; Venezia et al. 2007), cell adhesion (Chen and Schubert 2002; Zhang et al. 1998; Murayama et al. 1998), calcium homeostasis (Yu 2009), tau phosphorylation (Kang et al. 2005; Shepherd et al. 2004; Pigino et al. 2001), and lysosomal protein degradation (Lee et al. 2010; Neely et al. 2011; Zhang et al. 2012). In the case of protein trafficking, calcium homeostasis, and lysosomal function, there is evidence that PSEN may modulate these processes independently of γ-secretase activity. These will be discussed in more detail below. PSEN also affects metal homeostasis independently of its γ-secretase function, particularly copper and zinc (Greenough et al. 2013; Greenough et al. 2011; Southon et al. 2013). This is of personal interest to my research as one of our group’s overarching goals is focused on trying to elucidate the mechanisms that affect metal regulation in the brain, particularly during aging and in neurodegenerative diseases.
The Role of Presenilin in Protein Trafficking
Inhibitors of γ-secretase can disrupt movement of APP and the iron transporter transferrin (Tf) through the endosomal recycling compartment (Zhang et al. 2006). PSEN1 is involved in both pre- and post-Golgi trafficking of APP and mediates retention of full-length APP and APP C-terminal fragments (CTFs) in endosomal compartments at the expense of PM-bound APP (Rechards et al. 2006). Numerous other studies have confirmed that PSENs influence the localization of full-length APP and/or APP CTFs (Chen et al. 2000; Leem et al. 2002; Kaether et al. 2002; Cai et al. 2003; Cai et al. 2006; Wrigley et al. 2005; Wang et al. 2004; Gandy et al. 2007). The role that PSEN plays in APP CTF localization is of particular interest in the context of amyloidogenic processing of APP as Aβ generation is dependent on APP endocytosis from the PM (Small and Gandy 2006; Thinakaran and Koo 2008). Deletion or mutation of the APP cytoplasmic endocytic motif or inhibition of endocytosis reduces Aβ levels, providing convincing evidence that the final step of APP proteolysis by γ-secretase involves internalization of membrane-bound APP CTFs via endosomes (Koo and Squazzo 1994; Ring et al. 2007; Cirrito et al. 2008; Perez et al. 1999). In addition to modulation of APP trafficking, PSENs have been implicated in the intracellular trafficking and/or maturation of other proteins including neuronal cadherin (N-Cad) NMDA (N-methyl-d-aspartate) receptors and tyrosinase (Naruse et al. 1998; Saura et al. 2004; Uemura et al. 2003; Wang et al. 2006). In some cases, the role of PSEN in protein trafficking has been de-coupled from γ-secretase activity. For example, BCL-2 (B cell lymphoma), a negative regulator of apoptosis that requires mitochondrial localization to function, is retained by PSEN in the endoplasmic reticulum (ER)/Golgi apparatus (Wang et al. 2005). However, inhibitors of γ-secretase do not influence BCL-2 retention, indicating a γ-secretase-independent function for PSEN.
Naruse and colleagues first described a role for PSEN1 in the maturation and trafficking of APP, APLP1 (a homolog of APP), and TrkB using cultured PSEN1-null embryonic mouse neurons (Naruse et al. 1998). Metabolic pulse chase labeling with 35S-methionine and subsequent biotin/streptavidin capture of total surface polypeptides revealed that PSEN1 does not affect protein trafficking en masse, and hence, appeared to control the plasma membrane localization of a distinct set of proteins (Naruse et al. 1998). Naruse and colleagues postulated that the accumulation of APP CTFs in the PSEN1−/− neurons was consistent with a functional disturbance to degradative pathways, notably internalization of APP from the cell surface and subsequent recycling (Koo and Squazzo 1994; Koo et al. 1996; Yamazaki et al. 1996). Later work by the Haass group demonstrated that APP internalization is indeed slowed by expression of dominant negative aspartate mutant PSEN1 (D385N) or addition of the γ-secretase inhibitor DAPT (Kaether et al. 2002). Importantly, expression of mutant PSEN2 (D366A) also delayed APP endocytosis, indicating that both PSEN1 and PSEN2 serve a dual role in APP proteolysis and trafficking (Kaether et al. 2002). A study by Zhang and colleagues then demonstrated that PSEN/γ-secretase is required for effective recycling of both APP and transferrin from the endocytic recycling compartment (ERC) to the plasma membrane (Zhang et al. 2006). In the absence of PSEN, proteins that traffic via the ERC, which is a collection of tubular organelles associated with microtubules, accumulate in the ERC and do not recycle via the normal endo/lysosomal degradation pathway. Tellingly, disturbances to endocytic pathways were described as one of the first manifestations in AD many years ago (Cataldo et al. 2000).
PSEN conditional knockout mice that do not produce Aβ develop age-dependent memory and learning deficits that phenocopy those associated with AD (Shen and Kelleher 2007). This led researchers to describe a PSEN “loss of function” model as an alternative model to the amyloid cascade theory of AD in which elevated levels of Aβ cause a toxic gain of function (Shen and Kelleher 2007). However, AD is likely a combination of both loss and gain of function. To what extent either is more critical is still a matter of debate. However, it is important to highlight that PSEN impacts multiple cellular processes, including protein trafficking. Roles for PSEN in the maturation and/or trafficking of numerous other proteins have since been described. These include direct substrates of γ-secretase proteolysis such as APP (Kaether et al. 2002), N-cadherin (Uemura et al. 2003), neurotrophin-receptor-associated death domain (Gowrishankar et al. 2004), and tyrosinase (Wang et al. 2006). Intriguingly, tyrosinase is a Cu-dependent enzyme that is involved in pigment formation. PSEN-null embryos possess depigmented eyes, and PSEN heterozygote knockout adult mice have depigmented coats (Wang et al. 2006). Furthermore, interference of PSEN1 splicing in zebra fish embryos via injection with site-specific morpholino antisense oligonucleotides causes depigmentation (Nornes et al. 2008), consistent with a conserved role for PSEN in tyrosinase-dependent melanin biogenesis. Whether perturbed copper homeostasis due to loss of PSEN expression could partly explain reduced tyrosinase-dependent melanin formation has not been investigated. However, a depigmentation phenotype due to a functional loss of Cu import has been reported in a Drosophila melanogaster model overexpressing DmATP7, the sole orthologue of human copper transporters ATP7a and ATP7b in flies (Norgate et al. 2006).
PSEN has also been implicated in the Golgi/ER retention of BCL-2 (B cell lymphoma 2), sequestering it away from mitochondria where it has anti-apoptotic function (Wang et al. 2005). Interestingly, the localization of BCL-2 is not sensitive to chemical inhibitors of γ-secretase activity such as DAPT (Wang et al. 2005). The localization and/or maturation of several other proteins that are dependent on PSEN, regardless of its role in γ-secretase, have since been reported. These include the cell surface receptors epidermal growth factor receptor (EGFR) (Repetto et al. 2007), intercellular adhesion molecule-5 (ICAM-5) or telencephalin (Esselens et al. 2004), and β1 integrins (Zou et al. 2008). The defects in the trafficking of these receptors appear to be at the final stage of protein degradation with a concomitant accumulation in full-length protein in intracellular compartments (Coen and Annaert 2010). Lysosomal defects are a feature of AD and accumulating evidence points to a role for PSEN in autophagy (Lee et al. 2010; Neely et al. 2011), as discussed in the next section.
PSENs also physically interact with several proteins that participate in general protein trafficking. For example, PSENs interact with members of the Rab family of GTPases. One example is Rab11, which is localized to recycling endosomes where it regulates endocytic trafficking (McCarthy 2005). Another example is Rab8, which in contrast to Rab11, mediates traffic of cargo via select routes from the trans-Golgi network (TGN) to the plasma membrane (Kametani et al. 2004). PSEN also binds to RabGTPase dissociation inhibitor (RabGDI), a chaperone for Rab proteins in the cytosol (Scheper et al. 2000). PSEN1 knockout mouse neurons were found to have reduced membrane RabGDI, and thus, PSEN1 was postulated to act as membrane receptor for RabGDI in the ER, affecting vesicle transport at this location (Scheper et al. 2000). The same group later reported that membrane association of Rab6, a mediator of specific retrograde transport from the cell surface to the Golgi-ER, was reduced in PSEN1 knockout cells and dependent on phosphorylation events (Scheper et al. 2004). In the context of APP processing, expression of a Rab6 dominant negative mutant was shown to inhibit Aβ production in favor of sAPP formation (McConlogue et al. 1996). Aβ production is dependent on APP endocytosis and implies that PSEN1 may influence APP processing via retrograde transport pathways. Interestingly, PrPsc, the agent responsible for prion infection in mammals, is transported from the cell surface to the ER via a Rab6-mediated retrograde transport pathway (Beranger et al. 2002).
PSEN has also been shown to interact with several other proteins involved in vesicle trafficking and membrane fusion, including two members of the soluble N-ethyl maelimide sensitive factor attachment protein factor (SNARE) family, syntaxin 1A (McCarthy 2005), and syntaxin 5 (Suga et al. 2005; Suga et al. 2009; Suga et al. 2004). Syntaxin 5 has a profound influence on copper homeostasis in mammals and Drosophila (Norgate et al. 2010). Hence, PSENs have been shown to exert effects on a diverse range of molecules involved in protein trafficking, some of which are involved in metal transport pathways. With these effects in mind, it is feasible that changes to PSEN function could potentially alter the trafficking and localization of specific copper and/or zinc transporters. Any changes to the expression or cell surface localization of these transporters could alter Cu+ or Zn2+ import/export respectively and affect downstream pathways that require these metals for their activities. Work in this area is currently under investigation and will be discussed later in this review.
The Role of Presenilin in Autophagy-Lysosomal Protein Degradation
Autophagy (or macroautophagy) is the process of sequestration of cellular components by an autophagosome and subsequent fusion of the autophagosome with a lysosome for degradation of the cellular contents (Neely et al. 2011). Autophagy is a vital component of protein turnover that degrades organelles, long-lived proteins, and protein aggregates, and in the process, recycles amino acids for new protein synthesis (Mortimore and Poso 1987). Defects in autophagy are a feature of glycosphingolipidoses, a subset of rare lysosomal storage disorders that are characterized by the accumulation of glycosphingolipids (GSLs). GSLs are particularly enriched in the lipid rafts of neurons and are important for cholesterol trafficking (Lingwood 2010). Neuronal dysfunction is common amongst inherited lysosomal storage disorders including Niemann-Pick type C1 (NPC1) disease and Sandhoff disease (Lloyd-Evans et al. 2008; Sandhoff et al. 1968). APP CTFs have been shown to accumulate in neurons of NPC patient brains, as well as cultured mouse neurons treated with a compound known to induce NPC defects (Jin et al. 2004). Enlarged endosomes and increased levels of lysosomal proteases in AD patients are indicators of lysosomal dysfunction (Cataldo et al. 2000; Cataldo et al. 1996). Tellingly, PSEN mutations that cause FAD are associated with increased lysosomal pathology in humans and animal models of FAD (Cataldo et al. 2004). Annaert and colleagues described a putative role for PSEN in autophagy when they demonstrated that intercellular adhesion molecule 5 (ICAM-5; formerly called telencephalin) was misrouted in cultured PSEN1-null hippocampal neurons (Esselens et al. 2004). PSEN1 and ICAM-5 had previously been shown to interact but ICAM-5 is not a γ-secretase substrate (Annaert et al. 2001). In wild-type neurons, ICAM-5 localized to the somatodendritic plasmalemma but in PSEN1-null neurons, ICAM-5 accumulated in a compartment that co-stained with autophagic vacuole markers, LC3 and Apg12p (Esselens et al. 2004). Furthermore, the half-life of ICAM-5 was significantly increased in the PSEN1-null neurons, indicative of a disturbance to protein degradation (Esselens et al. 2004). Annaert and colleagues proposed that PSENs play a role in autophagy and protein turnover independent of γ-secretase function and speculated that the accumulation of ICAM-5 in autophagic vacuoles could be due to defective fusion with lysosomes. Similarly, the accumulation of α- and β-synuclein in cultured neurons from PSEN1-null mice was attributed to a defect in autophagy independent of γ-secretase activity (Wilson et al. 2004). Nakagawa and colleagues demonstrated that PSEN1 expression was upregulated with a concomitant increase in γ-secretase activity in cultured cell models of disrupted autophagy (Ohta et al. 2010). Hence, several lines of evidence point towards a link with altered PSEN activities and autophagy/lysosomal dysfunction. In 2010, Lee and colleagues reported that degradation of long-lived proteins was reduced in murine PSEN1-null blastocysts, PSEN1-deficient neurons, and FAD mutant patient fibroblasts, attributable to a loss of autophagic-lysosomal processing (Lee et al. 2010). Acidification is a critical factor for activating degradative enzymes within autolysosomes, and Lee and colleagues showed evidence for reduced lysosomal hydrolase, cathepsin D activity, and increased lysosomal pH values in PSEN1 KO cells vs. WT cells (Lee et al. 2010). Hence, a defect in acidification of autolysosomes was considered a likely culprit. A series of experiments demonstrated that trafficking of the vacuolar (H+)-ATPase (v-ATPase) responsible for lysosomal acidification was defective in PSEN1 KO cells (Lee et al. 2010). Mechanistically, the authors provided evidence to indicate that full-length PSEN1 holoproteins in the ER bind to the oligosaccharide responsible for v-ATPase trafficking to the lysosome, and facilitate its maturation via N-glycosylation (Lee et al. 2010). Although this remains an intriguing potential mechanism to explain the PSEN-dependent lysosomal defects in AD, several groups have since challenged it. Firstly, Neely and colleagues used PSEN knockout mouse embryonic fibroblast (MEF) cells and PSEN1 and PSEN2 RNAi-treated cells to demonstrate that both PSEN1 and PSEN2 were required for efficient lysosomal proteolysis; however, they could not replicate the acidification defects reported by Lee and colleagues (Neely et al. 2011). Instead, they proposed that loss of PSEN function causes a defect in autophagosome-lysosome fusion, a hypothesis previously postulated by others (Esselens et al. 2004; Wilson et al. 2004). The loss of lysosomal function in PSEN-null and PSEN-deficient cells was not replicated with γ-secretase inhibitors: the authors concluding that PSEN affects autophagy-lysosomal degradation independent of γ-secretase function (Neely et al. 2011). Two independent groups have been unable to replicate the glycosylation and/or acidification defect phenotypes reported by Lee and colleagues and have presented alternative hypotheses to explain the role of PSENs in lysosomal function (Zhang et al. 2012; Coen et al. 2012). In particular, the study by Coen and colleagues was very thorough in its attempt to detect differences in endolysosomal pH, hydrolytic activity, or v-ATPase subunit V0a1 trafficking (as a result of N-glycosylation) in PSEN knockout MEFs and blastocysts but could not detect such changes (Coen et al. 2012). Furthermore, mutation of the single conserved N-glycosylation site in the Drosophila homolog of V0a1, v100, did not alter its function in flies, providing in vivo evidence that v-ATPase subunit V0a1 trafficking is not dependent on N-glycosylation (Coen et al. 2012). Instead, the authors provide data to demonstrate that lysosomal Ca2+ storage/release is altered in PSEN-deficient cells, and propose that lysosomal deficits associated with loss of PSEN expression are linked to perturbed Ca2+-signaling (Coen et al. 2012), a vital step in lysosomal fusion (Saftig et al. 2009). However, recent studies using multiple cell types have demonstrated lysosomal acidification defects and subsequent impairment of autophagy attributable to loss of PSEN function (Avrahami et al. 2013; Dobrowolski et al. 2012), supporting the original findings of Lee and colleagues (Lee et al. 2010). The discrepancies in findings from the studies above could be due to differences in the fluorescent probes used to measure lyososmal pH changes, as often these changes are quite small and fluorescence intensity is organelle-dependent. A paper from the Nixon group assessed multiple probes and reported that Lysosensor Yellow/Blue-Dextran was optimal for measuring lysosomal pH changes across several cell types, particularly relative to the fluorescein-based dyes used in other studies (Wolfe et al. 2013). In the context of the Ca2+ deficits and lysosomal dysfunction evident in AD, a unifying hypothesis that links lysosome dysfunction with perturbed intracellular Ca2+ signaling due to PSEN loss of function is an intriguing possibility. Recently, Lee and colleagues went one step further to provide supporting evidence for their PSEN dysfunction/lysosomal acidification/defective autophagy model, demonstrating that lack of PSEN-dependent N-glycosylation of v-ATPase causes de-acidification of lysosomes which then causes a pH-dependent release of Ca2+ into the cytosol via the endolysosomal Ca2+ channel, transient receptor potential cation channel mucolipin subfamily member 1 (TRPML1) (Lee et al. 2015). Thus, defective Ca2+ calcium signaling associated with AD may be a downstream effect of de-acidification of lysosomes. This has important implications in terms of the regulation of other transition metal ions including Zn2+ and Cu+, which are sensitive to TRP channel activity and may have lysosomal function (Andersson et al. 2009; Gu and Lin 2010a; Mukhopadhyay et al. 2011).
Presenilin and Calcium Homeostasis
A wealth of evidence has accumulated showing disruption of intracellular calcium regulation in cells harboring FAD-associated mutations or loss-of-function mutations in PSEN. A consistent observation has been that induced release of calcium from ER stores is strongly increased by FAD-associated alleles (LaFerla 2002; Smith et al. 2005; Stutzmann 2005). PSEN binds to the calcium-binding proteins calsenilin and calmyrin, in addition to the cardiac ryanodine receptor (RYR2) and to sorcin (a RYR2-binding protein). These observations have led to the hypothesis that altered calcium homeostasis in patients with Alzheimer’s disease contributes to neuronal dysfunction and, ultimately, to cell death. A mechanism by which PSEN might increase ER calcium pools was suggested by Tu et al. (Tu et al. 2006). The authors demonstrated that full-length PSEN holoproteins form constitutive ER calcium leak channels (Tu et al. 2006). ER Ca2+ leak channels release Ca2+ from the ER and function to maintain cellular Ca2+ homeostasis (Camello et al. 2002). PSEN-mediated ER Ca2+ leak channel activity appeared to be independent of γ-secretase activity, or assembly of the γ-secretase complex, because it was retained by PSEN1 with a mutation in an active site aspartic acid residue (D257A) and was unaffected by the complete absence of γ-secretase activity in mice null for all three APH1 proteins (Tu et al. 2006). However, FAD-associated alleles exert strong dominant negative effects on leak channel function (Nelson et al. 2007). Bezprozvanny and colleagues proposed that passive PSEN-mediated calcium leakage, in equilibrium with SERCA (also known as ATP2A2) calcium re-uptake pumps, is fundamental to the establishment of intracellular cytoplasmic and ER calcium concentrations and, consequently, to physiological responses to calcium. A potential indication of the importance of this mechanism in another disease context is the reports of PSEN missense mutations and SNPs that segregate with familial dilated cardiomyopathy (Li et al. 2006; Li et al. 2011a). Molecular evidence supporting a role for PSEN in cardiac function came from studies using transgenic flies that either suppressed or overexpressed the Drosophila ortholog of presenilin (dPsn). Silencing of dPsn caused a reduced heart rate and overexpression of dPsn had the opposite effect, and these changes were attributed to changes in calcium receptor activities, namely dSERCA, dRyR, and dIP3R (Li et al. 2011b).
Molecular modeling of PSEN1 indicated that PSEN1 could potentially form a water-containing pore between the TMD6 and TMD7 region (Tolia et al. 2006; Sato et al. 2006), consistent with ion transport (and proteolytic) function. Cysteine-scanning mutagenesis of mouse PSEN1 TMDs later revealed that TMD6 is largely inaccessible to water, with only a few residues exposed, but TMD9, in addition to TMD7, could potentially form an ion conductance pore (Nelson et al. 2011). However, an attempt to recapitulate the effects of PSENs as passive ER Ca2+ leak channels using multiple Ca2+ imaging techniques provided conflicting results, with the authors of that particular study emphasizing the need to use ER-targeted Ca2+ probes (Shilling et al. 2012). Indeed, experiments using fluorescent probes such as Fura-2, which are widely used in the Ca2+ imaging field, should be interpreted with caution as they are not specific to Ca2+ but also bind other metals (Snitsarev et al. 1996). For example, Fura-2 has a higher affinity for Zn2+ than Ca2+, and as recommended by others, the Zn2+ chelator TPEN (N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine) can be used to quench the contributing Zn2+ signal in Fura-2 Ca2+ imaging experiments (Matias et al. 2010; Roe et al. 1990). It could be argued that the relative abundance of Zn2+ is insignificant compared to Ca2+. However, this is not the case in glutamatergic synapses where ZnT3-mediated [Zn]2+ release from presynaptic terminals can be as high as 300 μM (Frederickson et al. 2005). Zn2+ may also function as a neuromodulator of synaptic transmission that interacts with Ca2+-sensitive cell surface receptors such as NDMARs (Li et al. 2001; Lu et al. 2000; Pan et al. 2011; Adlard et al. 2010). Additionally, Ca2+ ion channels such as transient receptor potential A1 (TRPA1) are activated by Zn2+ and other transition metals (Andersson et al. 2009; Mukhopadhyay et al. 2011; Hung et al. 2009; Gu and Lin 2010b). This last point has important implications in terms of PSEN role in metal homeostasis because studies from our group revealed that zinc and copper transport is altered in PSEN-null and PSEN-deficient cultured cells and model organisms (Greenough et al. 2011; Southon et al. 2013). This will be discussed in the next section. In summary, PSENs play a role in calcium signaling, potentially via several mechanisms.
Presenilin as a Regulator of Transition Metals
An imbalance of biometal homeostasis is a feature of AD that has been linked with the pathogenesis of the disease (Bush 2012). The mammalian brain contains an intrinsically high concentration of copper, zinc, and iron compared to other tissues, which is reflective of its high requirement for these metals in numerous metal-dependent enzyme and metabolic processes (Greenough et al. 2013). The abundance and activity of specific metal transporters on plasma membranes control the majority of cellular copper import (via CTR1), zinc import (via ZIPs), and iron import (via transferrin/DMT1) as well as copper export (via ATP7a/b), zinc export (via ZnTs), and iron export (via ferroportin) respectively. Healthy cells are very good at sensing local metal concentrations and can regulate the abundance of transporters at the cell surface to limit or augment metal import/export. In the case of iron, a role for APP has been described whereby it stabilizes ferroportin on the cell surface (Wong et al. 2014) to facilitate iron export (Duce et al. 2010). Consistent with this function, the neuronal iron overload and oxidative stress that is a feature of AD may be attributable to APP dysfunction, potentially via altered processing by PSEN/γ-secretase. But what about copper and zinc? APP and Aβ are both metal-binding proteins, and overexpression of APP can reduce neuronal copper levels, whereas knockout of APP causes an elevation of copper, implying a role for APP in copper export (Bellingham et al. 2004). In more recent studies, knockout of PSEN in cultured mouse embroyonic fibroblasts (MEFs), as well as RNAi knockdown in human cells, caused a large reduction in copper and zinc uptake, together with a reduction in superoxide dismutase-1 (SOD1) activity, a Cu/Zn-dependent antioxidant enzyme (Greenough et al. 2011). A reduction in total copper and SOD1 activity was also observed in the brains of 6-month-old PSEN1 heterozygous knockout mice (PSEN1 KO is homozygous lethal), and a PSEN1 gene dose-dependent reduction in total copper and SOD1 activity was evident in E14 mouse embryos (Greenough et al. 2011). A key point from that particular study was that pharmacological inhibition of γ-secretase activity did not alter copper uptake or SOD1 activity in multiple cell types tested, and expression of a γ-secretase activity-deficient mutant of PSEN1 (D385A) in the PSEN knockout cells was able to rescue the copper uptake deficit and restore SOD1 activity. Together, these results strongly suggest that PSEN function can influence copper homeostasis independently of its role in APP proteolysis. But what is the mechanism? Can PSEN itself be a transporter of metals such as copper or zinc? Although there is no obvious metal-binding domains the presence of a potential ion conductance pore, as reported above to be coordinated within its transmembrane domains (Nelson et al. 2011), does raise it as an intriguing possibility. Does PSEN directly stabilize known metal transporters on the cell surface, akin to APP’s interaction with Ferroportin? Or does it affect copper and zinc transport in a non-direct way? For example, does PSEN, via its role in protein trafficking (as reviewed above), influence the abundance of specific metal transporters on the cell surface? Another possibility is that the lysosomal dysfunction caused by PSEN ablation or reduced activity could influence the availability of metal transporters on the cell surface due to reduced protein turnover. This would certainly fit with a functional PSEN holoprotein model, consistent with the experiments whereby D385A mutant human PSEN1, which is not efficiently endoproteolytically cleaved but rather accumulates as full-length holoproteins, can restore copper uptake and SOD1 activity more effectively than wild-type human PSEN1 in PSEN-deficient MEF cells (Greenough et al. 2011). Experiments to determine the exact role of PSEN- and FAD-associated PSEN mutations have on metal homeostasis are ongoing.
Concluding Remarks
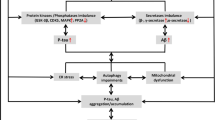
Since the late 1990s, PSEN has been implicated in the pathogenesis of AD due its role in the generation of Aβ peptides via γ-secretase and the association of PSEN mutations with FAD. The toxic gain of function or “amyloid cascade hypothesis” has been the dominant theory, and it was originally considered that a quantitative increase in Aβ, as evidenced by increased amyloid plaque load, was the primary cause of AD. This was subsequently altered to a more qualitative model, whereby the ratio of Aβ42/Aβ40 was more important as the longer form of the peptides is considered more pathogenic, particularly soluble oligomers. Despite the weight of evidence to indicate that Aβ is central to the disease, the dogma has been challenged in recent years. Extensive experiments conducted by numerous groups using animal and cell models as well in vitro assays have not reached a consensus on what constitutes the toxic species of Aβ. Furthermore, human clinical trials that have targeted Aβ reduction have overwhelmingly failed, despite positive animal trials. Many possible reasons for these failures have been described, including poor trial design and wrong dosing, and trial participants were too late stage in the disease. Whatever the case may be, AD is a pleiotropic and incredibly complex disease, making it hard to determine what has more weighted relevance. Is it oxidative stress? Is it mitochondrial dysfunction? Is it endocytic and/or protein trafficking defects or calcium signaling deficits? Or is it lysosomal/proteostasis dysfunction? What is clear is that PSEN can impact on all of these pathways, both in a γ-secretase-dependent and -independent manner. Moreover, brain biometals including copper, zinc, and iron play distinct roles in many of the pathways affected by AD. A recent critical review of the current literature on AD by an independent researcher concluded “…the etiology centers on the identification of the normal physiological function (a natural role has already been implied by many lines of research) of the APP/Aβ system and PSEN all related to maintaining relative metal ion levels within the neuron (Kepp 2016)”. The “metal ion theory of Alzheimer’s disease” that was postulated two decades ago is supported by a wealth of literature (a Pubmed search with the search term “metals & Alzheimer’s disease” returns >4400 publications) and certainly still carries weight in terms of explaining many of the causative factors of the disease. Maturing technologies for determining brain metal-protein interactions (Lothian et al. 2013) is ushering in a new age of “metalloproteomics” that will advance our understanding of AD and other neurodegenerative disorders that feature perturbed metal function.
Abbreviations
- AD:
-
Alzheimer’s disease
- APP:
-
Amyloid precursor protein
- ER:
-
Endoplasmic reticulum
- PSEN:
-
Presenilin
References
Adlard PA et al. (2010) Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J Neurosci 30(5):1631–1636
Andersson DA et al. (2009) Clioquinol and pyrithione activate TRPA1 by increasing intracellular Zn2+. Proc Natl Acad Sci U S A 106(20):8374–8379
Annaert WG et al. (2001) Interaction with telencephalin and the amyloid precursor protein predicts a ring structure for presenilins. Neuron 32(4):579–589
Avrahami L et al. (2013) Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem 288(2):1295–1306
Bellingham SA et al. (2004) Gene knockout of amyloid precursor protein and amyloid precursor-like protein-2 increases cellular copper levels in primary mouse cortical neurons and embryonic fibroblasts. J Neurochem 91(2):423–428
Beranger F et al. (2002) Stimulation of PrP(C) retrograde transport toward the endoplasmic reticulum increases accumulation of PrP(Sc) in prion-infected cells. J Biol Chem 277(41):38972–38977
Boonen RA, van Tijn P, Zivkovic D (2009) Wnt signaling in Alzheimer’s disease: up or down, that is the question. Ageing Res Rev 8(2):71–82
Bush AI The Metal Theory of Alzheimer’s Disease. J Alzheimers Dis, 2012
Cai D et al. (2003) Presenilin-1 regulates intracellular trafficking and cell surface delivery of beta-amyloid precursor protein. J Biol Chem 278(5):3446–3454
Cai D et al. (2006) Phospholipase D1 corrects impaired betaAPP trafficking and neurite outgrowth in familial Alzheimer’s disease-linked presenilin-1 mutant neurons. Proc Natl Acad Sci U S A 103(6):1936–1940
Camello C et al. (2002) Calcium leak from intracellular stores--the enigma of calcium signalling. Cell Calcium 32(5–6):355–361
Cataldo AM et al. (1996) Colocalization of lysosomal hydrolase and beta-amyloid in diffuse plaques of the cerebellum and striatum in Alzheimer’s disease and Down’s syndrome. J Neuropathol Exp Neurol 55(6):704–715
Cataldo AM et al. (2000) Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol 157(1):277–286
Cataldo AM et al. (2004) Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J Neuropathol Exp Neurol 63(8):821–830
Chen Q, Schubert D (2002) Presenilin-interacting proteins. Expert Rev Mol Med 4(19):1–18
Chen F et al. (2000) Carboxyl-terminal fragments of Alzheimer beta-amyloid precursor protein accumulate in restricted and unpredicted intracellular compartments in presenilin 1-deficient cells. J Biol Chem 275(47):36794–36802
Cirrito JR et al. (2008) Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 58(1):42–51
Coen, K., W. Annaert, Presenilins: how much more than gamma-secretase?! Biochem Soc Trans, 2010. 38(6): 1474–1478.
Coen K et al. (2012) Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol 198(1):23–35
Dobrowolski R et al. (2012) Presenilin deficiency or lysosomal inhibition enhances Wnt signaling through relocalization of GSK3 to the late-endosomal compartment. Cell Rep 2(5):1316–1328
Duce JA et al. (2010) Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142(6):857–867
Esselens C et al. (2004) Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J Cell Biol 166(7):1041–1054
Frederickson CJ, Koh JY, Bush AI (2005) The neurobiology of zinc in health and disease. Nat Rev Neurosci 6(6):449–462
Gandy S et al. (2007) Alzheimer’s presenilin 1 modulates sorting of APP and its carboxyl-terminal fragments in cerebral neurons in vivo. J Neurochem 102(3):619–626
Gowrishankar K, Zeidler MG, Vincenz C (2004) Release of a membrane-bound death domain by gamma-secretase processing of the p75NTR homolog NRADD. J Cell Sci 117(Pt 18):4099–4111
Greenough MA, Camakaris J, Bush AI (2013) Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem Int 62(5):540–555
Greenough MA et al. (2011) Presenilins promote the cellular uptake of copper and zinc and maintain copper chaperone of SOD1-dependent copper/zinc superoxide dismutase activity. J Biol Chem 286(11):9776–9786
Gu Q, Lin RL (2010a) Heavy metals zinc, cadmium, and copper stimulate pulmonary sensory neurons via direct activation of TRPA1. J Appl Physiol (1985) 108(4):891–897
Gu Q, Lin RL (2010b) Heavy metals zinc, cadmium, and copper stimulate pulmonary sensory neurons via direct activation of TRPA1. J Appl Physiol 108(4):891–897
Hung YH et al. (2009) Paradoxical condensation of copper with elevated beta-amyloid in lipid rafts under cellular copper deficiency conditions: implications for Alzheimer disease. J Biol Chem 284(33):21899–21907
Jin LW et al. (2004) Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am J Pathol 164(3):975–985
Kaether C et al. (2002) Presenilin-1 affects trafficking and processing of betaAPP and is targeted in a complex with nicastrin to the plasma membrane. J Cell Biol 158(3):551–561
Kametani F et al. (2004) Mutant presenilin (A260V) affects Rab8 in PC12D cell. Neurochem Int 44(5):313–320
Kang DE et al. (2005) Presenilins mediate phosphatidylinositol 3-kinase/AKT and ERK activation via select signaling receptors. Selectivity of PS2 in platelet-derived growth factor signaling. J Biol Chem 280(36):31537–31547
Kepp KP Alzheimer’s disease due to loss of function: a new synthesis of the available data. Prog Neurobiol, 2016
Koo EH, Squazzo SL (1994) Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem 269(26):17386–17389
Koo EH et al. (1996) Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. J Cell Sci 109(Pt 5):991–998
LaFerla FM (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci 3(11):862–872
Lee JH et al. (2010) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141(7):1146–1158
Lee JH et al. (2015) Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep 12(9):1430–1444
Leem JY et al. (2002) A role for presenilin 1 in regulating the delivery of amyloid precursor protein to the cell surface. Neurobiol Dis 11(1):64–82
Li Y et al. (2001) Induction of mossy fiber --> Ca3 long-term potentiation requires translocation of synaptically released Zn2+. J Neurosci 21(20):8015–8025
Li D et al. (2006) Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am J Hum Genet 79(6):1030–1039
Li H et al. (2011a) Polymorphisms of presenilin-1 gene associate with dilated cardiomyopathy susceptibility. Mol Cell Biochem 358(1–2):31–36
Li A et al. (2011b) Changes in the expression of the Alzheimer’s disease-associated presenilin gene in drosophila heart leads to cardiac dysfunction. Curr Alzheimer Res 8(3):313–322
Lingwood D, Simons K (2010) Lipid rafts as a membrane-organizing principle. Science 327(5961):46–50
Lloyd-Evans E et al. (2008) Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 14(11):1247–1255
Lothian A et al. (2013) Metalloproteomics: principles, challenges and applications to neurodegeneration. Front Aging Neurosci 5:35
Lu YM et al. (2000) Endogenous Zn(2+) is required for the induction of long-term potentiation at rat hippocampal mossy fiber-CA3 synapses. Synapse 38(2):187–197
Matias CM et al. (2010) Validation of TPEN as a zinc chelator in fluorescence probing of calcium in cells with the indicator Fura-2. J Fluoresc 20(1):377–380
McCarthy JV (2005) Involvement of presenilins in cell-survival signalling pathways. Biochem Soc Trans 33(Pt 4):568–572
McConlogue L et al. (1996) Differential effects of a Rab6 mutant on secretory versus amyloidogenic processing of Alzheimer’s beta-amyloid precursor protein. J Biol Chem 271(3):1343–1348
Mortimore GE, Poso AR (1987) Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr 7:539–564
Mukhopadhyay I et al. (2011) Expression of functional TRPA1 receptor on human lung fibroblast and epithelial cells. J Recept Signal Transduct Res 31(5):350–358
Murayama M et al. (1998) Direct association of presenilin-1 with beta-catenin. FEBS Lett 433(1–2):73–77
Naruse S et al. (1998) Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron 21(5):1213–1221
Neely KM, Green KN, LaFerla FM (2011) Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a gamma-secretase-independent manner. J Neurosci 31(8):2781–2791
Nelson O et al. (2007) Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest 117(5):1230–1239
Nelson O et al. (2011) Mutagenesis mapping of the presenilin 1 calcium leak conductance pore. J Biol Chem 286(25):22339–22347
Norgate M et al. (2006) Essential roles in development and pigmentation for the Drosophila copper transporter DmATP7. Mol Biol Cell 17(1):475–484
Norgate M et al. (2010) Syntaxin 5 is required for copper homeostasis in Drosophila and mammals. PLoS One 5(12):e14303
Nornes S et al. (2008) Interference with splicing of presenilin transcripts has potent dominant negative effects on presenilin activity. Hum Mol Genet 17(3):402–412
Ohta K et al. (2010) Autophagy impairment stimulates PS1 expression and gamma-secretase activity. Autophagy 6(3):345–352
Pan E et al. (2011) Vesicular zinc promotes presynaptic and inhibits postsynaptic long-term potentiation of mossy fiber-CA3 synapse. Neuron 71(6):1116–1126
Perez RG et al. (1999) Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J Biol Chem 274(27):18851–18856
Pigino G et al. (2001) Presenilin-1 mutations reduce cytoskeletal association, deregulate neurite growth, and potentiate neuronal dystrophy and tau phosphorylation. J Neurosci 21(3):834–842
Rechards M et al. (2006) Presenilin-1-mediated retention of APP derivatives in early biosynthetic compartments. Traffic 7(3):354–364
Repetto E et al. (2007) Presenilin 1 regulates epidermal growth factor receptor turnover and signaling in the endosomal-lysosomal pathway. J Biol Chem 282(43):31504–31516
Ring S et al. (2007) The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci 27(29):7817–7826
Roe MW, Lemasters JJ, Herman B (1990) Assessment of Fura-2 for measurements of cytosolic free calcium. Cell Calcium 11(2–3):63–73
Saftig P, Klumperman J (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10(9):623–635
Sandhoff K, Andreae U, Jatzkewitz H (1968) Deficient hexosaminidase activity in an exceptional case of Tay-Sachs disease with additional storage of kidney globoside in visceral organs. Pathol Eur 3(2):278–285
Sato C et al. (2006) Structure of the catalytic pore of gamma-secretase probed by the accessibility of substituted cysteines. J Neurosci 26(46):12081–12088
Saura CA et al. (2004) Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42(1):23–36
Scheper W et al. (2000) Alzheimer’s presenilin 1 is a putative membrane receptor for rab GDP dissociation inhibitor. Hum Mol Genet 9(2):303–310
Scheper W, Zwart R, Baas F (2004) Rab6 membrane association is dependent of presenilin 1 and cellular phosphorylation events. Brain Res Mol Brain Res 122(1):17–23
Shen J, Kelleher RJ III (2007) The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A 104(2):403–409
Shepherd CE et al. (2004) Positional effects of presenilin-1 mutations on tau phosphorylation in cortical plaques. Neurobiol Dis 15(1):115–119
Shilling D et al. (2012) Lack of evidence for presenilins as endoplasmic reticulum Ca2+ leak channels. J Biol Chem 287(14):10933–10944
Small SA, Gandy S (2006) Sorting through the cell biology of Alzheimer’s disease: intracellular pathways to pathogenesis. Neuron 52(1):15–31
Smith IF, Green KN, LaFerla FM (2005) Calcium dysregulation in Alzheimer’s disease: recent advances gained from genetically modified animals. Cell Calcium 38(3–4):427–437
Snitsarev VA, McNulty TJ, Taylor CW (1996) Endogenous heavy metal ions perturb fura-2 measurements of basal and hormone-evoked Ca2+ signals. Biophys J 71(2):1048–1056
Southon A et al. (2013) Presenilin promotes dietary copper uptake. PLoS One 8(5):e62811
Stutzmann GE (2005) Calcium dysregulation, IP3 signaling, and Alzheimer’s disease. Neuroscientist 11(2):110–115
Suga K et al. (2004) Syntaxin 5 interacts with presenilin holoproteins, but not with their N- or C-terminal fragments, and affects beta-amyloid peptide production. Biochem J 381(Pt 3):619–628
Suga K et al. (2005) Syntaxin 5 interacts specifically with presenilin holoproteins and affects processing of betaAPP in neuronal cells. J Neurochem 94(2):425–439
Suga K et al. (2009) The Syntaxin 5 isoforms Syx5 and Syx5L have distinct effects on the processing of {beta}-amyloid precursor protein. J Biochem 146(6):905–915
Thinakaran G, Koo EH (2008) Amyloid precursor protein trafficking, processing, and function. J Biol Chem 283(44):29615–29619
Tolia A, Chavez-Gutierrez L, De Strooper B (2006) Contribution of presenilin transmembrane domains 6 and 7 to a water-containing cavity in the gamma-secretase complex. J Biol Chem 281(37):27633–27642
Tu H et al. (2006) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126(5):981–993
Uemura K et al. (2003) Presenilin 1 is involved in maturation and trafficking of N-cadherin to the plasma membrane. J Neurosci Res 74(2):184–191
Venezia V et al. (2007) Amyloid precursor protein and presenilin involvement in cell signaling. Neurodegener Dis 4(2–3):101–111
Wang H et al. (2004) Presenilins and gamma-secretase inhibitors affect intracellular trafficking and cell surface localization of the gamma-secretase complex components. J Biol Chem 279(39):40560–40566
Wang HQ et al. (2005) Interaction of presenilins with FKBP38 promotes apoptosis by reducing mitochondrial Bcl-2. Hum Mol Genet 14(13):1889–1902
Wang R et al. (2006) Regulation of tyrosinase trafficking and processing by presenilins: partial loss of function by familial Alzheimer’s disease mutation. Proc Natl Acad Sci U S A 103(2):353–358
Wilson CA et al. (2004) Degradative organelles containing mislocalized alpha-and beta-synuclein proliferate in presenilin-1 null neurons. J Cell Biol 165(3):335–346
Wolfe DM et al. (2013) Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci 37(12):1949–1961
Wong BX et al. (2014) beta-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS One 9(12):e114174
Wrigley JD et al. (2005) Functional overexpression of gamma-secretase reveals protease-independent trafficking functions and a critical role of lipids for protease activity. J Biol Chem 280(13):12523–12535
Yamazaki T, Koo EH, Selkoe DJ (1996) Trafficking of cell-surface amyloid beta-protein precursor. II. Endocytosis, recycling and lysosomal targeting detected by immunolocalization. J Cell Sci 109(Pt 5):999–1008
Yu JT, Chang RC, Tan L (2009) Calcium dysregulation in Alzheimer’s disease: from mechanisms to therapeutic opportunities. Prog Neurobiol 89(3):240–255
Zhang Z et al. (1998) Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 395(6703):698–702
Zhang M et al. (2006) Presenilin/gamma-secretase activity regulates protein clearance from the endocytic recycling compartment. FASEB J 20(8):1176–1178
Zhang X et al. (2012) A role for presenilins in autophagy revisited: normal acidification of lysosomes in cells lacking PSEN1 and PSEN2. J Neurosci 32(25):8633–8648
Zou K et al. (2008) Novel role of presenilins in maturation and transport of integrin beta 1. Biochemistry 47(11):3370–3378
Acknowledgments
The Florey Institute of Neuroscience and Mental Health acknowledges the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant. This work was supported by the Australian National Health and Medical Research Council (NHMRC) and the Australian Research Council (ARC). MAG is the recipient of a NHMRC Dementia Research Fellowship. I would personally like to thank Professor Ashley Bush for his continued guidance and support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Greenough, M. The Role of Presenilin in Protein Trafficking and Degradation—Implications for Metal Homeostasis. J Mol Neurosci 60, 289–297 (2016). https://doi.org/10.1007/s12031-016-0826-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-016-0826-4