
Abstract
Previous studies revealed that patients with post-traumatic stress disorder (PTSD) have a smaller than normal medial prefrontal cortex (mPFC), and PTSD rats [single prolonged stress, (SPS)] have an increased mPFC neuron apoptosis, which are related to the severity of PTSD symptoms. Three signalling pathways [protein kinase RNA-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1)] in the endoplasmic reticulum (ER) play a critical role in resisting apoptosis. The aim of this study was to investigate whether the three branches of ER signalling are involved in SPS-induced mPFC neuron apoptosis. We used transmission electron microscopy (TEM) to detect morphological changes in ER and fluorescence spectrophotometry to detect the concentration of intracellular calcium in mPFC. We used molecular biological techniques to detect the expression levels of three branch signalling pathways of ER: phosphorylated PERK (p-PERK)/phosphorylated eukaryotic translation initiation factor 2A (p-eIF2a), ATF6a/X-box binding protein 1 (XBP1), and IRE1a. In addition, the ER molecular chaperone 78-kDa glucose-regulated protein (GRP78) and the ER-related apoptosis factors caspase family and Bax also were examined. Apoptosis neurons were detected by terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling. The results showed that the concentration of calcium in mPFC was increased in SPS rats. Using TEM, we found that mPFC neurons in SPS rats showed an expanded ER and chromatin margination. The increased expressions of p-PERK/p-eIF2a, ATF6a/XBP1, and IRE1 in response to SPS were also observed, although the degrees of increase were different. In addition, the protein and mRNA expression of GRP78 was increased in SPS rats; the upregulation of ER-related apoptosis factors and apoptosis neurons after SPS stimulation was observed. These results suggested that the three signalling pathways of unfolded protein response were involved in PTSD-induced, ER-dependent apoptosis in mPFC.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Post-traumatic stress disorder (PTSD) is one of the most common mental disorders that may develop after exposure to a serious life-threatening trauma. PTSD shows four characteristic symptoms: re-experiencing of the traumatic event, numbness, negative alteration in cognition and mood, and symptoms of avoidance and hyperarousal (American Psychiatric Association 2013). The symptoms can occur from a few days to years after the traumatic event. Using neuroimaging techniques (Frick et al. 1995; Sherin and Nemeroff 2011), researchers have found abnormal brain function and structure in patients with PTSD, and some studies have shown that patients with PTSD have a smaller medial prefrontal cortex (mPFC) volume and less activation during traumatic script-driven imagery (Hull 2002). The reports suggested that less activation of mPFC plays a key role in the developmental process of PTSD.
In our laboratory, a single prolonged stress (SPS) model rat was used to mimic PTSD. The SPS animal model was proposed by Liberzon (Liberzon and Martis 2006) and includes three stages: restraint stress, forced swim, and ether exposure. Our team found in previous studies (Wang et al. 2009; Zhang et al. 2012) that SPS changed the behaviours of rats, decreased corticosterone levels in their plasma, and enhanced negative feedback causing reduced hypothalamic–pituitary–adrenal(HPA) axis activity, which are hallmarks of the behaviour and neuroendocrinology of patients with PTSD. In recent years, our research group (li et al. 2013; Zhao et al. 2014) has focused on mPFC of SPS rats, and we have found increased apoptosis of neurons and changes in the endoplasmic reticulum (ER) of mPFC in SPS rats. The ER changes included changed expression of ER resident proteins and disturbed calcium homeostasis, which may be related to mPFC neuroapoptosis (Zhao et al. 2014; Wen et al. 2015). However, the exact role of ER in the neuroapoptotic process in the mPFC during PTSD remains poorly understood.
ER, as a key intracellular organelle, is responsible for the maintenance of calcium homeostasis and the synthesis and folding of proteins (secretion and cell surface proteins). The ER environment is strategically important for correct protein synthesis. Calcium disorders can lead to accumulation of misfolded or unfolded proteins. The accumulation of misfolded proteins leads to altered neuronal connectivity and even neuronal death (Soto 2003; Han et al. 2013; Majd et al. 2015), which have been related to several neuron-related diseases such as Alzheimer's disease, Parkinson's disease, and PTSD. A complex, three-pronged response [protein kinase RNA-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring enzyme 1 (IRE1)], called the unfolded protein response (UPR), has been found to correct aberrant folding in higher eukaryotes (Mesitov et al. 2013). UPR acts to restore ER homeostasis through upregulating the expression of ER molecular chaperone proteins (e.g. 78-kDa glucose-regulated protein [GRP78], calreticulin), which increases the probability of correct folding.
In mammals (Harding et al. 2000; Yoshida et al. 2001; Chen and Brandizzi 2013), PERK, ATF6, and IRE1 are the three ER transmembrane proteins and are also the ER stress sensors that respond to the accumulation of unfolded proteins in the ER lumen. GRP78, an ER molecular chaperone, plays a key role in the process of UPR. First, it controls the start of UPR through association and dissociation with ER stress sensors. Second, it promotes correct folding of proteins to decrease ER load. Third, it interacts with ER localized pro-caspase-7 to control the start of the ER-related apoptotic pathway (Reddy et al. 2003). When cells are in a state of ER stress, GRP78 will dissociate from these sensors, leading to their activation. Activated PERK phosphorylates eukaryotic initiation factor 2α (p-eIF2a), which inhibits protein translation (except for ATF4) to decrease the ER load (Mitsuda et al. 2011). However, the activation of ATF4 is preferentially translated upon the phosphorylation of eIF2a. ATF4 regulates the genes involved in amino acid metabolism and redox homeostasis. During conditions of ER stress (Yoshida et al. 2001), ATF6 translocates from ER to the Golgi, where it is processed by sites 1 and 2 proteases, thereby releasing the N-terminal transcriptional regulatory domain into the cytoplasm. The transcriptional regulatory domain of ATF6 translocates to the nucleus to regulate the gene expression of ER stress effectors such as X-box binding protein 1 (XBP1) and GRP78. Among these processes, IRE1 also dissociates from GRP78 and becomes autophosphorylated, and the activation of IRE1 is known to be an unconventional endoribonuclease that can excise a 26-nucleotide intron from XBP1 mRNA, resulting in a frame shift of reading codons in the XBP1 mRNA and the translation of active transcription factor XBP1s. Thus, activation of PERK, ATF6, and IRE1 initiates an ER-to-nucleus intracellular signalling cascade collectively termed as UPR. The most salient feature of the UPR is to increase the transactivation function of a gamut of basic leucine zipper transcription factors such as ATF6, ATF4, and XBP1. Once activated, these transcription factors coordinate transcriptional induction of ER chaperones and genes involved in ER-associated degradation to enhance the protein-folding capacity of the cell and decrease the unfolded protein load of ER, respectively.
The activation of the three-signal pathway restores the impaired function and structure of ER. However, when ER homeostasis cannot be restored, the proapoptotic process is induced by releasing pro-caspase-7 from GRP78 (Reddy et al. 2003), leading to caspase-7 activation, which promotes the activation of proapoptotic Bcl2 family members such as Bax (Ranganathan et al. 2006; Fu et al. 2007). Bax is localized to ER in addition to mitochondria (Zong et al. 2003), and ER apoptotic caspase-12 activation is Bax-dependent. Rao et al found, in an in vitro study, that caspase-12 activation was dependent on caspase-7 (Rao et al. 2001), and this process may occur through Bax (Lin et al. 2014). Activated caspase-12 enters the cytoplasm from ER, gradually activates caspase-3, and eventually leads to neuroapoptosis. Given the central role of ER, three signalling pathways switch from prosurvival to prodeath signalling. The aim of the present study was to investigate whether ER stress and UPR in mPFC neurons are involved in PTSD-induced apoptosis by examining ER morphology, the PERK/ATF6/IRE1 ER signalling pathway, the ER molecular chaperone GRP78, and the ER-related apoptotic proteins caspase-7, Bax, caspase-12, and caspase-3.
Material and Methods
Animals
All the experimental procedures of this study complied with Guidance Suggestions for the Care and Use of Laboratory Animals laid down by the Ministry of Science and Technology of the People’s Republic of China and also approved by Institutional Animal Care. The Experimental Center of China Medical University provided 40 Wistar rats (n = 20/group, males, weights 180–200 g). Before the start of experiments, the rats were pair-housed in cages with enough food and water for 7 days. Animal houses have air entrance and air handing facilities, which maintain a room temperature (23 ± 2 °C) environment with 55 ± 5 % humidity. The animal houses were maintained on a 12-h light–dark cycle (lights on 08:00 am).
Grouping and Single Prolonged Stress (SPS) Animal Model of PTSD
In this experiment, we assigned rats into a control group and an SPS group (7 days after the SPS procedure) randomly. The rats in the SPS group were exposed to the SPS procedure after a 7-day quiescent period. The control rats were left in the home cage during the SPS procedure. There are three stages (restraint stress, forced swimming, and ether exposures) in the SPS procedure (Ding et al. 2010). For restraint stress, rats were restrained for 2 h inside a clear plastic bag. The size of the bags was almost as big as the body of rats. In order to make sure the rats were able to breathe freely, a hole was made in each bag. For forced swimming, after restraint stress, they were immediately placed in a clear acrylic cylinder filled two thirds with water (24 ± 2 °C temperature) and forced to swim for 20 min. The acrylic cylinder had 500 mm height and 240 mm diameter. Then the rats were allowed to rest for 15 min. For ether exposures, the rats were exposed to ether until unconsciousness after rest, and then placed into the animal house without disturbing.
Morris Water Maze Test (MWM)
The MWM test, based on the classic Morris Protocol, was used to assess spatial memory performance (Morris 1984) and detect whether the SPS model of PTSD was successful. The experiment includes hidden platform tests and a probe test. The platform was placed in the first quadrant. The first 5 days consisted of hidden platform tests: the rats were placed in a quadrant facing the wall of the pool and allowed to find the target platform (2 cm below the surface) within 120 s. If the rat found the target platform, it was allowed to stay on the platform for 20 s. If not, experimenters had to guide them to find the target platform. Each rat was tested by the 120-s training test (with 20-s intervals) four times every day. We recorded the movement track of each rat. On the 6th day, the Probe Test was performed as follows: the target platform was removed in this test, and the rat was placed in the third quadrant. We recorded the swimming time that the rat spent in the first quadrant. The ratio of swimming time in the first quadrant/total time was the index used for capability of the rat's spatial memory.
Tissue Preparation
The rats of each group (control group and SPS group) were decapitated after anesthesia with urethane (50 mg/kg, i.P.). Then whole brains were removed and placed in a dish of crushed ice immediately. The brain was cut perpendicularly according to the Rat Brain Atlas. The dissected mPFC (bregma 2.2 mm and 3.7 mm) was frozen in liquid nitrogen immediately and then stored at −80 °C for preparation of western blot and PCR.
Preparation of Sections
The rats of each group were anesthetized (urethane, 50 mg/kg, i.P.) until sense of pain disappeared, then transcardially perfused through the ascending aorta with 300 mL preheated (37 °C) 0.9 % saline solution and 300 mL of 4 % paraformaldehyde [PFA, dissolved in 0.1 M phosphate buffer saline (PBS)] respectively. Whole brains were rapidly removed to 4 % PFA at 4 °C for 6 h. The brains were immersed in 30 % sucrose solution for 3 days. The brains were frozen in liquid nitrogen. The frozen brain tissue was cut into 14-μm thick sections on a cryostat and stored at −20 °C preparing for the immunohistochemistry and immunofluorescence techniques.
Detection of Free Calcium in mPFC Cells
The rats were decapitated after anesthesia with urethane; their brains were removed immediately. According to the Rat Brain Atlas, the mPFC (bregma 2.1 mm and 3.7 mm) was dissected. The cell suspension of mPFC was prepared by a routine method, and the concentration of the cell suspension was 109–1010/L. The cell suspension was incubated with 1 mmol/L fura-2-acetoxymethy ester (Fura-2/AM) (Beyotime Institute of Biotechnology, Haimen, China) for 35 min and the concentration was measured with a spectrofluorometer.
Assessing the ER and Nucleus Morphological Changes in the mPFC Using Transmission Electron Microscopy (TEM)
After anesthesia with urethane, the rats were perfused through the ascending aorta with 300 mL preheated (37 °C) 0.9 % saline solution, then perfused with a solution of 4 % paraformaldehyde and 2.5 % glutaraldehyde in PBS. The removed mPFC were immersed in 2.5 % glutaraldehyde (0.1 M PBS, pH 7.4) with shaking for 6 h at 4 °C. The tissues were washed by 0.1 M PBS then cut into blocks (1 mm wide, 5 mm long, and 1 mm thick). The blocks were immersed in 1 % osmium tetroxide for 2 h at 4 °C then rinsed in distilled water and dehydrated in graded series of ethanol (20–100 %) and propylene oxide. Following infiltration with Epon 812, the blocks were placed at 65 °C (48 h) for polymerization. After polymerization, the blocks were cut into ultra-thin sections by diamond knives. The ultra-thin sections were collected on copper grids. We used 4 % uranyl acetate and Reynolds lead citrate to stain the ultra-thin sections. A transmission electron microscope (JEM-1200EX; JEOL, Tokyo, Japan) was used to observe the sections (10 sections/each mPFC).
Immunofluorescent Assay for p-PERK, ATF6a, and Ire1a
After washing with PBS (three times, 5 min each), the tissue sections were treated with 5 % bovine serum albumin (BSA) in 0.1 M PBS at 37 °C for 30 min. Tthen the sections were incubated with I antibody (p-PERK, immuway, China, 1:200; ATF6a, Santa Cruz, CA, USA, 1:300; IRE1a, Santa Cruz, CA, USA, 1:200) in 2 % BSA-PBS overnight at 4 °C. After washing with PBS, the sections were incubated with II antibody (FITC-conjugated rabbit anti-p-PERK polyclonal antibody, Boster, China, 1:100; FITC-conjugated mouse anti-ATF6a antibody, Boster, China, 1:100; Cy3-conjugated goat anti-IRE1a antibody, Boster, China, 1:100) for 2 h at 37 °C. After washing with PBS, slices were mounted with glycerin and observed using a fluorescence microscope (LSM 510 Meta, Zeiss, Oberkochen, Germany) with a ×40 objective. Meanwhile, in order to assess nonspecific staining, a few sections were incubated in PBS instead of primary antibody in each experiment.
Western Blot to Detect the Expression of p-PERK,p-eIF2a, GRP 78, XBP1, and Apoptosis Factors
The protein fraction (50 μg/lane) extracted from prepared mPFC tissue was separated by 8 %, 10 %, or 12 % (w/v) SDS-PAGE (gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and then electroblotted to a PVDF membrane (Millipore, Bedford, MA, USA) using a semi-dry blotting apparatus (Bio-Rad, USA). The membranes was blocked with 5 % dried skim milk in TBST (Tris-Buffered Saline, Tween-20) and incubated with I antibody (anti-p-PERK, rabbit, Immuway, 1:500; anti-p-eIF2α, rabbit, Immuway, 1:500; anti-XBP1, mouse, Stanta Cruz, CA, USA, 1:500; anti-GRP 78, mouse, Santa Cruz, CA, USA, 1:500; anti-Caspase 7/12/3, mouse, Santa Cruz, CA, USA, 1:500; anti-Bax, mouse, Santa Cruz, CA, USA, 1:500; anti-GAPDH, Santa Cruz, CA, USA, 1:1000) overnight at 4 °C and II antibody (Boster Biological Technology Ltd., 1:1000) for 2 h at RT. The membrane was washed with TBST, and then the protein expression was identified using enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech, Buckinghamshire, UK). A gel image analysis system was used to analyze the bands and record the OD value. At last, we calculated the ratio of OD (target protein/GAPDH) as the data, which was analysed using SPSS software.
Total RNA Extraction and PCR
RNA Extraction
We used Trizol Kit (D312 LOT B2402-1, TaKaRa Bio, Japan) to extract the total RNA from prepared mPFC tissue according to the manufacturer’s instructions and the RNA purity was detected by measuring the ratio of A260/A280 (1.8–2.0).
RT-PCR
The total RNA was reverse transcribed into cDNA, which was amplified using an RNA PCR Kit (AM Ver.3.0, Takara bio, Otsu, Japan). The specific GRP78 primer was designed and synthesized by Sheng Gong Biological Engineering Technology (Shanghai, China), and the sequence of each gene is shown in Table 1. The amplification profile of GRP78 included: (1) denaturation at 94 °C for 4 min, (2) 30 additional cycles at 94 °C for 30 s and then 40 s at 57 °C, 72 °C for 40 s, and (3) extension at 72 °C for 10 min. The amplification profile of GAPDH included: (1) denatureion at 94 °C for 2 min, (2) 30 additional cycles at 95 °C for 30 s and then 40 s at 57 °C, 72 °C for 40 s, and (3) extension at 72 °C for 5 min. The PCR products were separated on 1.5 % agarose gel by electrophoresis, the density of each band was analyzed using a gel image analysis system (Tanon 2500R, Shanghai, China). The mRNA levels of GRP78 were normalized by GAPDH.
Quantitative Real-Time PCR (QT-PCR)
The total RNA of mPFC was reverse transcribed into cDNA using PrimerScript RT reagent Kit (Perfect Real Time, TAKARA, Japan). The quantitative real-time PCR was performed using SYBR® Premix Ex Taq™ II (Perfect Real Time). The genes of XBP1 and GAPDH were amplified and detected on the LightCycler 480 (Roche) of the China Medical University Experiment Center. The primers of XBP1 and GAPDH, shown in Table 1, were designed and synthesized by Sheng Gong (Shanghai, China). All measurements were in triplicate in a single assay. The fold change of the XBP1 gene was calculated by the comparative Ct method, and the fold change was the data of real-time PCR.
Statistical Analysis of Date
All data were analyzed by IBM SPSS 19.0 software. All data were expressed as mean ± SEM and analyzed by one-way analysis of variance (ANOVA), When P < 0.05, we considered that the data of the two groups had statistical significance.
Results
Morris Water Maze Test
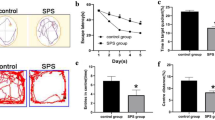
The Morris water maze (MWM) test was used to detect whether the SPS animal model was successful in detecting the rats’ behavioral changes. Figure 1 shows the results of the MWM test. The results show that the SPS rats swam a farther distance to find the target platform under water in comparison with the control group rats (Fig. 1a, b). SPS rats spent less time swimming in the platform quadrant as a percentage of total time than rats in the control group (P < 0.05) (Fig. 1c). The results show that SPS exposure changed the rats’ behavior.

The results of the MWM test. a 3D photo of tracked movements of rats in the control group, target platform with arrow. b 3D photo of tracked movements of rats in the SPS group, target platform with arrow. c The percentage of time in target quadrant (%) in MWM test (n = 10, F (1, 19) = 103.282). Statistical analysis was based on one-way ANOVA. *P < 0.001 versus the control group
Increased Free Calcium Level in mPFC Cells of SPS Rats
Figure 2 shows the results of analysis of the intracellular free calcium levels in all groups (control group and SPS group). These results show that the levels of intracellular free calcium in the mPFC neurons in the SPS groups were increased compared with the control group (P < 0.05) (Fig. 2). The results also show that SPS disturbed calcium homeostasis in mPFC neurons, which may have been caused by the imbalance of ER homeostasis, increasing the generation and accumulation of misfolded protein in ER.

The changes in intracellular calcium (Ca2+) levels (nmol/L). Data represent the means ± SD (n = 6 for each group). Statistical analysis was based on one-way ANOVA (n = 6, F(1,11) = 328.58), *P < 0.001 vs. the control group
SPS Changed the Morphology of Endoplasmic Reticulum in mPFC Neuron
Transmission electron microscopy (TEM) was performed to analyze the ER morphological changes in neurons in the mPFC of rats (control group and SPS group). As shown in Fig. 3, the mPFC neurons in the control rats exhibited normal morphology (Fig. 3a). Compared with neurons in control group rats, some neurons in the mPFC of SPS rats (Fig. 2b) exhibited pronounced dilation of the ER lumen, which was a characteristic change of ER morphology in states of ER stress. The expanded volume is indicated by the star in Fig. 3b. Researchers (Romisch 2005; Xu et al. 2005) previously reported that the expansion of ER volume is a necessary adjustment in response to increased accumulation of misfolded proteins induced by SPS.

TEM images of the mPFC in the two groups. TEM images of the mPFC. a Normal control mPFC neuron, displaying abundant endoplasmic reticulum (arrow). The nucleus (star) is large and round, density of chromatin is uniform. b mPFC neuron of SPS group rat. The expanded volume of ER (arrow) and chromatin margination in nucleus (star) are shown. bar = 300 nm
PERK Branch of Unfolded Protein Response was Activated
In cells, the UPR was a protective response against ER stress. The PERK branch signal is one of three branch signals of the UPR. Some researchers (Bell et al. 2016; Harding et al. 2000) have reported that the PERK-dependent pathway plays a key role in the UPR. In the PERK signalling pathway, PERK is a protein kinase of eIF2a, and the activity of PERK (p-PERK) phosphorylates eIF2a (p-eIF2a), reducing general protein synthesis to decrease the ER load. p-PERK and p-eIF2a are markers of activation of the PERK branch. The results of immunofluorescence experiments (Fig. 4a and b) to detect p-PERK showed that its expression was increased in SPS groups compared with the control group. The protein expression of p-PERK was significantly increased in the SPS group in comparison with the control group, which is consistent with the results of the immunofluorescence assay. In addition, in comparison with the control group, the protein expression of p-eIF2a, the downstream of PERK-dependent pathway, was also increased in the SPS group (Fig. 4d). Together, these results show that SPS-induced ER stress promoted PERK autophosphorylation, resulting in increased protein or immunofluorescence levels of p-PERK and p-eIF2a in mPFC.

The expression results of p-PERK and p-eIF2a in mPFC. a–b The p-PERK expression in mPFC by The immunofluescence staining (bar = 50 μm): a control group; b SPS group. The quantitative analysis (mean optical densities, n = 15, F(1, 29) = 54.342) of p-PERK positive cells detected by immunofluoresence staining is shown in c The protein expression of p-PERK and p-eIF2a detected by immunoblot assay is shown in d The analysis result of protein expression of p-PERK detected by western blot assay (n = 6, F (1, 11) = 112.64) is shown in e The analysis result of protein expression of p-eIF2a detected by western blot assay (n = 6, F (1, 11) = 49.316) is shown in f Data represents the means ± SD. All data were analysed using one-way ANOVA. *P < 0.001 versus the control group
ATF6a Branch of Unfolded Protein Response Were Activated
ATF6a, a transmembrane protein, was thought to be primarily responsible for gene transcription regulation of UPR. We used an immunofluorescence assay to detect ATF6a. The results for ATF6a are shown in Fig. 5a and b. Figure 5a and b show that ATF6a was present in the control rats and demonstrated diffused cytoplasmic localization, whereas SPS rats had an obvious increase of ATF6a immunofluorescence compared with control group rats. This suggested that ATF6a was activated in response to SPS. ATF6a was activated by ER stress induced by SPS through cleavage from the membrane and moved to the nucleus, leading to increased mRNA expression of XBP1 (Haze et al. 1999; Yoshida et al. 2001). Thus, we detected mRNA expression of XBP1 by quantitative reverse transcription polymerase chain reaction (RT-PCR), and the expression of XBP1 was normalized to the mRNA level of GAPDH from the same specimen. The results of analysis of mRNA expression of XBP1 are shown in Fig. 5d. We found that mRNA expression of total XBP1 was upregulated after SPS. All of these results suggest that the ATF6a-dependent pathway was activated by SPS.

The expression results of ATF6a in mPFC. a–b The ATF6a expression in mPFC by immunofluescence staining (bar = 50 μm): a control group; b SPS group. The quantitative analysis (mean optical densities, n = 15, F(1, 29) = 78.607) of ATF6a positive cells detected by immunofluoresence staining is shown in c The analysis result of XBP1 mRNA expression is shown in d (n = 3, F (1, 5) = 84.476). Data represent the means ± SD. All data were analysed using one-way ANOVA. *P < 0.05 versus the control group
IRE1a Branch of Unfolded Protein Response Were Activated
The IRE1a-dependent signalling pathway was the third branch of UPR studied. Using an immunofluorescence assay, we explored the expression of IRE1a immunofluorescence in mPFC. As shown in Fig. 6a and b, IRE1a immunofluorescence was present in the normal control group and mainly showed diffuse cytoplasmic localization. The results of analysis of IRE1a immunofluorescence are shown in Fig. 6c. We found that the intensity of IRE1a immunofluorescence was increased in mPFC neurons after stimulation by SPS. Upon ER stress, activated ATF6a increased the expression of XBP1 mRNA; however, XBP1 mRNA is cleaved by activated IRE1, which then generates the spliced form of XBP1 (XBP1s), which generates the activated XBP1s that is considered a marker of UPR (Yoshida et al. 2001; Yoshida et al. 2006). To confirm whether SPS induced the activation of XBP1, we examined the protein expression of XBP1 by Western blotting. The Western blot analysis results for XBP1 are shown in Fig. 6d. Figure 6d shows that XBP1u was constitutively expressed in mPFC neurons as a 29 kDa protein in the normal group. The active form XBP1 was a 50 kDa protein (XBP1s). The results of this analysis (Fig. 6e) show that SPS rats had decreased protein expression of XBP1u and increased protein expression of XBP1s in mPFC compared with the control group, which suggests that XBP1 was activated by IRE1a.

The expression results of IRE1a and XBP1in mPFC. a–b The IRE1a expression in mPFC by The immunofluescence staining(bar = 50 μm): a control group; b SPS group. The quantitative analysis (mean optical densities, n = 15, F(1, 29) = 46.444) of IRE1a positive cells detected by immunofluoresence staining is shown in c. The protein expression of XBP1detected by western blot assay is shown in d. The analysis result of protein expression of XBP1 detected by western blot assay [XBP1s: n = 6, f (1, 11) = 18.264; XBP1u: n = 6, F(1,11) = 22.177] is shown in e. Data represents the means ± SD. All data were analysed using one-way ANOVA. *P < 0.05 versus the control group
Expression of ER Molecular Chaperone GRP78 was Upregulated
Western blotting and RT-PCR were used to detect the expression of GRP78 in protein and mRNA levels in mPFC neurons of SPS rats. The molecular weights of GRP78 and GADPH were 78 kDa and 36 kDa, respectively. The results for GRP78 are shown in Fig. 7. The protein expression of GRP78 in the SPS group showed a marked increase compared with that of the control group (Fig. 7a and b). In addition, the RT-PCR results of GRP78 showed an increase in total transcript levels of GRP78 in mPFC (Fig. 7c and d). In control rats, very low mRNA levels of GRP78 were detected; however, SPS rats had a marked increase of GRP78 mRNA expression. These results were consistent with those of Western blot analysis and suggested that SPS triggered increased GRP78 in protein and mRNA levels.

The expression results of GRP78 in mPFC. a The protein expression of GRP 78 detected by western blot assay. b The analysis result of protein expression of GRP78 detected by Western blot assay (n = 6, F (1, 11) = 103.609). c The mRNA expression of GRP78 detected by RT-PCR. d The analysis result of mRNA expression of GRP 78 (n = 6, F (1, 11) = 56.372). All data were analysed using one-way ANOVA. *P < 0.001 versus the control group
Increased Apoptosis and Upregulation of Apoptosis-Related Factor Expression
Terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labelling (TUNEL) was used to detect apoptosis in mPFC of all groups (control and SPS groups), and the results are shown in Fig. 8. The nucleus of apoptosis-positive cells was seen as brown particles on microscopy. Figure 8 shows that the control group had a small amount of apoptosis-positive cells and a lighter color. However, the amount of apoptosis-positive cells was obviously increased, and the colour of the brown particles became deeper in the mPFC of SPS rats in comparison with the control group, which suggested the existence of mPFC apoptosis in SPS rats. In addition, TEM results showed that SPS rats had chromatin margination in the nucleus (Fig. 3), which represents the morphological changes of apoptotic neurons, consistent with the increased apoptosis in neurons (Fig. 8).

Apoptotic cells in the mPFC by TUNEL staining, a control group, b SPS group, c number of apoptotic cells in mPFC (n = 15, F (1, 29) = 416.743). Data are expressed as mean ± SD. One-way ANOVA was used for analysis.*P < 0.001 versus to the control group
In our experiments, we proved that SPS induced ER stress and activated the three branches of UPR. However, prolonged or serious stress induced increased accumulation of misfolded proteins, which exceeded the capability of UPR to deal with, leading to the activation of ER pathway apoptosis. We detected the protein expression of ER-related apoptosis factors (caspase-7, Bax, caspase-12, and caspase-3) by Western blot assay. These Western blot analysis results demonstrate that the protein levels of caspase-7, Bax, caspase-12, and caspase-3 were markedly increased (Fig. 9), suggesting that ER-related apoptosis factors were activated by SPS and revealing signs of ER stress-induced apoptosis. All of these results indicate that ER stress and ER-related apoptosis factor activation contribute to SPS and SPS-induced apoptosis.

The protein expression of ER-related apoptosis factors in mPFC. a The protein expression of ER-related apoptosis were detected by Western blot assay. b The analysis results of ER-related apoptosis factor expression [caspase 7: n = 6, F (1, 11) = 347.964; Bax: n = 6, F (1, 11) = 272.15; caspase 12: n = 6, F (1, 11) = 452.175; caspase 3: n = 6, F (1, 11) = 262.319]. Data are expressed as mean ± SD. One-way ANOVA was used for analysis.*P < 0.001 versus the control group
Discussion
PTSD is mental disorder often associated with behavioural symptoms, psychiatric comorbidity, and diminished quality of life (Klein et al. 2009). Its main clinical symptom characteristics are reexperiencing of traumatic events through flashbacks and nightmares, numbness, negative alteration in cognition, and mood and avoidance behaviours (DSM 5th, 2013). Using brain imaging methods, researchers (Frick et al. 1995; Shin et al. 2004) have identified changes in brain function and structure as characteristics of PTSD that are associated with severity of clinical symptoms. Consistent with this, in recent studies researchers have found a smaller hippocampus volume in PTSD. Dysfunction of the hippocampus affects contextual memory, and higher activation of the amygdala decreases the inhibition of the fear response (Morey et al. 2015). mPFC (Arruda-Carvalho and Clem 2015) is thought to exert a top-down regulatory influence on the amygdala, and dysfunction of mPFC may affect the connectivity between mPFC and amgydala, which is linked to amygdala dysfunction. In particular, mPFC is a higher-order structure that regulates the function of the amygdala and the hippocampus (Zelikowsky et al. 2014). With use of neuroimaging techniques, research on veterans with PTSD showed a failed mPFC activation (Bremner 2007), and morphometric magnetic resonance imaging studies have demonstrated that patients with PTSD have a smaller mPFC than healthy persons. In some studies, researchers have found that the activation and volume of mPFC are inversely related to the severity of PTSD symptoms (Bremner 2007; Zelikowsky et al. 2014). All of this evidence suggests a strong relationship between mPFC atrophy and the development of PTSD. Using TEM and TUNEL, our group (Ding et al. 2010; Han et al. 2013; li et al. 2013; Zhao et al. 2014; Li et al. 2015; Yu et al. 2014) found increased apoptosis of neurons in the mPFC of SPS model rats, which mimicked behavioural abnormalities and neuroendocrinological changes of PTSD. Previous studies showed that neuron apoptosis in mPFC can be induced by SPS, which may be a reason for mPFC dysfunction. In our experiments, we assessed the success of the SPS model using a behavioral test (the MWM), and, as shown in Fig. 1, SPS decreased the rats’ ability to find the target in the MWM.
We found three apoptosis pathways: a death receptor signalling pathway, a mitochondria-induced signalling pathway, and an ER-induced signalling pathway. The ER stress-induced signalling pathway has been described in recent studies, and it is thought to play an important role in cell fate because it mediates not only cell death through crosstalk of the other two apoptosis pathways (Volgyi et al. 2015), but also survival of cells through UPR, which can decrease the generation of misfolded proteins and promote correct protein folding. ER is a locus of folding and post-translational maturation of almost all secreted proteins and membrane proteins. In addition, ER serves as a calcium store and plays important roles in intracellular calcium homeostasis, energy metabolism, and redox homeostasis. ER homeostasis is essential for cellular activities and survival. However, alterations in calcium or accumulation of misfolded proteins both lead to a condition called ER stress. Some ER ultrastructural research has shown pronounced dilation of the ER lumen in response to ER stress (Romisch 2005; Xu et al. 2005), which was reflective of morphological changes in the process of ER stress. In our study, we found increased calcium levels and larger ER lumen volume in SPS rats’ mPFC in comparison with the control group, both of which suggested SPS-induced ER stress in mPFC neurons. In our previous studies, the ER stress and UPR were also been observed in the hippocampus and amygdala (Han et al. 2013; Xiao et al. 2015), all changes that may alter connectivity of amygdala-hippocampus-mPFC, which may be involved in the process of PTSD.
Three branches of UPR (PERK, ATF6, and IRE1) were activated against ER stress and restored ER to its normal state by increasing ER molecular chaperone proteins such as GRP78 involved in several neurological diseases, such as Alzheimer's disease and Parkinson's disease (Rao et al. 2002). The results of our research showed that the protein and immunofluorescence levels of p-PERK and p-eIF2a were both increased in the mPFC of SPS rats. The PERK branch of UPR transduces both prosurvival as well as proapoptotic signals following the accumulation of unfolded or misfolded proteins in ER. SPS-increased calcium dysfunction caused ER stress, leading to GRP78 dissociated from the N-terminus of PERK and inducing dimerization and autophosphorylation of PERK at T981 (p-PERK) (Kebache et al. 2004), which was the reason for the increased expression of p-PERK. p-PERK can promote phosphorylation of eIF2a at S51 (p-eIF2a) (Harding et al. 1999; Raven et al. 2008), which causes upregulation of p-eIF2a. In eukaryotes (Merrick 2004), the eIF2a protein with GTP-dependent start site recognition is vital to translation initiation. p-eIF2a inhibited translation initiation by blocking the exchange of guanidine diphosphate bound to eIF2a. p-PERK and p-eIF2a are key proteins of PERK-independent pathways against ER stress. Our experimental results showed that the protein and immunofluorescence levels of p-PERK and p-eIF2a were both increased in the mPFC of SPS rats, resulting of SPS-induced calcium disorder, which suggested that the PERK branch of UPR was activated against SPS-induced ER stress. p-PERK/p-eIF2a pathway signalling can diminish accumulation of misfolded proteins in ER through inhibition of translation initiation, decreasing ER load. Jiang et al reported on the activation of the PERK/eIF2a signalling pathway against ER stress-induced apoptosis in hepatocytes (Jiang et al. 2015). In other studies, researchers have also reported that inhibition of p-PERK and p-eIF2a increased apoptosis (Chang et al. 2002; Gao et al. 2015), consistent with their prosurvival effect.
In our present study, we found that the expression of ATF6a and XBP 1 in immunofluorescence or mRNA levels in mPFC neurons was increased after SPS exposure. It is well known that ATF6a is another typical ER stress transducer and that activation of the ATF6a signalling pathway involves complex steps of proteolytic translocation (Yoshida et al. 2001; Hashida et al. 2012). ER stress activated and increased the expression of ATF6a, which resulted in increased expression of ATF6a. Following the activation of the ATF6a pathway, the active nuclear form of ATF6a was transported into the nucleus and increased mRNA expression of XBP1 to deal with the ER stress, which is consistent with our finding that mRNA expression of XBP1 was upregulated in mPFC neurons in SPS rats.
SPS rats had increased expression of IRE1a, XBP1, and GRP78 as determined by immunofluorescence, protein, and mRNA levels. IRE1a, with both an endoribonuclease and a serine/threonine kinase domain (Kaufman et al. 2002), was the third ER transducer protein. Similarly to PERK and ATF6a, IRE1a was activated after becoming dissociated from GRP78 in the presence of accumulation of misfolded proteins in ER. The activation of IRE1a splices XBP1 mRNA using the RNase domain, leading to the generation of the active transcription factor XBP1s, which enhances the expression of UPR target gene GRP78, confirming our results that the mRNA and protein expression of GRP78 were upregulated in mPFC neurons in response to SPS. Both the inhibited translation initiation by the p-PERK/p-eIF2a signalling pathway and increased UPR target protein expression induced by IRE1 and ATF6a signalling pathways are essential for decreasing ER load of accumulation of misfolded proteins induced by SPS.
The PERK, ATF6, and IRE1 pathways not only promote ER adaptation to misfolding, but also switch survival signalling to proapoptotic signalling when ER stress is prolonged or severe. We also detected ER-related apoptosis factors such as caspase-7, Bax, Caspase-12, and caspase-3, and the experimental results showed a markedly increased expression of these factors in the SPS rats’ mPFC, which suggested that SPS induced the ER-related apoptotic pathway. In normal cells, GRP78 binds not only to three ER stress sensors, but also to apoptosis factor pro-caspase-7 (Reddy et al. 2003). In conditions of ER stress, GRP78 dissociated from the PERK/ATF6a/IRE1 pathways, and activated them to copy with accumulation of misfolded proteins; however, when the misfolded proteins accumulated beyond the capability of UPR, GRP78 dissociated from pro-caspase-7 in ER, leading to activation of caspase-7, which is consistent with our observation that the protein expression of caspase-7 in the mPFC of SPS rats increased. Activated caspase-7 can promote the activation of Bax in ER (Fu et al. 2007; Ranganathan et al. 2006), leading to ER apoptosis-related activation of caspase-12 (Lin et al. 2014). The activated caspase-12 will enter the cytoplasm from ER, gradually activate caspase-3, and eventually lead to neuroapoptosis.
Conclusions
In the present study, we found changes in expression levels of PERK, ATF6a, and IRE1, indicating that three ER pathways participate in SPS-induced apoptosis. The increase in the expression of GRP78 and the caspase family suggests that activation of three pathways initiates ER stress-induced apoptosis and neuronal protection.
References
American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders, 5th edn. American Psychiatric Association, Arlington
Arruda-Carvalho M, Clem RL (2015) Prefrontal-amygdala fear networks come into focus. Front Sys Neurosci 9:145
Bell MC, Meier SE, Ingram AL, Abisambra JF (2016) PERK-opathies: an endoplasmic reticulum stress mechanism underlying neurodegeneration. Curr Alzheimer Res 13(2):150–163
Bremner JD (2007) Neuroimaging in posttraumatic stress disorder and other stress-related disorder. Neuroimaging Clin N Am 17(4):523–538
Chang RC, Wong AK, Ng HK, Hugon J (2002) Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 13:2429–2432
Chen Y, Brandizzi F (2013) IRE1: ER stress sensor and cell fate executor. Trends Cell Biol 23(11):547–555
Ding JL, Han F, Shi YX (2010) Single-prolonged stress induces apoptosis in the amygdala in a rat model of post-traumatic stress disorder. J Psychiatr Res 44:48–55
Frick KM, Baxter MG, Markowaska AL, Olton DS, Price DL (1995) Age-related spatial reference and working memory deficits assessed in the water maze. Neurobiolo Aging 16:149–160
Fu Y, Li J, Lee AS (2007) Grp78/bip inhibits endoplasmic reticulum bik and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res 67:3734–3740
Gao H, Yan P, Zhang S, Nie S, Huang F, Han H, Deng Q, Huang Q, Yang W, Wu H, Yao P, Ye K, Xu JM, Liu L (2015) Chornic alpha-linolenic acid treatment alleviates age-associated neuropathology: roles of PERK/eIF2a singling pathway. Bran Behav Immun. Pil:S0889-1591(15)30019-2. doi:10.1016/j.bbi.2015.09.012. [Epub ahead of print] [PubMed: 26399745]
Han F, Yan S, Shi Y (2013) Single-prolonged stress induces endoplasmic reticulum-dependent apoptosis in the hippocampus in a rat model of post-traumatic stress disorder. PLoS ONE 8(7):e69340
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271–274
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5:897–904
Hashida K, Kitao Y, Sudo H, Awa Y, Maeda S, Mori K, Takahashi R, Iinuma M, Hori O (2012) ATF6 alpha promotes astroglial activation and neuronal survival in a chronic mouse model of Parkinson’s disease. Plos one 7(10):e47950
Haze K, Yoshida H, Yanagi H, Yura T, Mori K (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10:3787–3799
Hull AM (2002) Neuroimaging findings in post-traumatic stress disorder systematic review. Br J Psychiatry 181:102–110
Jiang ZQ, Ma YX, Li MH, Zhan XQ, Zhang X, Wang MY (2015) 5-Hydroxymethylfurfural protects against ER stress-induced apoptosis in GalN/TNF-α-injured L02 hepatocytes through regulating the PERK-eIF2α signaling pathway. Chin J Nat Med 13(12):896–905
Kaufman RJ, Scheuner D, Schroder M, Shen X, Lee K, Liu CY, Arnold SM (2002) The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol 3:411–421
Kebache S, Cardin E, Nguyen DT, Chevet E, Larose L (2004) Nck-1 antagonizes the endoplasmic reticulum stress-induced inhibition of translation. J Biol Chem 279:9662–9671
Klein B, Mitchell J, Gilson K et al (2009) A therapist-assisted Internet-based CBT intervention for posttraumatic stress disorder: preliminary results. Cogn Behav Ther 38(2):12–31
Li Y, Han F, Shi Y (2013) Increased neuronal apoptosis in medial prefrontal cortex is accompanied with changes of Bcl-2 and Bax in a rat model of post-traumatic stress disorder. J Mol Neurosci 51(1):127–137
Li XY, Han F, Shi YX (2015) IRE1a-XBP1 pathway is activated upon induction of single-prolonged stress in rat neurons of the medial prefrontal cortex. J Mol Neurosci 57:63–72
Liberzon I, Martis B (2006) Neuroimaging studies of emotional responses in PTSD. Ann NY Acad Sci 1071:87–109
Lin ML, Chen SS, Huang RY, Lu YC, Liao YR, Reddy MV, Lee CC, Wu TS (2014) Suppression of PI3K/Akt signaling by synthetic bichalcone analog TSWU-CD4 induces ER stress- andBax/Bak-mediated apoptosis of cancer cells. Apoptosis 19(11):1637–1653
Majd S, Power JH, Grantham HJ (2015) Neuronal response in Alzheimer’s and parkinson’s disease: the effect of toxic proteins on intracellular pathways. BMC Neurosci 16:69
Merrick WC (2004) Cap-dependent and cap-independent translation in eukaryotic systems. Gene 332:1–11
Mesitov MV, Moskovtesev AA, Kubatiev AA (2013) Molecular logic of the endoplasmic reticulum stress signal pathway: the system of unfolded protein response. Patol Fiziol Eksp Ter 4:97–108
Mitsuda T, Omi T, Tanimukai H, Sakagami Y, Tagami S, Okoxhi M, Kuso T, Takeda M (2011) Sigma-1Rs are upregulated via PERK/eIF2a/ATF4 pathway and execute protective function in ER stress. Biochem Biophys Res Commun 415(3):519–525
Morey RA, Haswell CC, Hooper SR, De Bellis MD (2015) Amygdala, hippocampus, and ventral medial prefrontal cortex volumes differ in maltreated youth with and without chronic posttraumatic stress disorder. Neuropsychopharmacology 41(3):791–801
Morris R (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11:47–60
Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA (2006) Functional coupling of p38-induced up-regulation of bip and activation of rna-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res 66:1702–1711
Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE (2001) Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem 276:33869–33874
Rao RV, Peel A, Logvinova A, del Rio G, Hermei E, Yokota T, Goldsmith PC, Ellerby LM, Ellerby HM, Bredesen DE (2002) Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett 514:122–128
Raven JF, Baltzis D, Wang S, Mounir Z, Papadakis AI, Gao HQ, Koromilas AE (2008) Pkr and pkr-like endoplasmic reticulum kinase induce the proteasome-dependent degradation of cyclin d1 via a mechanism requiring eukaryotic initiation factor 2alpha phosphorylation. J Biol Chem 283:3097–3108
Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS (2003) Endoplasmic reticulum chaperone protein grp78 protects cells from apoptosis induced by topoisomerase inhibitors: role of atp binding site in suppression of caspase-7 activation. J Biol Chem 278:20915–20924
Romisch K (2005) Endoplasmic reticulum-associated degradation. Annu Rev Cell Dev Biol 21:435–456
Sherin JE, Nemeroff CB (2011) Post-traumatic stress disorder: the neurobiological impact of psychological trauma. Dialogues Clin Neurosci 13:263–278
Shin LM, Orr SP, Carson MA, Rauch SL, Macklin ML, Lasko NB, Peters PM, Metzger LJ, Dougherty DD, Cannistraro PA, Alpert NM, Fischman AJ, Pitman RK (2004) Regional cerebral blood flow in amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry 6:168–176
Soto C (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4:49–60
Volgyi K, Juhász G, Kovacs Z, Penke B (2015) Dysfunction of endoplasmic reticulum(ER) and mitochondria(MT) in Alzheimer’s disease: the role of the ER-MT cross-talk. Curr Alzheimer Res 12(7):655–672
Wang HT, Han F, Shi YX (2009) Activity of the 5-HT1A receptor is involved in the alteration of glucocorticoid receptor in hippocampus and corticotrophin-releasing factor in hypothalamus in SPS rats. Int J Mol Med 24:227–231
Wen L, Han F, Shi Y (2015) Changes in the glucocorticoid receptor and Ca2+/Calreticulin-dependent signaling pathway in the medial prefrontal cortex of rats with post-traumatic stress disorder. J Mol Neurosci 56(1):24–34
Xiao B, Yu B, Liu DJ, Han F, Shi YX (2015) Single prolonged stress induces dysfunction of endoplasmic reticulum in a rat model of post-traumatic stress disorder. Molecul Med Rep 12(2):2015–2020
Xu C, Bailly-Maitre B, Reed JC (2005) Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115:2656–2664
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881–891
Yoshida H, Oku M, Suzuki M, Mori K (2006) pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol 172:565–575
Yu B, Wen LL, Xiao B, Han F, Shi YX (2014) Single prolonged stress induces ATF6 alpa-dependent endoplasmic reticulum stress and the apoptotic process in medial frontal cortex neurons. BMC Neurosci 15:115
Zelikowsky M, Hersman S, Chawla MK et al (2014) Neuronal ensembles in amygdala, hippocampus, and prefrontal cortex track differential components of contextual fear. J Neurosci 34(25):8462–8466
Zhang JH, Han F, Shi YX (2012) Single prolonged stress induces changes in the expression of mineralocorticoid receptor in the medial prefrontal cortex in a rat model of post-traumatic stress disorder. Molecul Med Rep 6:330–334
Zhao D, Han F, Shi Y (2014) Effect of glucose-regulated protein 94 and endoplasmic reticulum modulator CasPase-12 in medial prefrontal cortex in a ratmodel of posttraumatic stress disorder. J Mol Neurosci 54(2):147–155
Zong WX, Li C, Hatzivassiliou G, Lindstrn T, Yu QC, Yuan J, Thompson CB (2003) Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol 162(1):59–69
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (No. 81571324;No.31200772) and Education Department of LiaoNing Province, China (No. LJQ2014083), What’ more, our feel grateful for technical support from the China Medical University Experiment Center. And our thanks professor Han Fang and professor Shi Yuxiu for guidance and discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wen, L., Han, F., Shi, Y. et al. Role of the Endoplasmic Reticulum Pathway in the Medial Prefrontal Cortex in Post-Traumatic Stress Disorder Model Rats. J Mol Neurosci 59, 471–482 (2016). https://doi.org/10.1007/s12031-016-0755-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-016-0755-2